
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemical softness as a predictor for reactivity at metal surfaces
Amy L.
Gunton
 and
Stephen J.
Jenkins
*
and
Stephen J.
Jenkins
*
Yusuf Hamied Department of Chemistry, University of Cambridge, Lensfield Road, Cambridge CB2 1EW, UK. E-mail: sjj24@cam.ac.uk
First published on 3rd March 2026
Abstract
The problem of identifying and characterising the active sites for heterogeneous catalysis can be addressed by employing descriptors of reactivity, such as the local softness. A new method of calculating the local softness for metal surfaces was recently developed and in the present work has been applied to eighteen flat and stepped metal surfaces. The local softness has been visualised and shown to give insight into the potential reactivity of different sites. A more granular local measure of the softness – the atomic softness – has also been calculated and used to predict CO adsorption trends.
I Introduction
The quantification of selectivity in surface chemistry is important for designing better heterogeneous catalysts. Benefits of gaining insight into selectivity may include higher yields of desired products for lower energy cost, with resulting environmental benefits. There are various approaches that can be used to study selectivity, such as by controlling and varying the temperature, pressure and flow of reactants across a catalyst, and studying the synthesis of products.1 The surface science approach to catalysis involves modelling a catalyst surface (which would generally consist of nanoparticles on a support) using a single facet of a given metal.2 This has been shown to yield good results for a range of reactions.2,3 One of the reasons that this approach has been so successful may be that many reactions are structure-sensitive, which means that the majority of catalytic turnover occurs on only a few types of surface feature, known as active sites.4 If a facet containing the active sites is identified, therefore, then instead of modelling an entire nanoparticle (which may be experimentally complex and/or computationally expensive) it is possible to model only the facet that exhibits the sites of interest. For individual reactions on a particular surface, computational methods such as density functional theory (DFT) can be employed to calculate orbitals and transition states that can then yield mechanistic insight.5–7 However, this approach is specific to one metal surface and reaction at a time, and it is difficult to generalise to predicting what might occur on different metal surfaces and for different reactions.In order to gain a general insight into broad trends in catalytic activity between different facets of different metals, reactivity indices can be used. One of the simplest forms of reactivity index would be to look at the reaction energy on the surface at a given site. It has been shown that there is a correlation between the reaction energy and the catalytic activity.8–10 Specifically, the activation energy of a reaction has been shown to scale linearly with the reaction energy.8–10 These linear correlations are known as Brønsted–Evans–Polanyi (BEP) relations.11 The adsorption energy of reactants and products can also be used as a reactivity index,12 and it has been shown that maximum catalytic activity will occur for metal surfaces that neither bind the products too strongly (therefore not allowing the products to leave) nor bind the reactants too weakly (which would mean the reaction would not be catalysed).13,14 These relations may be summarised in volcano plots, highlighting that optimal catalytic activity is obtained for catalysts of intermediate reactivity.11
However, all the measures discussed above are specific to a given metal and adsorbate system. A different type of reactivity index could give general insight into trends between different surfaces and reactions. For example, it would be helpful to have a reactivity index that could be used to predict the adsorption behaviour of a range of adsorbates based on the properties of the clean metal surface alone. One popular descriptor for the surface science approach to catalysis is the d-band centre model.15,16 This uses the energy separation between the Fermi level of a metal surface and the 'centre of mass' of the d band (i.e. the first moment of the d-band density of states) to gauge reactivity. This measure has been much used to predict catalytic activity,17–19 but it is a global measure and therefore cannot easily be used to predict which sites on a surface will be most active for catalysis. Also the d-band centre is necessarily restricted to transition metals.
In contrast, the more general concept of hard and soft acids and bases (HSAB), developed by Pearson,20 has been used to formulate various reactivity indices. An example of such an index related to HSAB is the Fukui function,21–23 which gives a relative measure of reactivity within a molecule or nanoparticle. The chemical softness is a similar descriptor with local and atomic variants and wide applicability to different catalytic systems.24 This reactivity index has been extensively used for molecules and nanoparticles, and a new method was recently presented by us to extend its use to metal surfaces.25 In that prior work, however, we restricted ourselves to a single surface – Pt{111} – and focussed upon the spatial variation of local and atomic softness within and around three aromatic adsorbates. Here, in contrast, we wish to address a range of different surfaces, with a focus upon how variations in softness may be related to the adsorption heat of carbon monoxide – the prototypical test molecule of surface science. This, we believe, addresses the potential applicability of softness as a reactivity index for heterogeneous catalysis.
A Calculating the chemical softness
The local softness was defined by Berkowitz and Parr24 as | (1) |
The local softness can be integrated over space to find the global softness, S, of an atom, molecule or site:
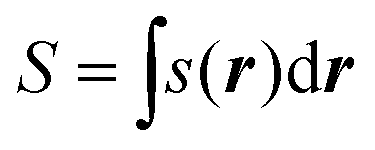 | (2) |
The local softness is related to the Fukui function, f(r), via the global softness, which acts as a normalisation factor:
| s(r) = Sf(r) | (3) |
While the chemical softness can be relatively easily approximated for molecules or nanoparticles, which are of finite extent and have a band gap, the calculation for metal surfaces is less straightforward.
