
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Associative vs. dissociative binding of CO2 on M5 transition metal clusters
Nguyen T. T. Le,
Alireza Nazari,
Yash Rele,
Mighila Rixon,
Ishudeep Singh Narula and
Matthew A. Addicoat *
*
School of Science and Technology, Nottingham Trent University, Clifton Lane, Nottingham, NG11 8NS, UK. E-mail: matthew.addicoat@ntu.ac.uk
First published on 9th December 2025
Abstract
Reaction paths were calculated using density functional theory for the reaction of carbon dioxide with a series of transition metal pentamers, M5 + CO2, (M = Nb, Mo, Ru, Rh, Pd, Ag, Pt). A stochastic search algorithm was used to identify geometries with intact CO2, as well as geometries where the CO2 molecule was partly (O + CO) and fully dissociated (O + C + O). Nb5 and Mo5 clusters were found to thermodynamically dissociate CO2. Pd5 and Ag5 were found to leave the CO2 molecule intact, Ru5 could partly dissociate CO2, while for Rh5 and Pt5, the fate of the adsorbed CO2 was dependent on the cluster geometry. The change in the CO2 πu orbital energy in the capture species on initial reaction with the M5 cluster was found to distinguish clusters where CO2 fully dissociated, but could not distinguish clusters where CO2 was found to partly dissociate. The charge transfer to the CO2 molecule at the first transition state, did however, distinguish clusters that fully dissociate CO2, those that partly dissociate CO2 to O + CO, and those that leave CO2 fully intact.
1. Introduction
In recent years, there has been increasing interest in the activation of CO2. Especially in the context of the environment, the ability to activate captured CO2 towards further reaction, turns a problem of needing to store the captured CO21 into a solution – a cheap C1 feedstock.2,3 CO2 is unfortunately well-known for its thermodynamic and kinetic stability, and it is unreactive in the gas phase,4 thus requiring activation via some catalyst species in order to weaken and/or break the CO bonds. Activation of CO2 typically proceeds via introducing electron density into the antibonding π* orbitals of CO2, weakening the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds and allowing the CO2 the molecule to bend away from linear, simultaneously creating a dipole and increasing the reactivity of CO2.5–7
O bonds and allowing the CO2 the molecule to bend away from linear, simultaneously creating a dipole and increasing the reactivity of CO2.5–7
Several manufactured materials including foams,8 CaCO3 microspheres,9 and novel cements10 have been proposed and used to capture and store CO2. More recently, novel materials including, Metal- and Covalent Organic Frameworks,11–13 carbon nanotubes,14 MXenes,15–20 and nanofilms21 have been proposed to not only capture, but to valorize CO2 for further use. CO2 is activated by electron donation into the antibonding π* orbitals of CO2, weakening the CO bonds allowing the CO2 molecule to bend.5–7,22,23 In the majority of these materials above, the active site of the catalyst is a metal atom24 and thus there is considerable interest in the mechanism of CO2 activation by various metals25 and there has been a great volume of spectroscopic and theoretical work over decades26–29 investigating the interaction of CO2 with single transition metal atoms/ions, M+/0/−. A range of CO2 binding motifs have thus been identified, including η1 coordination (via the C atom), bidentate η2 binding (via C, O), and dissociative addition where binding produces CO and O species. Where multiple CO2 molecules adsorb, oxalate formation has also been identified.7,30
In between single atoms and bulk surfaces, transition metal clusters have long been an avenue for the investigation of gas phase reactions, with a variety of substrates including CO,22,31 N2O32 and notably CO2.32,34,35 Size effects are well-known in the cluster regime, with several experimental36 and computational studies37 noting properties varying over several orders of magnitude upon single atom addition.
One study from Mackenzie group illustrated these size effects with respect to CO2 activation, employing small Pt4− and Pt5− anionic clusters. They showed that while Pt4− + CO2 has a dissociative global minimum (i.e. Pt4−(CO)O), the spectroscopically observed species possessed intact CO2 (Pt4−OCO), whereas for the one atom larger cluster anion, Pt5−, infrared multiphoton dissociation (IR-MPD) spectroscopy showed the computationally predicted global minimum, dissociatively bound Pt5−(CO)O exists and in contrast to Pt4−, no evidence of molecularly bound CO2 was seen, despite such a species being predicted within the energy of their cluster source.38
Inspired by this work of Mackenzie et al.,38 and following on from our work on M4 clusters,39 we report a “horizontal” study, investigating the reaction of CO2 with M5 neutral transition metal clusters from niobium through silver and additionally including platinum.
2. Computational method
The computational method employed here is the same as in our previous work, and recapped here.39 Structures of M5 clusters, for M = Nb–Ag (excluding Tc) and Pt were generated using the Kick stochastic structure search procedure with five individual M atoms supplied.40,41 Full searches were undertaken on the lowest possible multiplicity (singlet or doublet) and all minima identified were re-optimized at higher multiplicities. For all metal species, the lowest four multiplicities were calculated, but for ruthenium and rhodium, the search was extended up to the 15-tet and the 12-tet, respectively. No symmetry was imposed at any point in the search, nevertheless, several clusters adopted clear point group symmetry, as evidenced by geometric parameters and frequencies. We chose not to confirm the symmetry by further calculation, as the addition of CO2 would immediately break symmetry.The lowest energy structure of each M5 cluster was then adopted as a fragment in a further stochastic search process. Kick runs were undertaken with the following configurations: M5 + CO2(linear); M5 + CO2(bent); M5 + CO + O; M5 + C + O + O. In order to easily identify the transition state where the CO2 molecule first began to dissociate, a Kick run was also undertaken explicitly searching for transition states, employing the M5 cluster and the bent CO2 molecule as fragments. Additional starting geometries were generated by hand (e.g. CO2 bound to different symmetry-distinct metal atoms, end-on/side-on, linear/bent, µ1/µ2/µ3-bound). From these calculations, the physisorbed “capture” species and the M5CO2 global minimum were identified, and the reaction pathway was then filled in and confirmed by a series of Quasi-Synchronous Transit (QST) and Intrinsic Reaction Coordinate (IRC) calculations.
The zero energy for each M5 + CO2 system is defined as the sum of the energies of the M5 metal cluster in the lowest possible multiplicity and the CO2 molecule. Thus structures with a negative relative energy (below the zero energy) are more stable than the separated reactants; structures with a positive relative energy (energy higher than the zero energy) are unstable with respect to the infinitely separated reactants. basis set superposition error (BSSE) was disregarded, as were zero-point energies and entropic contributions, as previous work that the effect of BSSE and ZPE on relative energies is minimal and that adding entropic effects was found to consistently raise relative energies by ≈0.5 eV at 298 K.31 All structure searches were undertaken with the B3P86 density functional42 and Stuttgart Relativistic Small Core (SRSC) basis set,43–45 as previous studies31,46 have shown this to be an accurate and computationally efficient combination. The final pathways were re-optimized at all relevant multiplicities using the TPSS functional47 with the Def2TZVP basis set48,49 and employing the D3-BJ empirical dispersion term.50 This latter combination, while more expensive, has also been shown to reproduce energetic ordering and vibrational data for reactions of small molecules on gas phase transition metal clusters including Rhn51 and Ptn.52 All calculations were undertaken using the Gaussian 16 software.53 All structures presented are included in the SI (xyz, zip) and absolute and relative energies for each structure at all multiplicities studied are presented in the SI (xlsx).
