
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Integrative reverse-screening approaches for target discovery: the case of hydroxytyrosyl punicate
James
Stewart
a,
Meriem
Chayah
ab and
Carmen
Domene
 *a
*a
aDepartment of Chemistry, University of Bath, Claverton Down, BA2 7AY Bath, UK. E-mail: C.Domene@bath.ac.uk
bDepartment of Medicinal and Organic Chemistry and Excellence Research Unit of Chemistry Applied to Biomedicine and the Environment, Faculty of Pharmacy, University of Granada, Campus Cartuja s/n, 18071, Granada, Spain
First published on 31st March 2026
Abstract
Reverse-screening methodologies have emerged as powerful tools for identifying molecular targets of bioactive compounds, complementing experimental approaches and accelerating drug discovery. Recent developments in integrative strategies combining multiple databases of protein–ligand interactions, gene expression profiles, and structural information, offer improved accuracy and broader coverage in mapping compound–target networks. In this review, we highlight the principles, strengths, and limitations of these integrative reverse-screening approaches, with particular attention to their application in natural product research. As an illustrative case study, we discuss hydroxytyrosyl punicate (HT–PA), a synthetic phenolipid derived from hydroxytyrosol and punicic acid, which exhibits antiproliferative and antiparasitic effects. The application of multi-database reverse screening to HT–PA identified potential targets, including arachidonate 5-lipoxygenase (ALOX5), transient receptor potential channels (TRPs), and peroxisome proliferator-activated receptors (PPARs), which are central to inflammation, metabolism, and pain regulation. This case exemplifies how integrative computational frameworks can provide mechanistic insights, prioritize targets for experimental validation, and guide the therapeutic development of natural product derivatives. More broadly, we argue that multi-database reverse screening represents a versatile platform for uncovering the molecular basis of bioactivity and advancing rational drug discovery from complex natural compounds.
Introduction
Molecular docking is a computational technique widely used in drug discovery and structural biology to predict the preferred orientation of a small molecule, the ligand, when bound to a target protein, the receptor. By estimating the binding mode and affinity, docking provides crucial insights into molecular interactions that can guide the rational design and optimisation of new therapeutics. In practice, docking simulates the interaction between a ligand and a target protein, typically applied to pre-selected compound sets rather than exhaustive chemical libraries. It can therefore accelerate early-stage drug discovery by highlighting promising candidates prior to experimental testing, reducing both time and cost.In addition to conventional docking studies focused on limited ligand–receptor pairs, high-throughput virtual screening (HTVS) has become a central computational strategy in target-driven drug discovery, one in which many compounds are screened against a single defined target. By enabling the rapid in silico evaluation of large chemical libraries, often comprising millions of compounds, against a defined protein structure, HTVS integrates streamlined docking workflows with increasingly sophisticated scoring functions, including machine-learning-accelerated approaches. Recent benchmarking efforts have highlighted both the scalability of HTVS and the sensitivity of its performance to target selection, structural quality, and scoring methodology.1–5
The applicability of HTVS has been further expanded by advances in protein structure prediction, particularly deep-learning-based modelling and co-folding strategies capable of generating structural templates for targets lacking experimental data. These approaches now provide access to three-dimensional models of individual proteins as well as protein–protein and protein–ligand complexes, thereby broadening the range of computationally addressable targets. The ability of modern structure-prediction frameworks to capture alternative conformations and potential binding sites has begun to mitigate some long-standing limitations of rigid-receptor docking, although challenges related to dynamic flexibility and scoring reliability remain. Crucially, the growing availability of proteome-wide structural models has also begun to expand the scope of reverse screening, enabling interrogation across increasingly comprehensive target panels.
Although the present review focuses on ligand-centric reverse screening rather than forward HTVS, this evolving computational landscape provides essential context. Forward strategies assess many ligands against a predefined target, whereas reverse screening adopts an inverted paradigm in which a single compound is interrogated across a panel of potential protein targets, an approach particularly well suited to elucidating mechanisms of action, identifying off-target interactions, and supporting drug repurposing efforts.
In parallel, experimental high-throughput screening (HTS) remains a cornerstone of target-based drug discovery, enabling the large-scale empirical evaluation of compound libraries against defined biological systems. While HTS offers direct validation of bioactivity, it requires substantial experimental infrastructure and resources. Reverse screening, whether implemented computationally or experimentally, complements both HTVS and HTS by facilitating the identification of off-target interactions, the characterisation of polypharmacological profiles, and the uncovering of previously unrecognised biological activities associated with a given small molecule, applications that collectively define the scope of this review.