Specifically, it is necessary to disentangle shifts in chemical potential that come simply from adding a constant to the external potential (which should have no physical consequence) from those that occur due to charge inflow/outflow (which is what we are interested in). In particular, there is a practical issue with using a plane-wave DFT computer code to calculate energies as they will be obtained relative to a reference potential that varies with supercell geometry for charged calculations. It may, of course, be possible to circumvent this issue by making use of localised basis functions in a code that does not impose periodicity in the surface-normal direction, but such approaches are not without their own limitations. For discussion of the advantages and disadvantages of selected such methods, the reader is referred to articles from García-Gil et al.,26 Smidstrup et al.,27 and Bennett et al.28 Given the current dominance of fully periodic calculations throughout the surface science literature, the incorporation of softness calculations within such a context is much to be desired.
A method to solve the problems implied by periodic boundary conditions was suggested by the current authors in a recent paper25 and will be described in detail in Section II B. This new approach involves separating eqn (1) into a numerator and denominator,
 | (4) |
| σ = Q/A | (5) |
 | (6) |
B Predicting CO adsorption energy
There has been a lot of work within the field of surface science on predicting the adsorption of simple species such as diatomic nitrogen30 and oxygen14,31 based on a reactivity index.12,14,30,31 In particular, CO is often used as a test molecule in surface science.31,32 For example, Hammer and co-workers have used the d-band centre reactivity index in order to predict CO adsorption energy on a range of platinum surfaces.32 Similarly, Nilsson and co-workers have used the d-band centre to predict CO adsorption on a range of transition metal surfaces.33 The d-band centre is generally thought to be a good predictor of the adsorption energies of small molecules on a given surface.18 However, as mentioned above, there would be advantages to being able to calculate a local reactivity index based on HSAB for metal surfaces in order to predict CO adsorption trends.II Methodology
The local and atomic softness were calculated for eighteen different metal surfaces. These comprised the {111}, {100} and {110} facets of copper, silver, gold, rhodium, palladium and platinum. Our selection of elements was motivated by the use of these metals in important heterogeneous catalytic applications (three-way automotive catalysis, methanol synthesis, ethylene epoxidation, water–gas shift, etc.) while the choice of facet orientation encompassed the most stable surfaces for materials crystallising in the face-centred cubic structure. CASTEP, a plane-wave code for implementing DFT using periodic boundary conditions, was used for all calculations.34,35 The exchange and correlation functional used within CASTEP was that of Perdew, Burke and Ernzerhof (PBE).36 The plane-wave-basis-set kinetic energy cutoff was 340 eV. The standard CASTEP library ultrasoft pseudopotentials were used to model electron–ion interactions.Geometry optimisations were carried out using the BFGS quasi-Newton optimisation method37 implemented in CASTEP, first on clean supercells and then also with CO adsorbed. In each case the geometry optimisations were performed with a force tolerance of 0.05 eV Å−1 and an energy tolerance of 2 × 10−5 eV per atom. The outermost layers on both sides of the double-sided slab were allowed to relax, but the inner layers of atoms were fully constrained. For more detail of exactly how many layers of atoms were allowed to relax for each surface, see the SI. The supercell geometry and Brillouin-zone sampling mesh varied according to the type of calculation and according to the surface (detailed in the SI and in Subsection II.B below). For the coinage metals (Cu, Ag, Au) the CO adsorption energy calculations were performed using the Tkatchenko-Scheffler (TS) van der Waals correction – a semi-empirical method that has been found to improve the binding strength of adsorbates on coinage metals.38 For the transition metals (Rh, Pd, Pt) the best course of action is less clear because the PBE functional already tends slightly to over-bind CO in these cases (see the literature39,40 and further discussion in the SI). Below, we present results where the dispersion correction has been applied to these elements, in addition to results where it has not. Furthermore, although the imposition of a finite surface excess charge could, in principle, give rise to a surface magnetic moment for these otherwise paramagnetic materials, our intention to extrapolate results back to the limit of infinitesimal charge is best suited by performing all calculations without spin polarisation; once again, the SI contains a more detailed discussion of this point.