3. Results and discussion
3.1. Nb5
The Nb5 cluster is an odd-electron species and therefore may be a doublet, quartet,…. The doublet multiplicity was found to be the lowest in energy, in line with previous calculations.54 In slight contrast to the early calculations of Salahub et al. on neutral Nb5, we identified the minimum energy structure to be of C2v symmetry with equatorial–equatorial bond lengths of 2.63 (2) and 2.87 Å and axial–equatorial bond lengths of 2.49 (2) and 2.61 Å. This cluster was used for the CO2 pathway without constraint.Two pathways were identified for the reaction of Nb5 + CO2, shown in Fig. 1 and 2. Corresponding geometric data is shown in Table S1. Both pathways begin with the CO2 molecule approaching the equatorial niobium atoms, and the CO2 molecule bending over one of the triangular faces of the Nb5 cluster. Structures I–V of both pathways are therefore the same. The two pathways diverge with a choice of which CO bond breaks first. In the pathway shown in Fig. 1, the CO bond that is closest to the equatorial plane of the Nb5 cluster breaks first resulting in a CO molecule bound to an axial niobium atom and the lone oxygen atom attached to an equatorial niobium atom. The second pathway, shown in Fig. 2 breaks the other CO bond, resulting in the CO molecule bound to equatorial niobium atoms and the dissociated oxygen atom bound to an axial niobium atom. Both pathways result in very similar global minima, with the CO2 molecule completely dissociated, at relative energies of −6.76 and −6.68 eV respectively. These two structures, Fig. 1-IX and 2-XI, may interconvert, via a transition state shown on the first pathway, Fig. 1-X.
 | ||
| Fig. 1 Stationary points on the Nb5 + CO2 potential energy surface. The doublet multiplicity is shown in bold and the quartet multiplicity is shown with thin lines. Relative energies are given in eV. Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
 | ||
| Fig. 2 Second reaction pathway on the Nb5 + CO2 potential energy surface. The doublet multiplicity is shown in bold and the quartet and sextet multiplicities are shown with thin lines. Relative energies are given in eV. Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
3.2. Mo5
The molybdenum atom is and even-electron species and therefore the Mo5 cluster is also an even-electron species and we investigate the singlet, triplet,… surfaces. Several authors have calculated the structure and properties of the Mo5 cluster using a variety of density functional methods, basis sets and structure search approaches. Plá and Diez identified a singlet global minimum of a capped out-of-plane rhombus, but with singlet and triplet trigonal bipyramid structures both only 0.03 eV per atom higher in energy.55 Vega and coworkers predicted a triplet bipyramid with C2v symmetry,56 while Yin and Chen identified a singlet trigonal bipyramid with C1 symmetry.57 Kantorovich and coworkers used the AIRSS approach58 to identify 9 Mo5 structures within 1 eV of the global minimum, which they predict to be a C2v singlet trigonal bipyramid.59 Sumer and Jellinek identified a similar set of low energy structures, but their global minimum had only C2 symmetry.60 Lei predicted a singlet trigonal bipyramid structure.61 In general agreement with these studies, we identified the global minimum to be a singlet trigonal bipyramid with approximately D3h symmetry and bond lengths of 2.34 Å (axial–equatorial) and 2.83 Å (equatorial–equatorial) and we employed this structure, without symmetry constraint, in our reaction with CO2.A single pathway was identified for the reaction of Mo5 + CO2, it is shown in Fig. 3 and Table S2. The pathway begins with the CO2 molecule approaching the axial molybdenum atoms, and the CO2 molecule breaks over the axial atom of the Mo5 cluster in the first transition state (structure Fig. 3-II). The remaining CO molecule binds in a µ3 fashion to a triangular face of the Mo5 cluster, before twisting, rotating parallel to the face and then dissociating. The Mo5CO2 global minimum therefore has the CO2 molecule fully dissociated with the carbon atom µ3-bound, and the oxygen atoms µ2 and µ1-bound, with a relative binding energy of −4.91 eV with respect to the singlet Mo5 + CO2.
 | ||
| Fig. 3 Reaction pathway of Mo5 + CO2. The singlet potential energy surface is shown in bold and the triplet and quintet multiplicities are shown with thin lines. Relative energies for the singlet surface are given in eV, energies of higher multiplicities are included in SI (xlsx). Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
3.3. Ru5
Ruthenium also has an even number of electrons and therefore we consider the singlet, triplet, quintet and septet multiplicities for the Ru5 cluster. The Ru5 cluster has been calculated previously by different authors and DFT methods,62–65 but there is good agreement amongst researchers that the ground state structure of Ru5 is a square pyramid structure with singlet multiplicity. We similarly identify a square pyramidal singlet structure with bond distances of 2.28 and 2.49 Å for the base–base and base–apex bonds respectively.Two pathways were identified for the Ru5 + CO2 reaction, they are shown in Fig. 4 and 5. Corresponding geometric data is shown in Table S3. The singlet to nonet multiplicities were quite close in energy and so the pathway calculations were extended up to the 15-tet. The CO2 molecule may approach either the apex ruthenium atom (Fig. 4) or one of the base ruthenium atoms (Fig. 5). In both cases, the first CO bond breaks with a barrier below the zero energy defined by E[Ru5(singlet)] + E[CO2], and leaving the CO molecule bound to the apex ruthenium atom. Two candidates for the global minimum Ru5CO2 structure are identified: The first structure has the CO molecule bound in a µ1 geometry to the apex Ru atom and the dissociated oxygen atom µ1-bound to a base Ru atom and in the Ru4 plane. The second geometry has the CO molecule µ2-bound to the Ru(apex)–Ru(base) bond opposite the µ1-bound oxygen atom. These two geometries are interconvertible by a transition state at −2.15 eV (i.e. a 0.49 eV barrier) with an imaginary frequency of 134i cm−1. The barrier to dissociating the second CO bond is 0.27 eV above zero energy for the singlet pathway, but below zero energy for the triplet to the 13-tet multiplicities. The lowest energy structure identified with CO2 completely dissociated has a µ1 and a µ2 oxygen atom and a µ2-bound carbon atom. This structure has an energy of −2.17 eV, 0.47 eV higher in energy than the Ru5OCO global minima.