Categories of reverse screening methods
Having established the conceptual distinction between forward and reverse screening, we now describe the principal methodological categories through which reverse screening is implemented. Reverse screening has become particularly valuable for natural products, drug repurposing candidates, and bioactive small molecules of complex or unknown polypharmacology, where the mechanistic basis of biological activity may be incompletely understood. By mapping potential targets of a compound, reverse screening not only aids drug discovery but also enhances mechanistic understanding of how a molecule exerts its biological effects. Such insights can guide subsequent experimental validation, inform therapeutic development strategies, and help anticipate potential off-target effects or adverse drug reactions. In this study, reverse screening serves as the primary tool to explore the biological targets of hydroxytyrosyl punicate (HT–PA), a novel compound derived from hydroxytyrosol and punicic acid, exemplifying how computational methods can complement experimental approaches in modern drug discovery.Reverse screening methods can be broadly divided into four main categories based on the underlying computational strategy: ligand-based similarity or shape screening, pharmacophore-based screening, structure-based reverse docking, and hybrid approaches that integrate multiple sources of information, such as ligand similarity, protein structural data, and network- or machine-learning-based models, to improve target prediction accuracy.6,7 These categories differ substantially in their data requirements, computational cost, and performance characteristics, and the appropriate choice of method depends heavily on the nature of the query compound and the availability of structural or bioactivity data for the target space of interest.
Performance evaluation of reverse screening methods
To assess the effectiveness of reverse screening approaches, several performance metrics are commonly employed. The area under the receiver operating characteristic curve (AUC) measures the overall ability of a method to discriminate true protein targets from non-targets across all possible score thresholds, with values approaching 1.0 indicating near-perfect discrimination. Enrichment factors (EF) quantify the fold-increase in the proportion of true targets recovered within a defined top-ranked fraction of predictions relative to random selection; early enrichment metrics such as EF at 1% or 5% are particularly informative in the context of target fishing, where only a small number of candidates are typically taken forward for experimental validation.8,9 The Boltzmann-enhanced discrimination of ROC (BEDROC) metric addresses a known limitation of the standard AUC by placing greater weight on early enrichment, making it better suited to scenarios where the prioritisation of true targets near the top of ranked lists is critical.8 Top-k recall, the fraction of known targets recovered within the top k predictions, is widely used as an intuitive, application-relevant performance measure, with k values of 1, 5, and 10 being the most commonly reported.10In addition to these ranking-based metrics, other commonly used measures include precision, recall, and the F1 score, which are essential for evaluating the balance between false positives and false negatives in target prediction. Precision quantifies the proportion of correctly identified targets among all predicted targets, while recall reflects the ability of a method to recover known or validated targets. The F1 score, defined as the harmonic mean of precision and recall, provides a single summary statistic that is particularly informative when datasets are imbalanced, as is often the case in reverse-screening benchmarks. Further performance indicators include specificity and false positive rate, which assess how effectively a method excludes non-interacting proteins from its predictions.
Finally, mean average precision (MAP) is increasingly reported to evaluate overall ranking quality by integrating precision values across multiple cutoff thresholds. MAP is particularly valuable when screening against large and diverse target sets, as it reflects how consistently true targets are prioritized near the top of prediction lists. Collectively, the use of complementary metrics enables a more comprehensive and reliable assessment of reverse-screening performance and facilitates fair comparison across ligand-based, structure-based, and hybrid methodologies.
It should be noted, however, that several authors have cautioned that AUC and enrichment-based metrics may not fully capture the practical utility of target prediction tools for researchers, who are typically more concerned with whether the true target appears within the top few predictions rather than with global ranking quality.9,11 This perspective has prompted wider adoption of top-k recall as a primary performance indicator in more recent benchmarking studies. Furthermore, benchmarking studies have consistently shown that the performance of all reverse screening methods is sensitive to the quality and composition of the underlying datasets, and that commonly used benchmark sets may contain biases, including analogue bias, artificial enrichment, and false negatives, that can lead to overestimated performance in retrospective evaluations.10 These considerations are critical when interpreting reported metrics across studies, as performance differences between methods may partly reflect differences in benchmark composition rather than true differences in predictive power.