A CO adsorption geometry optimisations
Geometry optimisations were performed for the clean surface first, with three layers on each side of the slab allowed to relax for the {111} and {100} surface, but four layers on each side for the {110} surface in light of its smaller interlayer spacing. CO was then placed on one side of the slab and further relaxation permitted. We restricted ourselves to atop adsorption sites, even when the experimental literature (rarely) indicates that a different site may be preferred, in order to focus upon differences in the inherent binding strength rather than contingent structural differences. For more details and a review of the relevant experimental literature, see the SI. As the layer spacing is different for different surfaces it would not be possible to have exactly the same slab thickness for all metals and facets. We therefore ensured that the slabs contained the first integer number of layers that would achieve a thickness above 18 Å. This was equivalent to 8–10 layers of {111} or {100} and about 13–15 layers of {110}, depending on the specific metal (see SI for details). The vacuum thickness was set equal to the slab thickness for the unrelaxed slab in each case.B Calculating the denominator of local softness
Here, we must evaluate the partial derivative | (7) |
First, the energies produced by CASTEP, including the chemical potential, are given relative to an energy reference which is not itself specified but is related to the average potential over the entire supercell. For different neutral supercells of the same surface, therefore, with different ratios of slab to vacuum, the chemical potential will appear to differ due only to the change in energy reference. This will arise as the potential of the slab is different from the potential of the vacuum, so changing the slab-to-vacuum ratio will change the average potential over the supercell.
Second, even were the problem of the reference potential to be resolved, there is a more serious issue with adding charge while trying to keep the electric potential the same. This arises from the periodic boundary conditions used by plane-wave codes such as CASTEP to treat periodic systems such as metal surfaces. The energy of charged supercells in periodic boundary conditions would diverge if no special steps were taken to avoid this. In common with other plane-wave codes, CASTEP applies a corrective potential in order to ensure convergence.41,42 This unphysical potential means that the condition of the derivative in eqn (7) is broken. However, by considering the nature of the corrective potential it is possible to find a solution to this problem. The charge added to the supercell can be expected to localise predominantly at the surfaces, therefore acting as an infinite array of charged plates that will be separated by the slab and vacuum thickness. For a given ratio of slab-to-vacuum, the corrective potential converges as the separation between surfaces increases.25 Therefore, by extrapolating to infinite surface-normal supercell dimension a single value of the denominator can be obtained. It should be noted, however, that this method is dependent on first correcting for the change in reference energy so that the actual change in the chemical potential can be found, excluding the unphysical component due to changing energy reference.
In order to solve the problem of the CASTEP reference energy it is helpful to consider what physical function of the system's electronic energies might be expected to stay constant when varying not only the charge but also the slab and vacuum thicknesses. A convenient example of just such a function is the electronic density of states (DOS) in the region well below the Fermi level. This region of the DOS is unlikely ever to change much upon varying the system's charge (apart for a rigid shift) because the occupancies of its contributing electronic states will remain virtually unchanged. The shift in reference potential between different charges can be then calculated by performing a cross-correlation of the DOS over some suitable range, which we have standardised to comprise the region within 5 eV above the lowest-energy calculated eigenvalue. Details of the cross-correlation procedure can be found in the Supporting Information of a recent paper by the current authors.25 Had we been using an all-electron code, the position of deep-lying core states might provide an even better and/or more convenient reference than the bottom of the valence band, but the cost of such calculations for the thick slabs that we use would likely be prohibitive.
In order to practically implement this method, geometry optimisations were first performed for a range of supercell geometries, keeping the slab-to-vacuum ratio as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The total surface-normal supercell dimension was varied from about 40–180 Å. In each case the smallest possible lateral dimensions of (1 × 1) were used and the Brillouin zone sampling was 8 × 8 × 1 for the {111} and {100} surfaces and 8 × 6 × 1 for the {110} surfaces. Then for each of the supercell surface-normal lengths, a DOS calculation was performed for small test charges of σ = ±0.02 e Å−2. These were performed keeping all the atoms fixed at their zero-charge positions, since we believe that polaronic effects should be negligible in the presence of such small surface charges, where the magnitude of excess charge per surface atom never exceeds the number of valence electrons in the neutral atom by more than 1.2% (usually much less). For this DOS calculation the Brillouin zone k-point mesh was 30 × 30 × 1 for the {111} and {100} surfaces and 30 × 21 × 1 for the {110} surfaces.
:1. The total surface-normal supercell dimension was varied from about 40–180 Å. In each case the smallest possible lateral dimensions of (1 × 1) were used and the Brillouin zone sampling was 8 × 8 × 1 for the {111} and {100} surfaces and 8 × 6 × 1 for the {110} surfaces. Then for each of the supercell surface-normal lengths, a DOS calculation was performed for small test charges of σ = ±0.02 e Å−2. These were performed keeping all the atoms fixed at their zero-charge positions, since we believe that polaronic effects should be negligible in the presence of such small surface charges, where the magnitude of excess charge per surface atom never exceeds the number of valence electrons in the neutral atom by more than 1.2% (usually much less). For this DOS calculation the Brillouin zone k-point mesh was 30 × 30 × 1 for the {111} and {100} surfaces and 30 × 21 × 1 for the {110} surfaces.
Although a fine sampling was used, it was still necessary to smooth the DOS. This was done using the OptaDOS code43,44 to apply a Gaussian broadening, with widths of 0.1, 0.2, 0.3 and 0.4 eV. The mean of the denominator over all four smearing widths was then used as the nominal value for each data point, with the standard deviation plotted on the resulting graphs as an error bar (see SI). This method removes the problem of changing energy reference but the problem of the CASTEP applied potential still remains. In order to solve this, the denominator was extrapolated to infinite surface-normal supercell dimension, and the result was taken as the standard denominator of local softness. This was done by finding the asymptotic fit using an exponential model. For graphs of this extrapolation, see the SI, where fitting parameters are also tabulated.