 | ||
| Fig. 4 Stationary points on the Ru5 + CO2 reaction pathway. The singlet potential energy surface is bolded and the triplet - 15-tet multiplicities are shown with thin lines. Relative energies are given in eV for the singlet multiplicity and included in the SI (xlsx) for all multiplicities. Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
 | ||
| Fig. 5 Alternate reaction path on the Ru5 + CO2 potential energy surface. The singlet multiplicity is shown in bold and the triplet - 15-tet multiplicities are shown with thin lines. Relative energies are given in eV. Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
3.4. Rh5
The Rh5 cluster is an odd-electron species and therefore we consider the doublet – 12-tet multiplicities. Rhodium is of intense interest as a catalyst and many researchers have calculated the Rh5 cluster. In an early study, Kelin and co-workers chose to model a trigonal bipyramid Rh5, but did not investigate other geometries due to computational expense.66 Rubio-Arroyo and coworkers predicted a twisted bowtie structure and two trigonal bipyramidal strucutres.67 Aguilera-Granja et al. predict a square pyramidal Rh5 cluster.62 Futschek et al. predict three Rh5 isomers, a C4v square pyramid and both a “tall” and a “flat” trigonal bipyramid structure, both of D3h symmetry.68 Similarly, Pederson and coworkers present a sextet square pyramid as the ground state, with a trigonal bipyramid being higher in energy at all spin multplicities.69 Nguyen and Pham70 and Poulain and coworkers,32 both employ a square pyramidal Rh5 structure in their reactivity studies with N2O.Our initial search on the doublet surface, yielded a square pyramid ground state, but had a trigonal bipyramid structure only 0.12 eV higher in energy. Re-optimizing these structures at higher multiplicities (up to the 12-tet) showed that the sextet square pyramid was the lowest energy Rh5 structure and that it is isoenergetic with the sextet and octet trigonal bipyramid structures. Therefore both square pyramid and trigonal bipyramid structures were considered for reaction with CO2. In the reaction pathways below, pathways for the doublet – decet surfaces are presented as the 12-tet was higher in energy.
 | ||
| Fig. 6 Stationary points on the Rh5 + CO2 potential energy surface for the square pyramidal Rh5 isomer. Doublet multiplicity is shown in bold and the quartet – decet multiplicities are shown with thin lines. Relative energies of doublet geometries are given in eV and included for all multiplicities in the SI (xlsx). Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
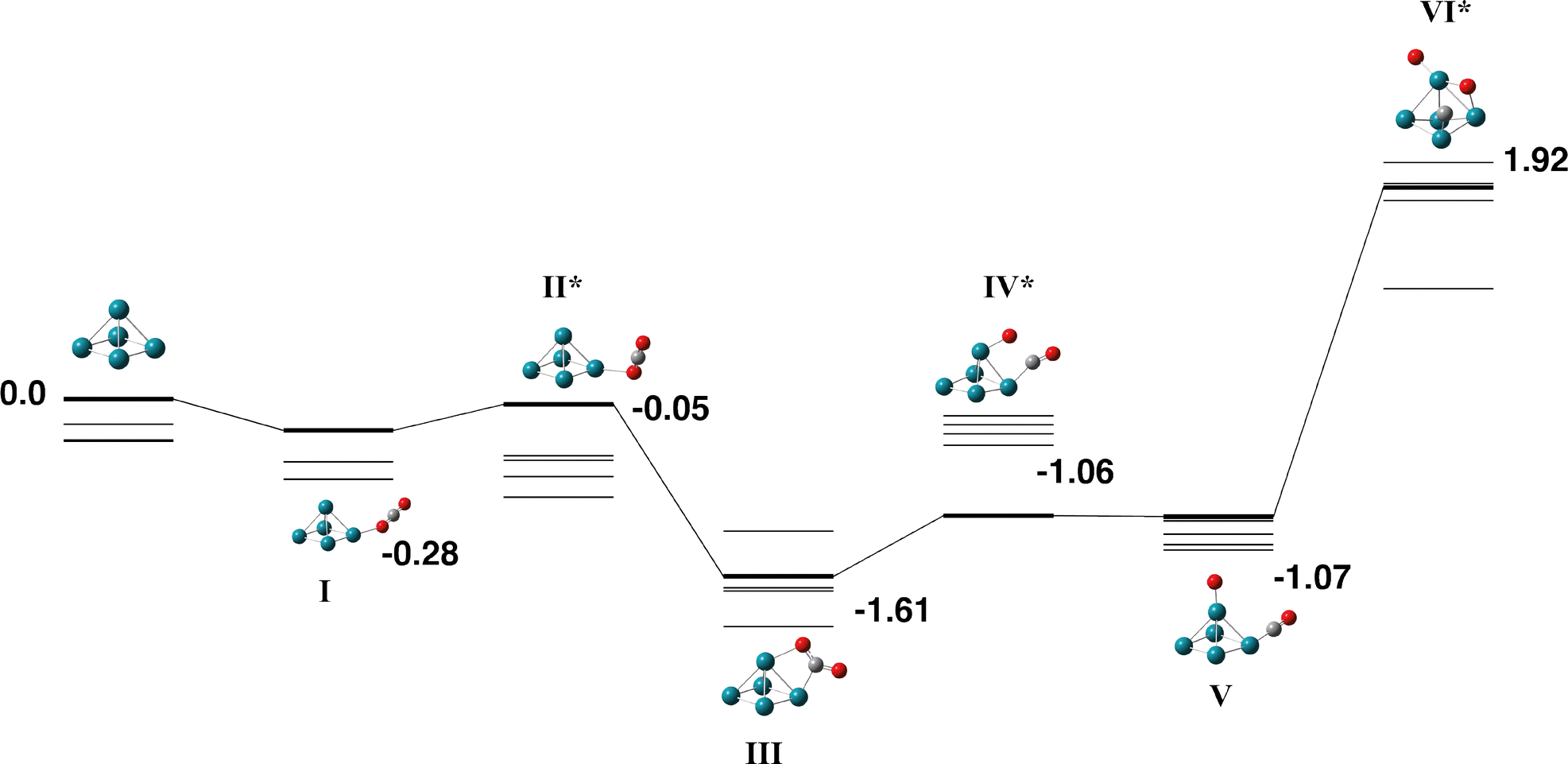 | ||
| Fig. 7 Alternate pathway on the Rh5 + CO2 potential energy surface for the square pyramidal Rh5 isomer. In this pathway, the first CO bond breaking in transition state VI leaves the CO molecule attached to the base Rh atom. The doublet multiplicity is shown in bold and the quartet – decet multiplicities are shown with thin lines. The final transition state leads to the same final structure as Fig. 6 as the CO breaks across the triangular Rh3 face. Relative energies are given in eV. Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
 | ||
| Fig. 8 Reaction pathway for trigonal bipyramidal Rh5 + CO2. The doublet multiplicity is shown in bold and the quartet – decet multiplicities are shown with thin lines. Relative energies shown are for the doublet surface and are given in eV, relative energies for all multiplicities are included in the SI (xlsx). Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
3.5. Pd5
The Pd5 cluster is an even-electron species and therefore we consider the singlet – septet multiplicities. Small Pd clusters have also been calculated by a variety of different methods over many years: Morokuma and coworkers predicted a Pd5 ground state of a triplet trigonal bipyramid Pd5 structure with D3h symmetry but noted a C2v trigonal bipyramid only 0.4 kcal mol−1 higher in energy, and also a triplet C4v square pyramid only 2.3 kcal mol−1 higher in energy.71 Several other authors predict a triplet trigonal bipyramid structure.72–76 Our search also identified a triplet trigonal bipyramid as the global minimum, and while we did not constrain symmetry, the structure has C2v symmetry. We did also identify the square pyramidal structure, the triplet was the lowest energy multiplicity for this structure and was 0.11 eV higher than the triplet trigonal bipyramid. While these two structures are sufficiently close in energy such that they are likely to coexist, we choose only the trigonal bipyramid to react with CO2.The Pd5 + CO2 reaction pathway is shown in Fig. 9 and Table S6, the quintet and septet pathways were high in energy and so only the singlet and triplet are shown. The capture species and adjacent transition state (structures Fig. 9-I and II) are shown for both an apex atom approach and an equatorial atom approach. Both approaches result in a bent CO2 molecule weakly bound to the central Pd(apex)–Pd(equatorial) bond with a binding energy of −1.34 eV relative to the zero energy. This is the global minimum structure for the pathway, breaking the first of the two CO bonds requires +1.08 eV and the barrier to breaking the second CO bond is +4.45 eV.