C Calculating the numerator and the local softness
Having explained how to calculate the denominator of eqn (4), we shall now turn our attention to the numerator, that must also be calculated in order to find the local softness; this quantity is defined as | (8) |
In terms of practically implementing eqn (8), the local electron density was calculated by CASTEP for small test charges (σ = ±0.02 e Å−2). The difference between the local electron densities for positive and negative charge was then calculated in order to estimate ∂ρ(r)/∂σ, as
 | (9) |
III Results
A Planar averages of local softness
Fig. 1 presents values of the local softness for silver and rhodium, averaged over the surface-parallel plane, as a function of distance from the centre of the slab (similar plots for the other studied surfaces are provided in the SI). It can readily be seen that the local softness close to the slab centre is relatively small and broadly independent of the chosen surface facet for each metal, even allowing for slabs of slightly differing thickness. In contrast, local softness in regions just outside of each slab (relative to the positions of the outermost nuclei) is not only comparatively large but also sensitive to the surface orientation. Further from the surface, we might expect the local softness to decay toward zero, but in fact we observe that it rebounds toward a finite value in the centre of the vacuum region. A similar effect has been noted very recently by Barrera et al.45 in relation to the nucleophilic Fukui function at a range of metallic and non-metallic surfaces, attributed to the existence of “ghost states” that the authors consider to be non-physical. We concur with this assessment, attributing the accumulation of electrons within the vacuum region to depression of the work function at negatively charged surfaces (see SI). At any rate, whilst vacuum states account for around 40% of the integrated Fukui function for the surfaces studied by Barrera et al.,45 here they typically account for only around 5–10% of the global softness in our systems. | ||
| Fig. 1 Planar-averaged softness values for Ag and Rh slabs as a function of distance from the slab centre. Black, blue, and red curves correspond to the {100}, {111}, and {110} surface orientations, respectively. Horizontal bars indicate the separation of the outermost layers of each slab, measured from the nuclear coordinates. | ||
Aside from these observations, the most striking feature of our planar-averaged softness plots is that the local softness achieves much higher values at the transition metal surfaces than at the coinage metal surfaces. This is only to be expected, in view of the presence of a partially occupied d-band in the former cases that is depressed well below the Fermi level in the latter. Less predictably, however, it is also worth noting that all of our transition metal calculations reveal locations just beneath the top-layer nuclei where the planar-averaged local softness achieves negative values, whereas the same is not true for any of the coinage metals. Nevertheless, further insight into the variation of local softness throughout the system will require visualisation that goes beyond planar averages.
B Colourplots of local softness
Fig. 2 shows the local softness of silver and rhodium surfaces as a colourmap plotted on an isosurface of 1/3 the bulk valence electron density in each case (similar plots for the other studied metals being presented in the SI). Such an isosurface cuts only through regions well separated from the unphysical accumulation of local softness in the vacuum regions of our supercells. The local softness for transition metals (e.g. rhodium) is significantly higher than for the coinage metals (e.g. silver). In all cases the local softness within the slab is low (below 0.1 eV−1 Å−3 in magnitude), which corresponds to dark blue on the colourscale. The local softness of second-layer atoms for the {110} surface is always much lower than that for the top-layer, step-edge atoms. For the coinage metals there is no visible difference between the softness of the second layer and the lower layers. In contrast, for the transition-metal {110} surfaces the second layer of atoms is slightly softer (more reactive) than the third layer of atoms and below, with a difference of about 0.1 eV−1 Å−3. | ||
| Fig. 2 Side views of the local softness for three surface facets of silver and rhodium, projected onto an isosurface corresponding to a valence electron density of one-third of its bulk value. | ||
The local softness above the surface is higher, reaching a maximum of about 3.7 eV−1 Å−3 (red) for the transition metals and about 1.2 eV−1 Å−3 (teal) for the coinage metals. This difference in surface local softness suggests higher peak reactivity for the former over the latter. The “hot spots” are found on top of the top-layer atoms for all surfaces. However, the shape of regions of higher (but not maximum) local softness varies between different surface facets. The local softness generally is slightly higher along close-packed rows for all surfaces.
There is much more variation in the values of local softness for the transition metals, and this is particularly pronounced for the stepped surfaces. For example Pd{110} displays the greatest range of values, from about zero to 3.7 eV−1 Å−3 and might therefore be expected to show high reactivity. At the other end of the scale, copper and silver surfaces typically show the lowest softness values at the chosen isosurface, with gold surfaces surprisingly showing slightly higher values on average. Indeed, on this basis, Au{111} would be predicted as the most reactive amongst the coinage metal surfaces, which is not entirely convincing in light of the literature.