 | ||
| Fig. 9 Stationary points on the Pd5 + CO2 potential energy surface. The singlet multiplicity is shown in bold and the triplet multiplicity is shown with thin lines. Relative energies corresponding to singlet geometries are given in eV, all relative energies are included in SI (xlsx). Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
3.6. Pt5
The 5d transition metal, platinum was also studied due to its intense interest and frequent use as a catalyst. The Pt5 cluster is studied at the singlet – septet multiplicities. A variety of structures have been obtained for the Pt5 cluster with significant differences in predicted energetics and several authors debating whether the cluster is planar or three-dimensional.77 Sumer and Jellinek identified several Pt5 low-energy isomers, including a planar edge-capped square, trigonal bipyramid, edge-capped tetrahedron, square pyramid, planar trapezoidal and planar bowtie. On inclusion of spin–orbit effects, only the first three structures remained.60 Kleinman and coworkers also studied the effect of spin–orbit coupling identifying the planar edge-capped square, followed by square pyramid, trigonal bipyramid, trapezoidal and bowtie structures.78 Wang et al. identified a quintet trigonal bipyramid as their gas-phase global minimum.79 Sebetci predicts a edge-capped tetrahedron global minimum, followed by a distorted rhombus, a trigonal bipyramid, a bowtie and trapezoidal structures.80 Grönbeck and Andreoni noted that the class of density functional had a strong effect on the predicted isomer, with a planar edge-capped square favoured by BLYP whereas a distorted square pyramid was predicted by LSDA.81 Singh et al. and Kumar and Kawazoe both predict the planar edge-capped square as the global minimum,82,83 but Cao identified a square pyramidal structure as the basis of their reactivity study.84 MRSDCI calculations of Majumdar et al. identified a distorted tetragonal pyramid.85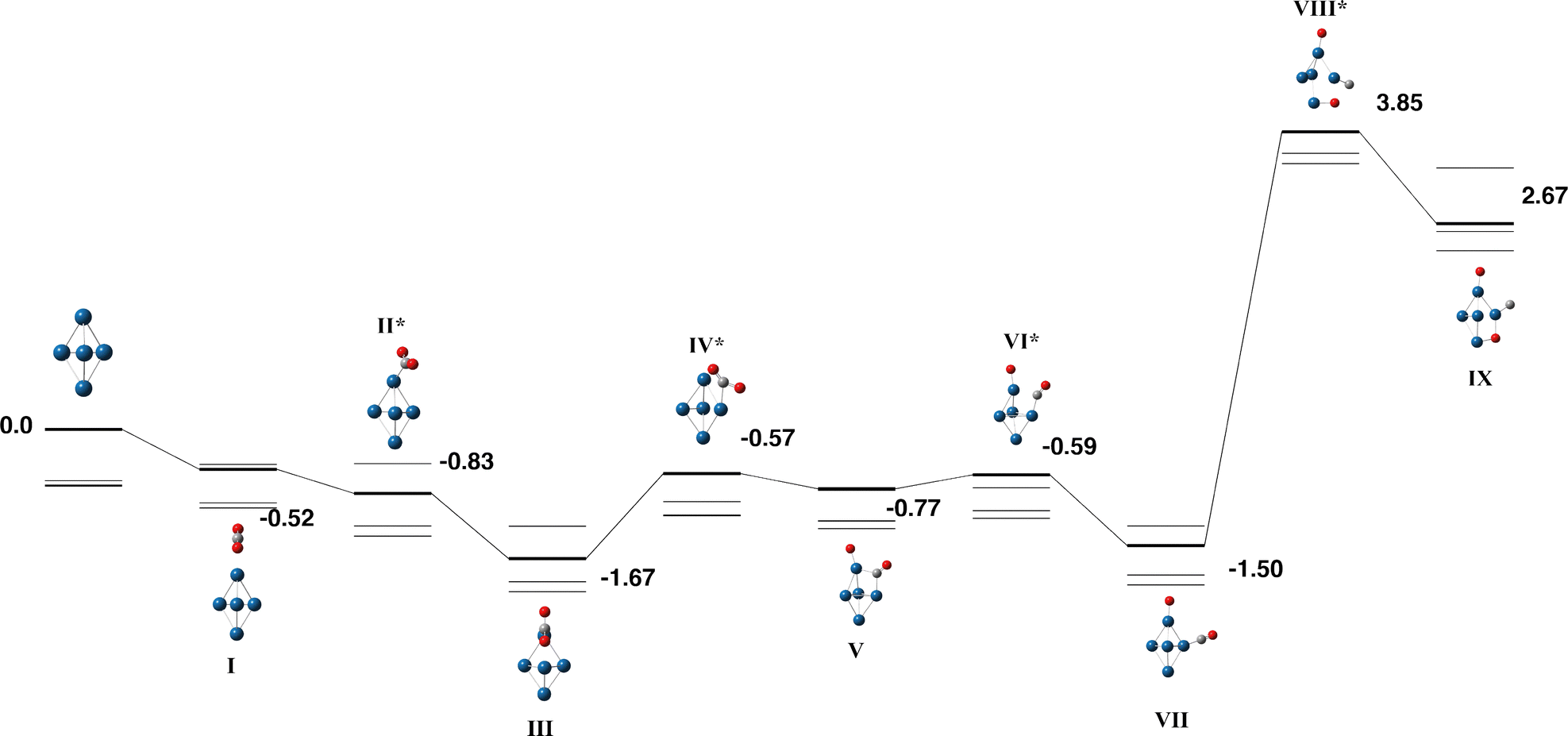 | ||
| Fig. 10 Stationary points on the Pt5 + CO2 potential energy surface beginning with the trigonal bipyramidal Pt5 isomer. The singlet multiplicity is shown in bold and the triplet – septet multiplicities are shown with thin lines. Relative energies for the singlet surface are given in eV, relative energies for all multiplicities are included in SI (xlsx). Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
 | ||
| Fig. 11 Stationary points on the Pt5 + CO2 potential energy surface for the square pyramidal Pt5 isomer. The singlet multiplicity is shown in bold and the triplet – septet multiplicities are shown with thin lines. Singlet relative energies are given in eV, other multiplicities are included in SI (xlsx). Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
3.7. Ag5
The Ag5 cluster is an odd-electron species and we consider the doublet – octet multiplicities. Atolabi et al. conducted one of several studies into the gas-phase Ag5 structure and predicted that the doublet planar trapezoidal structure was 0.49 eV lower in energy than the doublet trigonal bipyramid structure.86 The B3PW91 calculations of Hisayoshi and coworkers predicted a similar energy difference of 0.53 eV.87 Fournier,88 Wang and coworkers,89 and Koutecký and coworkers all predict a C2v doublet trapezoidal structure.90 Fazli and coworkers also employed a trapezoidal Ag5 cluster in their DFT study. Recent synthetic advances have produced a ligand-free trigonal bipyramid Ag5 cluster,91 which has been tested as an anti-tumour agent.92 The same cluster was employed by Atolaibi et al. in their DFT-study with CO2, CH4, and H2O molecules. While these authors considered the trapezoidal Ag5 isomer for reaction with CH4 and indicated that the trapezoidal isomer was more stable, they only calculated the trigonal bipyramid for their calculations with CO2, showing weak interaction of the CO2 molecule with both the apex and equatorial silver atoms.93The trapezoidal Ag5 structure was chosen as the basis for the reaction path search, and the resulting pathway for the reaction of Ag5 + CO2 is shown in Fig. 12 with geometric data tabulated in Table S9. The doublet surface only is presented as all higher multiplicities were more than 1 eV higher in energy. CO2 interacts only very weakly with the Ag5 cluster, the capture species has the CO2 molecule interacting with one of the atoms on the short edge of the trapezoid and is only −0.13 eV below zero energy. Only one other structure, Fig. 12-III was below zero energy, with an energy of −0.29 eV. Attempting to break the first CO2 bond requires +1.77 eV. Searches were made for structures with the CO2 molecule dissociated (e.g. CO + O, C + 2(O)), and an IRC was started from the transition state Fig. 12-IV, but in all these calculations the Ag5 cluster did not remain intact and therefore we did not continue the reaction path.