Nevertheless, we do expect that the local softness may be used to predict how strongly an adsorbate might stick to the surface at different positions on the same metal surface. The colourplots suggest that the most reactive site on the {110} surfaces would be the atop step-edge site, followed by the step-edge short bridge site. Likewise for the {100} surfaces, the colourplots suggest that the most reactive sites would be the atop site, followed closely by the bridge site. As mentioned in the introduction, CO is a good example of a small adsorbate which can be used to model adsorption on metal surfaces. The preference for atop binding, which can be predicted from the local softness, matches that found in the literature for the CO molecule.46 For the {111} surfaces, the atop site has the highest softness but there is less difference between the softness of the bridge sites compared to the three-fold-hollow sites. Therefore, while the atop site is predicted to be the most favourable for all the facets, if an adsorbate were found to bind on the hollow site it would be most likely to happen on a transition-metal {111} facet. In the SI, the literature adsorption site of CO on the same eighteen surfaces is discussed in detail, but here we simply note that the only hollow-site binding on all eighteen surfaces was observed for Pd{111},46 which is in line with the predictions that could be made based upon local softness. The local softness is a useful measure, therefore, but it would nevertheless be helpful to have a more granular index of reactivity, which could be used to predict the adsorption energy trends across different sites. This we shall now explore.
C Atomic softness as a predictor for CO adsorption
As mentioned above, a thorough literature review of the adsorption sites favoured in the experimental literature was conducted and is presented in the SI. However, in terms of predicting CO adsorption energy from the atomic softness, the sites observed in the experimental literature relate to a range of different coverages and therefore it is difficult to distinguish between the effect of a change in reactivity as opposed to a change in coverage, which would increase repulsive interactions between adsorbates. For this reason, while we were guided by the experimental literature,46–64 we chose ultimately to focus only upon CO adsorption at the atop site for a coverage of 0.5 ML or less (see SI).Eqn (6) can be used to calculate the softness of individual atoms on a surface, permitting a granular measure of the softness of top-layer atoms. In view of the unphysical accumulation of softness in the vacuum regions of our supercells, however, we truncated the volumes associated with top-layer atoms at a height above the surface corresponding to the minima in our plots of planar-averaged softness (see Section III A). Fig. 3 reveals a plausible trend whereby metal surfaces with very soft top-layer atoms tend to have higher CO adsorption energy for moderate coverage at the atop site. The atomic softness can therefore be used to predict reactivity in broad terms, such as the difference between coinage metals and transition metals. Within each of these two groups individually, however, the atomic softness fails to accurately predict the differences in reactivity of similar metal surfaces as there is too much scatter on the graph. A rather stronger correlation is obtained when averaging results for specific facet orientations across the coinage metals separately from the transition metals but this is partly to be expected simply because the number of data-points is reduced. Notably, however, an increasing trend in CO binding from {111} through {100} to {110} does seem to emerge from the transition metal data.
 | ||
| Fig. 3 Calculated CO adsorption energy (at the atop site, for a coverage of 0.5 ML or below) versus top-layer atomic softness (s1) of the clean surface. In the left-hand panels, data-points are provided for all eighteen surfaces studied, while in the right-hand panels we average separately over the coinage metals and over the transition metals. In the upper panels, dispersion correction is applied in calculating all of the adsorption energies, while in the lower panels it is applied only to the coinage metals (see text for our rationale in doing so). The straight-line fits are −0.3169–0.0952s1 (R2 = 0.639) for the upper-left panel; −0.1515–0.1091s1 (R2 = 0.925) for the upper-right panel; −0.4106–0.0774s1 (R2 = 0.595) for the lower-left panel; and −0.2721–0.0890s1 (R2 = 0.921) for the lower-right panel. | ||
IV Conclusions
The local softness above a metal surface is related to reactivity and might be expected to predict how strongly an adsorbate will stick at different positions, such as the atop, bridge or hollow sites. It is also possible to develop a more granular version of the local softness that nevertheless retains important local information, which we call the atomic softness. We have shown that this can be used to predict trends in CO adsorption energies. In particular, the nine transition metal surfaces studied all had more negative adsorption energy, and also higher atomic softness, than any of the nine coinage metal surfaces. In terms of predicting the difference in CO adsorption energy between metal surfaces of similar reactivity, however, the performance of the atomic softness was less impressive. Nevertheless, despite this limitation, we believe that the atomic softness is a promising reactivity index for predicting the broad reactivity of metal surfaces, especially perhaps in the case of alloys and/or nanostructured materials.Conflicts of interest
There are no conflicts to declare.Data availability
The data underlying this study are openly available in the University of Cambridge Data Repository (Apollo) at DOI: 10.17863/CAM.127873.Supplementary information (SI): literature review of experimentally determined CO adsorption sites. Supercell dimensions and Brillouin zone sampling details for CO atop adsorption. Comments on PBE adsorption energies, work function, and spin polarisation. Details of softness denominator calculations. Planar-averaged and isosurface plots of local softness for all systems studied. Tables of atomic softness and CO adsorption energy. See DOI: https://doi.org/10.1039/d5cp03378k.
Acknowledgements
Computational resources were provided by the Cambridge Service for Data-Driven Discovery (CSD3). One of us (ALG) is grateful to EPSRC for a research studentship (Grant No. 1502496). For the purpose of open access, the authors have applied a Creative Commons Attribution (CC BY) licence to any Author Accepted Manuscript version arising.References
- G. Ertl, Handbook of Heterogeneous Catalysis, Wiley, 2nd edn, 2008 Search PubMed.
- G. A. Somorjai, The surface science of heterogeneous catalysis, Surf. Sci., 1994, 299–300, 849–866 CrossRef CAS.
- G. A. Somorjai, Catalysis and surface science, Surf. Sci., 1979, 89, 496–524 CrossRef CAS.
- F. H. Ribeiro, A. E. Schach von Wittenau, C. H. Bartholomew and G. A. Somorjai, Reproducibility of turnover rates in heterogeneous metal catalysis: Compilation of data and guidelines for data analysis, Catal. Rev. Sci. Eng., 1997, 39, 49–76 CrossRef CAS.
- Y. P. Tan, S. Khatua, S. J. Jenkins, J. Q. Yu, J. B. Spencer and D. A. King, Catalyst-induced changes in a substituted aromatic: A combined approach via experiment and theory, Surf. Sci., 2005, 589, 173–183 CrossRef CAS.
- L.-Y. Chen, T.-C. Kuo, Z.-S. Hong, M.-J. Cheng and W. A. Goddard, Mechanism and kinetics for both thermal and electrochemical reduction of N2 catalysed by Ru(0001) based on quantum mechanics, Phys. Chem. Chem. Phys., 2019, 21, 17605–17612 RSC.
- K. Honkala, A. Hellman, I. N. Remediakis, A. Logadottir, A. Carlsson, S. Dahl, C. H. Christensen and J. K. Nørskov, Ammonia synthesis from first-principles calculations, Science, 2005, 307, 555–558 CrossRef CAS PubMed.
- J. Nørskov, T. Bligaard, A. Logadottir, S. Bahn, L. B. Hansen, M. Bollinger, H. Bengaard, B. Hammer, Z. Sljivancanin, M. Mavrikakis, Y. Xu, S. Dahl and C. J. H. Jacobsen, Universality in heterogeneous catalysis, J. Catal., 2002, 209, 275–278 CrossRef.
- F. Calle-Vallejo, D. Loffreda, M. T. M. Koper and P. Sautet, Introducing structural sensitivity into adsorption-energy scaling relations by means of coordination numbers, Nat. Chem., 2015, 7, 403–410 CrossRef CAS PubMed.
- J. K. Nørskov, T. Bligaard, B. Hvolbæk, F. Abild-Pedersen, I. Chorkendorff and C. H. Christensen, The nature of the active site in heterogeneous metal catalysis, Chem. Soc. Rev., 2008, 37, 2163 RSC.
- S. J. Jenkins, Foundations of Surface Science, Foundations of Surface Science, Oxford University Press, 2023 Search PubMed.
- M. M. Montemore and J. W. Medlin, Predicting and Comparing C-M and O-M Bond Strengths for Adsorption on Transition Metal Surfaces, J. Phys. Chem. C, 2014, 118, 2666–2672 CrossRef CAS.
- P. Sabatier, Hydrogénations et déshydrogénations par catalyse, Ber. Dtsch. Chem. Ges., 1911, 44, 1984–2001 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Towards the computational design of solid catalysts, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- B. Hammer and J. K. Nørskov, Why gold is the noblest of all the metals, Nature, 1995, 376, 238–240 CrossRef CAS.
- B. Hammer and J. Nørskov, Electronic factors determining the reactivity of metal surfaces, Surf. Sci., 1995, 343, 211–220 CrossRef CAS.
- S. Jiao, X. Fu and H. Huang, Descriptors for the Evaluation of Electrocatalytic Reactions: d-Band Theory and Beyond, Adv. Funct. Mater., 2022, 32, 2107651 CrossRef CAS.
- M. T. Gorzkowski and A. Lewera, Probing the Limits of d-Band Center Theory: Electronic and Electrocatalytic Properties of Pd-Shell-Pt-Core Nanoparticles, J. Phys. Chem. C, 2015, 119, 18389–18395 CrossRef CAS.
- M. V. Ganduglia-Pirovano, V. Natoli, M. H. Cohen, J. Kudrnovský and I. Turek, Potential, core-level, and d band shifts at transition-metal surfaces, Phys. Rev. B, 1996, 54, 8892–8898 CrossRef CAS PubMed.
- R. G. Pearson, Hard and soft acids and bases, J. Am. Chem. Soc., 1963, 85, 3533–3539 CrossRef CAS.
- K. Fukui, T. Yonezawa and H. Shingu, A Molecular Orbital Theory of Reactivity in Aromatic Hydrocarbons, J. Chem. Phys., 1952, 20, 722–725 CrossRef CAS.