 | ||
| Fig. 12 Reaction pathway for the Ag5 + CO2 doublet potential energy surface. Other multiplicities are high in energy and are not shown. Relative energies are given in eV, and are included for higher multiplicities in the SI (xlsx). Metal atoms are shown in blue/green, oxygen atoms are shown in red and the carbon atom is shown in grey. | ||
3.8. Periodic trends
A previous computational study on the reaction of carbon monoxide, CO, with second-row transition metal timers showed that capture species were very similar in energy across the periodic table, with the exception of silver clusters, which bound CO only weakly.31 This was observed similarly here for the M5 clusters with CO2, and is shown in Fig. 13. The consistent energy of the capture species can be rationalized in both cases that the capture species is defined as the first interaction of the two species and represents physisorption, where the chemical/electronic structure of the two species is barely perturbed. This is also consistent with the axial and equatorial, or apex and base capture species having similar interaction energies, where both were identified for the one M5 cluster. | ||
| Fig. 13 Plot of the relative energies of capture, lowest energy associative (OCO), lowest energy partly dissociated (O,CO) and fully dissociative (O,C,O) structures of M5 metal clusters in the lowest possible multiplicity. | ||
In the previous study of the interaction of M3 + CO, there were only two other species of interest, the associative and dissociatively bound species.31 In the case of CO2 as a reactant, there are now three species of interest: the lowest energy bound OCO, the lowest energy O + CO and the lowest energy fully dissociated 2(O) + C structure. The energies of the associatively bound OCO species are relatively similar across the periodic table, ranging from −2.463 eV (Nb5) to −1.340 eV (Pd5), the two exceptions were Ag5, which binds weakly, with a Ag5⋯OCO binding energy of only −0.289 eV, and Mo5, where CO2 molecule dissociates via interaction with the axial atom of the cluster only. In structure Fig. 3-II, the C⋯O distance is 1.80 Å and the θOCO angle is 122°. The lowest energy associative structure for Mo5CO2 is therefore the capture species.
Similar trends are observed for the energies of the partly dissociative (O + CO) and fully dissociative (2(O) + C) structures. Both energies rise moving left to right across the second row of transition metals (Nb–Ag). The M5O + CO energies become disfavourable (positive) for Pd5, while the fully dissociated structures are disfavoured to the right of Rh5 inclusive. The lone third row pentamer in this study, Pt5, favourably dissociates CO2 to CO + O, but the fully dissociative structure is disfavoured with respect to the zero energy, Pt5 + CO2.
Table 1 shows the calculated vibrational frequencies of CO2 in the capture species for each M5 cluster. The two bending frequencies, now non-degenerate, are red-shifted by ≈60 cm−1, but no trend was observed for this reduction. The frequencies of the symmetric and asymmetric CO2 stretches do not change significantly on interaction with the M5 cluster. To attempt a diagnostic for the fate of CO2, the energy of the CO2 πu orbital and the ΔE, with respect to the calculated value for free CO2 (−0.03280 a.u.) is also shown in Table 1. For Nb5 and Mo5, a strong reduction in the orbital energy of ≈0.05 a.u. is observed. Both of these clusters dissociate CO2 fully. All other M5 clusters lowered the CO2 πu orbital energy by ≈0.02 a.u. and no distinction between clusters that possess a fully intact CO2 in their minimum energy structure vs. clusters that partly dissociate CO2 to O + CO was observed. Nevertheless, the orbital energy does indicate those clusters that fully dissociate CO2 into atoms.
| System | Bend 1 | Bend 2 | Symm stretch | Asym stretch | E(M5,HOMO) | E(M5,LUMO) | E(πu) | ΔE(πu) | Eads | Eint |
|---|---|---|---|---|---|---|---|---|---|---|
| (cm−1) | (eV) | |||||||||
| CO2 | 622 | 622 | 1283 | 2319 | −0.893 | 0.0 | ||||
| Nb5 | 554 | 577 | 1277 | 2341 | −4.332 | −2.730 | −2.351 | −1.458 | −0.483 | −0.484 |
| Mo5 | 560 | 560 | 1279 | 2340 | −4.388 | −2.406 | −2.383 | −1.491 | −0.438 | −0.439 |
| Ru5 apex | 521 | 557 | 1259 | 2323 | −5.655 | −2.817 | −1.394 | −0.502 | −0.361 | −0.370 |
| Ru5 base | 462 | 585 | 1254 | 2326 | −4.637 | −2.808 | −1.476 | −0.583 | −0.462 | −0.466 |
| Rh5 sq. py. | 546 | 571 | 1268 | 2325 | −4.970 | −3.471 | −1.455 | −0.563 | −0.282 | −0.452 |
| Rh5 tri. bipy. | 528 | 571 | 1266 | 2328 | −5.028 | −3.226 | −1.446 | −0.554 | −0.188 | −0.499 |
| Pd5 a | 583 | 587 | 1281 | 2326 | −5.304 | −4.239 | −1.512 | −0.620 | −0.301 | −0.346 |
| Pd5 b | 581 | 587 | 1278 | 2325 | −5.373 | −4.252 | −1.609 | −0.716 | −0.368 | −0.369 |
| Ag5 | 593 | 605 | 1281 | 2314 | −5.074 | −2.813 | −1.379 | −0.487 | −0.133 | −0.133 |
| Pt5 tri. bipy. | 552 | 564 | 1277 | 2336 | −5.606 | −4.416 | −1.742 | −0.849 | −0.516 | −0.547 |
| Pt5 sq. py. | 551 | 570 | 1261 | 2310 | −5.466 | −4.272 | −1.536 | −0.643 | −0.360 | −0.377 |
Table 2 shows the Hirshfeld charges on CO2 molecule in the capture species and the first transition state for each of the reaction pathways. The q(CO2) for the physisorbed capture species, as in the previously presented M4 clusters,39 is consistent and positive (≈0.1) for all M5 clusters, except Ru5 with CO2 bound to the apex atom, suggesting that this species is actually a chemisorbed species. Considering the charge transfer observed in the first transition state, the same diagnostic holds for M5 clusters also as the M4 clusters; For the left-most three clusters, full CO2 dissociation is thermodynamically possible, though for Ru5, this would require surmounting a +0.27 eV barrier on the singlet surface, but a below zero energy barrier on higher multiplicity surfaces (see SI spreadsheet for details) having q(CO2) < −0.35 e−. Right-most clusters, Pd5 and Ag5, do not dissociate either CO2 bond and have low back-donation to CO2, < 0.2e−, and clusters that likely activate CO2 without fully dissociating it, Rh4, Pt4, have intermediate q(CO2) values.
| System | Capture species q(CO2) | TS 1 q(CO2) |
|---|---|---|
| Nb5 | 0.142 | −0.418 |
| Mo5 | 0.118 | −0.494 |
| Ru5 apex | −0.381 | −0.3747 |
| Ru5 base | 0.098 | −0.434 |
| Rh5 sq. py. | 0.094 | −0.336 |
| Rh5 tri. bipy. | 0.106 | −0.314 |
| Pd5 a | 0.086 | 0.037 |
| Pd5 b | 0.088 | 0.008 |
| Ag5 | 0.043 | −0.112 |
| Pt5 tri. bipy. | 0.121 | −0.247 |
| Pt5 sq. py. | 0.095 | −0.332 |
Fig. 14 shows the barrier heights (transition state energies) for two key transition states in each pathway, corresponding to the breaking of the first and second CO bond. The trends seen are broadly similar to the equivalent M4 clusters.39 The left-right divide previously seen for both barriers between Ru/Rh, is slightly less clear for the M5 clusters, with both Ru5 and Rh5 being likely to break the first CO bond, but not the second. The softer barriers for Pt5 compared to Pd5 also suggest that the first CO bond would break on this cluster, leading to O + CO products on the cluster surface in this case also. Han and coworkers studied a range of mono and bimetallic metal surfaces and made similar conclusions, that activation energies for CO2 dissociation increase left to right across the periodic table, with Au, Ag, and Pd based alloys having CO2 dissociation barriers >1.50 eV.18
 | ||
| Fig. 14 Plot of the relative energies of barriers to dissociation of the first (blue) and second (orange) C⋯O bonds for M5CO2 reaction pathways on the lowest multiplicity (singlet or doublet) surface. | ||
3.9. M4 vs. M5 clusters
Each reaction path presented here and for the equivalent M4 clusters39 may be distilled into a profile containing six species; the capture species, lowest energy MnCO2, TSO⋯CO, lowest energy MnO·CO, TSC⋯O and lowest energy MnO·C·O. These abbreviated reaction profiles are shown in Fig. S1–S7. Reactions on M5 surfaces are typically more exothermic than the equivalent M4 clusters. Excluding palladium, increasing the cluster size lowers the energy of the first barrier by ≈0.5 eV. This is consistent with the B3LYP results for Zr4 and Zr5 clusters computed by Ghanty and coworkers.33 The height of the second barrier on adding the fifth metal atom is less consistent, lowering for Nb, Mo and Rh, but rising for the other metals. This is consistent with the left hand metals (Nb, Mo) dissociating CO2 fully, right side metals (Pd, Ag, Pt) keeping CO2 intact, and central metals (Ru, Rh) showing intermediate behaviour, which is therefore the most tunable by alteration of the cluster size.4. Conclusions
We have explored the chemistry of CO2 reaction on a series of M5 transition metal clusters, by deriving reaction pathways using Density Functional Theory. Rh5 and Pt5 clusters had multiple competitive ground state geometries, and reaction paths were derived for each, noting that the trigonal bipyramid and square pyramidal geometries may interconvert.Moving from left to right across the M5 series, the energies of the capture species remained relatively constant as expected for the minimally interacting, physisorbed species. The energies of the lowest energy associatively-bound species with CO2 fully intact also fell within a narrow range of −2.463 to −1.340 eV, generally rising from Nb5 to Ag5. The energetics of the partly (O + CO), and fully (O + C + O) dissociated species define the outcome of the reaction, with both rising strongly as one moves to the right of the periodic table. The partly dissociated species was found to be disfavoured with respect to the separated M5 +CO2 reactants for Pd5, Ag5 and Pt5, while the fully dissociated species was disfavoured from Rh5 and could not be located for Ag5.
The energy of the CO2 πu orbital was found to distinguish those structures that dissociate CO2 fully (Nb5 and Mo5) from those that do not, but could not resolve clusters that partly dissociate CO2 from those that leave CO2 fully intact. The magnitude of charge transfer to the CO2 fragment was found to be diagnostic with strong (>0.35e), weak (<0.2e) and intermediate values indicating full CO2 dissociation, no dissociation, and activation respectively.
Author contributions
NTTY, AN, YR, MR, IN: investigation, writing – original draft MAA: conceptualization of this study, methodology, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: geometric parameters (pdf), structures (xyz) and energies, vibrational data (xlsx) for all pathways. See DOI: https://doi.org/10.1039/d5cp03418c.Acknowledgements
MAA is grateful for HPC time via the UK Materials and Molecular Modelling Hub via grant no. (EP/T022213).References
- Y. Peng, J. Gao, Y. Zhang, J. Zhang, Q. Sun, Q. Du, Z. Tang and T. Zhang, J. Energy Storage, 2023, 58, 106286 CrossRef.
- X. Zhang, G. Liu, K.-H. Meiwes-Broer, G. Ganteför and K. Bowen, Angew. Chem., Int. Ed., 2016, 55, 9644–9647 CrossRef CAS PubMed.
- Z. Yang, C. Yue, W. Pang, H. Wang, X. Cong, S. Huang, J. Qi, J. Lv, M.-Y. Wang and X. Ma, Chem. Eng. Sci., 2025, 317, 122104 CrossRef CAS.