- K. Fukui, T. Yonezawa, C. Nagata and H. Shingu, Molecular Orbital Theory of Orientation in Aromatic, Heteroaromatic, and Other Conjugated Molecules, J. Chem. Phys., 1954, 22, 1433–1442 CrossRef CAS.
- K. Fukui, Role of Frontier Orbitals in Chemical-Reactions, Science, 1982, 218, 747–754 CrossRef CAS PubMed.
- M. Berkowitz and R. G. Parr, Molecular hardness and softness, local hardness and softness, hardness and softness kernels, and relations among these quantities, J. Chem. Phys., 1988, 88, 2554 CrossRef CAS.
- A. L. Gunton and S. J. Jenkins, Chemical Softness in Aromatic Adsorption: Benzene, Nitrobenzene and Anisole on Pt{111}, J. Phys. Chem. A, 2024, 128, 6296–6304 CrossRef CAS PubMed.
- S. García-Gil, A. García, N. Lorente and P. Ordejøn, Optimal strictly localized basis sets for noble metal surfaces, Phys. Rev. B, 2009, 79, 075441 CrossRef.
- S. Smidstrup, D. Stradi, J. Wellendorff, P. A. Khomyakov, U. G. Vej-Hansen, M.-E. Lee, T. Ghosh, E. Jónsson, H. Jonsson and K. Stockbro, First-principles green's-function method for surface calculations: A pseudopotential localized basis-set approach, Phys. Rev. B, 2017, 96, 195309 CrossRef.
- D. Bennett, M. Pizzochero, J. Junquera and E. Kaxiras, Accurate and efficient localized basis sets for two-dimensional materials, Phys. Rev. B, 2025, 111, 125123 CrossRef CAS.
- R. F. W. Bader, Atoms in Molecules: A Quantum Theory, Oxford University Press, 1 edn, 1990 Search PubMed.
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, From the Sabatier principle to a predictive theory of transition-metal heterogeneous catalysis, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- J. Greeley, J. K. Nørskov and M. Mavrikakis, Electronic structure and catalysis on metal surfaces, Annu. Rev. Phys. Chem., 2002, 53, 319–348 CrossRef CAS PubMed.
- B. Hammer, O. H. Nielsen and J. K. Nørskov, Structure sensitivity in adsorption: CO interaction with stepped and reconstructed Pt surfaces, Catal. Lett., 1997, 46, 31–35 CrossRef CAS.
- A. Nilsson, L. G. M. Pettersson, B. Hammer, T. Bligaard, C. H. Christensen and J. K. Nørskov, The electronic structure effect in heterogeneous catalysis, Catal. Lett., 2005, 100, 111–114 CrossRef CAS.
- S. J. Clark, M. D. Segall, C. J. Pickard, P. J. Hasnip, M. I. J. Probert, K. Refson and M. C. Payne, First principles methods using CASTEP, Z. Kristallogr. Cryst. Mater., 2005, 220, 567–570 CrossRef CAS.
- M. C. Payne, M. P. Teter and D. C. Allan, Iterative minimization techniques for ab initio total-energy calculations: Molecular dynamics and conjugate gradients, Rev. Mod. Phys., 1992, 64, 1045–1097 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized gradient approximation made simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- B. G. Pfrommer, M. Côté, S. G. Louie and M. L. Cohen, Relaxation of crystals with the quasi-Newton method, J. Comput. Phys., 1997, 131, 233–240 CrossRef CAS.
- A. Tkatchenko and M. Scheffler, Accurate molecular van der Waals interactions from ground-state electron density and free-atom reference data, Phys. Rev. Lett., 2009, 102, 6–9 CrossRef PubMed.
- A. Stroppa and G. Kresse, The shortcomings of semi-local and hybrid functionals: What we can learn from surface science studies, New J. Phys., 2008, 10, 063020 CrossRef.
- A. Patra, H. Peng, J. Sun and J. P. Perdew, Rethinking co adsorption on transition-metal surfaces: Effect of density-driven self-interaction errors, Phys. Rev. B, 2019, 100, 035442 CrossRef CAS.
- G. Makov and M. C. Payne, Periodic boundary conditions in ab initio calculations, Phys. Rev. B, 1995, 51, 4014–4022 CrossRef CAS PubMed.
- I. Dabo, B. Kozinsky, N. Singh-Miller and N. Marzari, Electrostatics in periodic boundary conditions and real-space corrections, Phys. Rev. B, 2008, 77, 115139 CrossRef.
- R. J. Nicholls, A. J. Morris, C. J. Pickard and J. R. Yates, OptaDOS - a new tool for EELS calculations, J. Phys.: Conf. Ser., 2012, 371, 012062 CrossRef.
- A. J. Morris, R. J. Nicholls, C. J. Pickard and J. R. Yates, OptaDOS: A tool for obtaining density of states, core-level and optical spectra from electronic structure codes, Comput. Phys. Commun., 2014, 185, 1477–1485 CrossRef CAS.