- M. Aresta, A. Dibenedetto and E. Quaranta, Reaction Mechanisms in Carbon Dioxide Conversion, Springer Berlin, Heidelberg, 2015, pp. 1–409 Search PubMed.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef CAS PubMed.
- J. M. Weber, Int. Rev. Phys. Chem., 2014, 33, 489–519 Search PubMed.
- M. C. Thompson, J. Ramsay and J. M. Weber, J. Phys. Chem. A, 2017, 121, 7534–7542 CrossRef CAS PubMed.
- T. Foyen, Z. P. Alcorn, M. A. Ferno, A. Barrabino and T. Holt, J. Pet. Sci. Eng., 2021, 196, 107651 CrossRef.
- X. Meng, L. Zhao, H. Guo, F. Sha, H. Shi, Z. Wu and J. Zhang, Crystals, 2019, 9, 433 CrossRef CAS.
- R. Shahbaz and F. Bahadori, Sustain. Chem. Pharm., 2025, 46, 102099 CrossRef CAS.
- S. Kumari, M. Gusain, B. Y. Lamba and S. Kumar, J. Mater. Chem. A, 2025, 13, 21352–21388 RSC.
- J. Jang, E. P. Delmo, W. Chen, Z. Sun, D. H. C. Wan, Y. Liu, S. Zhu, Y. Wang, T. Li, H. Huang, J. Ge and M. Shao, Carbon Energy, 2025, e70019 CrossRef CAS.
- Y. Ren, H. Liu, F. Duan, S. Lu, X. Chen and M. Du, J. Mater. Chem. A, 2025, 13, 24988 RSC.
- B. Liu, J. A. Wang and D. Niu, Carbon, 2025, 243, 120562 CrossRef CAS.
- Y.-X. Yu, J. Colloid Interface Sci., 2025, 695, 137799 CrossRef CAS PubMed.
- Á. Morales-Garca, A. Fernández-Fernández, F. Viñes and F. Illas, J. Mater. Chem. A, 2018, 6, 3381–3385 RSC.
- I. Persson, J. Halim, H. Lind, T. W. Hansen, J. B. Wagner, L.-Å. Näslund, V. Darakchieva, J. Palisaitis, J. Rosen and P. O. Å. Persson, Adv. Mater., 2019, 31, 1805472 CrossRef PubMed.
- J. Ko, B.-K. Kim and J. W. Han, J. Phys. Chem. C, 2016, 120, 3438–3447 CrossRef CAS.
- J. Yang, L. Sun, Z. He, W. Xu and J. Peng, J. Mater. Sci., 2025, 60, 4669–4686 CrossRef CAS.
- A. Jurado, K. Ibarra, Á. Morales-Garca, F. Viñes and F. Illas, ChemPhysChem, 2021, 22, 2456–2463 CrossRef CAS PubMed.
- J. Li, J. Jia, X. Wang, X. Wang, J. Wang, C. Zhang, Y. Bai and X. Zhang, Appl. Surf. Sci., 2025, 710, 163887 CrossRef CAS.
- V. K. Ocampo-Restrepo, L. Zibordi-Besse and J. L. F. Da Silva, J. Chem. Phys, 2019, 151, 214301 CrossRef PubMed.
- J. M. Weber, Int. Rev. Phys. Chem., 2014, 33, 489–519 Search PubMed.
- H. Liu, L. An, P. Wang, C. Yu, J. Zhang, H. Shin, B. Peng, J. Li, M. Li, H. An, J. Yu, Y. Chen, P. Wang, K.-S. Lee, K. Lalit, Z. Liu, O. K. Farha, W. Huang, J. Z. Liu, L. Qi, K. Xie and E. H. Sargent, Nat. Commun., 2025, 16, 6185 Search PubMed.
- H. Schwarz, Coord. Chem. Rev., 2017, 334, 112–123 CrossRef CAS.
- M. Sievers and P. Armentrout, Int. J. Mass Spec., 1998, 179–180, 103–115 CrossRef.
- A. M. Ricks, A. D. Brathwaite and M. A. Duncan, J. Phys. Chem. A, 2013, 117, 11490–11498 CrossRef CAS PubMed.
- Z. Zhao, X. Kong, D. Yang, Q. Yuan, H. Xie, H. Fan, J. Zhao and L. Jiang, J. Phys. Chem. A, 2017, 121, 3220–3226 CrossRef CAS PubMed.
- A. Iskra, A. S. Gentleman, A. Kartouzian, M. J. Kent, A. P. Sharp and S. R. Mackenzie, J. Phys. Chem. A, 2017, 121, 133–140 CrossRef CAS PubMed.
- M. C. Thompson, L. G. Dodson and J. M. Weber, J. Phys. Chem. A, 2017, 121, 4132–4138 CrossRef CAS PubMed.
- M. A. Addicoat, M. A. Buntine, B. Yates and G. F. Metha, J. Comput. Chem., 2008, 29, 1497–1506 CrossRef CAS PubMed.
- O. Olvera-Neria, R. Avilés, H. Francisco-Rodrguez, V. Bertin, R. Garca-Cruz, J. C. González-Torres and E. Poulain, J. Mol. Mod., 2015, 21, 80 CrossRef PubMed.
- Megha, K. Mondal, A. Banerjee and T. K. Ghanty, Phys. Chem. Chem. Phys., 2020, 22, 16877–16886 RSC.
- M. N. Collacique, V. K. Ocampo-Restrepo and J. L. F. Da Silva, J. Chem. Phys, 2022, 156, 124106 CrossRef CAS PubMed.
- V. K. Ocampo-Restrepo, L. G. Verga and J. L. F. Da Silva, J. Phys. Chem. C, 2021, 125, 26296–26306 CrossRef CAS.
- A. Yamada, K. Miyajima and F. Mafune, Phys. Chem. Chem. Phys., 2012, 14, 4188–4195 RSC.
- P. C. D. Mendes, V. K. Ocampo-Restrepo and J. L. F. Da Silva, Phys. Chem. Chem. Phys., 2020, 22, 8998–9008 RSC.
- A. E. Green, J. Justen, W. Schöllkopf, A. S. Gentleman, A. Fielicke and S. R. Mackenzie, Angew. Chem., Int. Ed., 2018, 57, 14822–14826 CrossRef CAS PubMed.
- S. Sherif, B. A. Velmurugan, N. Abbas, M. Shaikh and M. A. Addicoat, Phys. Chem. Chem. Phys., 2025, 27, 1 RSC.
- M. A. Addicoat and G. F. Metha, J. Comput. Chem., 2009, 30, 57–64 CrossRef CAS PubMed.
- M. A. Addicoat, S. Fukuoka, A. J. Page and S. Irle, J. Comput. Chem., 2013, 34, 2591–2600 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys, 1993, 98, 5648–5652 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys, 1985, 82, 270–283 CrossRef CAS.
- W. R. Wadt and P. J. Hay, J. Chem. Phys, 1985, 82, 284–298 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys, 1985, 82, 299–310 CrossRef CAS.
- M. A. Addicoat, K. F. Lim and G. F. Metha, J. Chem. Phys, 2012, 137, 034301 CrossRef PubMed.
- J. Tao, J. P. Perdew, V. N. Staroverov and G. E. Scuseria, Phys. Rev. Lett., 2003, 91, 146401 CrossRef PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- E. M. Cunningham, A. S. Gentleman, P. W. Beardsmore and S. R. Mackenzie, Phys. Chem. Chem. Phys., 2019, 21, 13959–13967 RSC.
- G. Meizyte, A. E. Green, A. S. Gentleman, S. Schaller, W. Schöllkopf, A. Fielicke and S. R. Mackenzie, Phys. Chem. Chem. Phys., 2020, 22, 18606–18613 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian-16 Revision B.01, 2016, Gaussian Inc., Wallingford CT Search PubMed.
- L. Goodwin and D. R. Salahub, Phys. Rev. A, 1993, 47, R774–R777 CrossRef CAS PubMed.
- J. Del Pla and R. Pis Diez, J. Phys. Chem. C, 2016, 120, 22750–22755 CrossRef CAS.
- M. Ziane, F. Amitouche, S. Bouarab and A. Vega, J. Nano. Res., 2017, 19, 380 CrossRef.
- Y.-H. Yin and J. Chen, Comput. Theor. Chem., 2022, 1212, 113720 CrossRef CAS.
- C. J. Pickard and R. J. Needs, J. Phys. Cond. Mat., 2011, 23, 053201 CrossRef PubMed.
- Y. Wei, V. Veryazov and L. Kantorovich, APL Mater., 2024, 12, 031127 CrossRef CAS.
- A. Sumer and J. Jellinek, J. Chem. Phys, 2022, 157, 034301 CrossRef CAS PubMed.
- L. Xue-Ling, Chin. Phys. B, 2010, 19, 107103 CrossRef.
- F. Aguilera-Granja, L. C. Balbás and A. Vega, J. Phys. Chem. A, 2009, 113, 13483–13491 CrossRef CAS PubMed.
- Y.-C. Bae, H. Osanai, V. Kumar and Y. Kawazoe, Mater. Trans., 2005, 46, 159–162 CrossRef CAS.
- W. Zhang, H. Zhao and L. Wang, J. Phys. Chem. B, 2004, 108, 2140–2147 CrossRef CAS.
- G.-X. Ge, H.-X. Yan, Q. Jing and Y.-H. Luo, J. Cluster Sci., 2011, 22, 473–489 CrossRef CAS.
- Y. Jinlong, F. Toigo and W. Kelin, Phys. Rev. B, 1994, 50, 7915–7924 CrossRef CAS PubMed.
- M. A. Mora, M. A. Mora-Ramírez and M. F. Rubio-Arroyo, Int. J. Quant. Chem., 2010, 110, 2541–2547 CrossRef CAS.
- T. Futschek, M. Marsman and J. Hafner, J. Phys. Condens. Matter, 2005, 17, 5927 CrossRef CAS.
- C.-H. Chien, E. Blaisten-Barojas and M. R. Pederson, Phys. Rev. A, 1998, 58, 2196–2202 CrossRef CAS.
- H. M. T. Nguyen and N. T. T. Pham, J. Phys. Chem. C, 2014, 118, 28562–28571 CrossRef CAS.
- J. Moc, D. G. Musaev and K. Morokuma, J. Phys. Chem. A, 2003, 107, 4929–4939 CrossRef CAS.
- X. Xing, A. Hermann, X. Kuang, M. Ju, C. Lu, Y. Jin, X. Xia and G. Maroulis, Sci. Rep., 2016, 6, 19656 CrossRef CAS PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. B, 2002, 66, 144413 Search PubMed.
- M. Moseler, H. Häkkinen, R. N. Barnett and U. Landman, Phys. Rev. Lett., 2001, 86, 2545–2548 CrossRef CAS PubMed.
- B. Kalita and R. C. Deka, J. Chem. Phys, 2007, 127, 244306 CrossRef PubMed.
- Y. Mu, Y. Han, J. Wang, J.-G. Wan and G. Wang, Phys. Rev. A: At., Mol., Opt. Phys., 2011, 84, 053201 CrossRef.
- L. Xiao and L. Wang, J. Phys. Chem. A, 2004, 108, 8605–8614 Search PubMed.
- M. N. Huda, M. K. Niranjan, B. R. Sahu and L. Kleinman, Phys. Rev. A, 2006, 73, 053201 CrossRef.
- Y. Wang, B. Xiang, H.-Q. Yang and C.-W. Hu, ACS Omega, 2017, 2, 3250–3259 Search PubMed.
- A. Sebetci, Chem. Phys., 2006, 331, 9–18 Search PubMed.
- H. Grönbeck and W. Andreoni, Chem. Phys., 2000, 262, 1–14 CrossRef.
- N. B. Singh and U. Sarkar, J. Mol. Mod., 2014, 20, 2537 CrossRef PubMed.
- V. Kumar and Y. Kawazoe, Phys. Rev. B, 2008, 77, 205418 CrossRef.
- P. Cao and H. Wang, J. Phys. Conf. Ser., 2021, 1992, 032151 CrossRef CAS.
- D. Majumdar, D. Dai and K. Balasubramanian, J. Chem. Phys., 2000, 113, 7928–7938 Search PubMed.
- M. Alotaibi, Q. Wu and C. Lambert, Appl. Surf. Sci., 2023, 613, 156054 CrossRef CAS.
- T. Yumura, M. Kumondai, Y. Kuroda, T. Wakasugi and H. Kobayashi, RSC Adv., 2017, 7, 4950–4959 Search PubMed.
- R. Fournier, J. Chem. Phys., 2001, 115, 2165–2177 Search PubMed.
- Y. Wang and X. G. Gong, Eur Phys. J. D, 2005, 34, 19–22 CrossRef.
- V. Bonačič-Koutecký, L. Češpiva, P. Fantucci and J. Koutecký, J. Chem. Phys., 1993, 98, 7981–7994 CrossRef.
- I. R. Arias, D. Buceta, G. Barone, M. C. Giménez-López, H. Lozano, M. Lazzari and M. Arturo López-Quintela, J. Colloid Interface Sci., 2022, 628, 437–447 CrossRef CAS PubMed.
- V. Porto, D. Buceta, B. Domínguez, C. Carneiro, E. Borrajo, M. Fraile, N. Davila-Ferreira, I. R. Arias, J. M. Blanco, M. C. Blanco, J. M. Devida, L. J. Giovanetti, F. G. Requejo, J. C. Hernández-Garrido, J. J. Calvino, M. López-Haro, G. Barone, A. M. James, T. García-Caballero, D. M. González-Castaño, M. Treder, W. Huber, A. Vidal, M. P. Murphy, M. A. López-Quintela and F. Domínguez, Adv. Func. Mater., 2022, 32, 2113028 CrossRef CAS.
- M. Alotaibi, T. Alotaibi, M. Alshammari and A. K. Ismael, Crystals, 2023, 13, 1691 CrossRef CAS.
| This journal is © the Owner Societies 2026 |