- N. F. Barrera, J. Cabezas-Escares, F. Muñoz, W. A. Muriel, T. Gómez, M. Calatayud and C. Cárdenas, Fukui function and fukui potential for solid-state chemistry: Application to surface reactivity, J. Chem. Theory Comput., 2025, 21, 3187–3203 CrossRef CAS PubMed.
- D. Wei, D. Skelton and S. Kevan, Desorption and molecular interactions on surfaces: CO/Rh (110), CO/Rh (100) and CO/Rh (111), Surf. Sci., 1997, 381, 49–64 CrossRef CAS.
- R. Marbrow and R. Lambert, Chemisorption, Surface Structural Chemistry, and Electron-Impact Properties of Carbon-Monoxide on Rhodium (110), Surf. Sci., 1977, 67, 489–500 CrossRef CAS.
- J. Batteas, A. Barbieri, E. Starkey, M. Van Hove and G. A. Somorjai, A tensor LEED analysis of the Rh(110)-p2mg(2 x 1)-2CO structure, Surf. Sci., 1994, 313, 341–348 CrossRef CAS.
- R. L. Park and H. H. Madden, Annealing changes on the (100) surface of palladium and their effect on CO adsorption, Surf. Sci., 1968, 11, 188–202 CrossRef CAS.
- R. J. Behm, K. Christmann, G. Ertl, M. A. Van Hove, P. A. Thiel and W. H. Weinberg, The structure of CO adsorbed on Pd(100): A LEED and HREELS analysis, Surf. Sci., 1979, 88, L59–L66 CrossRef CAS.
- R. Raval, S. Haq, M. Harrison, G. Blyholder and D. King, Molecular Adsorbate-Induced Surface Reconstruction - CO/Pd(110), Chem. Phys. Lett., 1990, 167, 391–398 CrossRef CAS.
- R. J. Behm, P. A. Thiel, P. R. Norton and G. Ertl, The interaction of CO and Pt(100). I. Mechanism of adsorption and Pt phase transition, J. Chem. Phys., 1983, 78, 7437–7447 CrossRef CAS.
- P. A. Thiel, R. J. Behm, P. R. Norton and G. Ertl, The interaction of CO and Pt(100). II. Energetic and kinetic parameters, J. Chem. Phys., 1983, 78, 7448–7458 CrossRef CAS.
- C. M. Comrie and R. M. Lambert, Chemisorption and surface structural chemistry of carbon monoxide on Pt(110), J. Chem. Soc., Faraday Trans., 1976, 72, 1659–1669 RSC.
- F. Abild-Pedersen and M. P. Andersson, CO adsorption energies on metals with correction for high coordination adsorption sites - A density functional study, Surf. Sci., 2007, 601, 1747–1753 CrossRef CAS.
- G. McElhiney, H. Papp and J. Pritchard, The adsorption of Xe and CO on Ag(111), Surf. Sci., 1976, 54, 617–634 CrossRef CAS.
- W. Hansen, M. Bertolo and K. Jacobi, Physisorption of CO on Ag(111): Investigation of the monolayer and the multilayer through HREELS, ARUPS, and TDS, Surf. Sci., 1991, 253, 1–12 CrossRef CAS.
- U. Burghaus, L. Vattuone, P. Gambardella and M. Rocca, HREELS study of CO oxidation on Ag(001) by O2 or O, Surf. Sci., 1997, 374, 1–8 CrossRef CAS.
- S. Krause, C. Mariani, K. C. Prince and K. Horn, Screening effects in photoemission from weakly bound adsorbates: CO on Ag(110), Surf. Sci., 1984, 138, 305–318 CrossRef CAS.
- S. A. C. Carabineiro and B. E. Nieuwenhuys, Adsorption of small molecules on gold single crystal surfaces, Gold Bull., 2009, 42, 288–301 CrossRef CAS.
- L. Piccolo, D. Loffreda, F. Aires, C. Deranlot, Y. Jugnet, P. Sautet and J. C. Bertolini, The adsorption of CO on Au(111) at elevated pressures studied by STM, RAIRS and DFT calculations, Surf. Sci., 2004, 566, 995–1000 CrossRef.
- M. S. Pierce, K.-C. Chang, D. C. Hennessy, V. Komanicky, A. Menzel and H. You, CO-induced lifting of Au(001) surface reconstruction, J. Phys. Chem. C, 2008, 112, 2231–2234 CrossRef CAS.
- I. Nakamura, A. Takahashi and T. Fujitani, Selective Dissociation of O-3 and Adsorption of CO on Various Au Single Crystal Surfaces, Catal. Lett., 2009, 129, 400–403 CrossRef CAS.
- Y. Jugnet, F. J. Cadete Santos Aires, C. Deranlot, L. Piccolo and J. C. Bertolini, CO chemisorption on Au(110) investigated under elevated pressures by polarized reflection absorption infrared spectroscopy and scanning tunneling microscopy, Surf. Sci., 2002, 521, L639–L644 CrossRef CAS.
| This journal is © the Owner Societies 2026 |