Ligand-based shape screening compares the three-dimensional shapes or chemical features of a query compound with those of known ligands, based on the principle that structurally similar molecules are likely to bind similar targets. This approach, commonly referred to as ligand-based virtual screening (LBVS), is particularly useful when high-quality receptor structures are unavailable. It enables rapid, large-scale computational screening by leveraging the structural and chemical similarity between molecules, allowing researchers to prioritize compounds for further experimental evaluation. In benchmarking comparisons across multiple protein families, ligand-based methods have generally performed competitively with or better than structure-based approaches in terms of AUC and early enrichment, particularly for well-characterised target classes with large bioactivity datasets.12,13 However, performance varies considerably across targets: ligand-based methods tend to underperform for targets with limited known ligand data or for structurally novel scaffolds dissimilar to training compounds.10,12 Among the available online tools, SwissTargetPrediction, a hybrid 2D/3D ligand similarity method that combines Tanimoto-based fingerprint comparison with electroshape 3D similarity and logistic regression scoring, has been evaluated in multiple independent benchmarking studies. In a large-scale evaluation, a machine-learning-augmented reverse screening approach correctly identified the true target with the highest predicted probability for over 51% of external compounds across more than 2000 protein targets.11 In an independent comparison of nine widely used ligand-based target fishing tools, SwissTargetPrediction was found to produce the most reliable predictions overall, while the similarity ensemble approach (SEA) recovered true targets for the greatest proportion of query compounds.14 SwissTargetPrediction achieved at least one correct human target in the top 15 predictions for over 70% of external compounds in validation experiments.15 In a separate benchmarking study comparing five target prediction servers, the hybrid LigTMap server achieved a top-10 success rate of 86%, followed by SEA at 83% and SwissTargetPrediction at 78%, although SwissTargetPrediction showed the highest top-1 precision at 66%.16
Pharmacophore-based screening focuses on identifying the spatial arrangement of key functional groups, such as hydrogen bond donors or acceptors, hydrophobic regions, and aromatic rings, that are essential for biological activity. The predicted pharmacophore model of a query compound is then matched to known protein targets to identify potential interactions. This approach provides a detailed understanding of the critical chemical features necessary for binding and can guide both target prediction and ligand optimization, even in the absence of complete structural information about the target proteins. PharmMapper is the principal publicly available tool for pharmacophore-based reverse screening,17 operating through flexible alignment of query molecules against a database of protein-derived pharmacophore models with Z-score normalisation for target ranking. It has been widely applied in the literature, including in studies of chemopreventive natural products: for example, PharmMapper and ReverseScreen3D were used to predict that eucalyptol, the active component of cardamom, targets CASP-3 and cAMP-dependent protein kinase (PKA), providing a mechanistic basis for its reported anti-inflammatory and anti-proliferative activities.6,18 In another application, Ge et al. combined PharmMapper with idTarget to predict that dihydropyrimidine dehydrogenase and spindle checkpoint kinase Bub1 are off-target binding partners of the antithrombotic agent dipyridamole, offering a computational explanation for its previously observed anticancer activity.19 Despite its utility, PharmMapper has known limitations, including its dependence on the quality and completeness of its underlying pharmacophore database and periodic server unavailability.
In structure-based reverse docking, the query compound is systematically docked across a panel of protein structures. By estimating binding affinities using appropriate scoring functions, reverse docking predicts potential protein targets based on the likelihood of stable molecular interactions. Structure-based reverse docking offers mechanistic insight into compound–target interactions, revealing how and where a molecule may bind. However, it requires high-quality receptor structures, accurate docking algorithms, and substantial computational resources. Limitations in scoring functions or structural flexibility can sometimes result in false positives or false negatives, highlighting the need for careful interpretation of docking results.
TarFisDock,20 idTarget,12 and INVDOCK21 represent the principal tools for structure-based reverse docking. The ACID tool, a consensus inverse docking server integrating results from multiple docking programmes, reported an AUC of 0.84 for drug repurposing predictions, recovering 62 of 91 known drug–target pairs within the top 2% of ranked predictions across a test set of 51 drugs.22 A representative application involves reverse docking of phenolic natural compounds against a panel of 163 cancer-related proteins, which correctly identified protein kinases PDK1 and PKC as the targets of xanthohumol and isoxanthohumol, subsequently confirmed by in vitro biological testing.23 In a further example, the targets of three herbal ingredients, acteoside, quercetin, and epigallocatechin gallate (EGCG), were successfully predicted across the human structural proteome using a combined pharmacophore and reverse docking pipeline, with the majority of known targets recovered and mechanistic pathway analyses performed via KEGG enrichment.12,21
Finally, hybrid approaches combine elements of ligand- and structure-based methods and frequently incorporate additional data sources, such as chemogenomic information, protein–protein interaction networks, or machine learning algorithms. By integrating multiple sources of information, hybrid methods can enhance the coverage, accuracy, and reliability of target prediction. These approaches leverage the complementary strengths of individual methods, capturing interactions that might be overlooked by any single strategy, and providing a more comprehensive and robust framework for identifying potential protein targets for bioactive compounds. The LigTMap server exemplifies this strategy, combining fingerprint-based ligand similarity search with docking and binding pose similarity analysis across 17 therapeutic protein classes, achieving a top-10 success rate of approximately 70% in validation experiments and outperforming purely ligand-based tools in several class-specific benchmarks.16 More broadly, consensus-based approaches that aggregate predictions from multiple tools, such as the combined use of SwissTargetPrediction and SEA, have been shown to improve both precision and target coverage relative to either tool alone.14 Despite their advantages, hybrid methods typically involve greater computational complexity and may be less interpretable than single-strategy approaches.
Among the available approaches, ligand-based shape screening currently offers the greatest number of publicly accessible online tools.7 In contrast, only a single pharmacophore-based reverse screening tool (PharmMapper17) and three structure-based reverse docking platforms (TarFisDock,20 idTarget,12 and INVDOCK21) were identified; however, at the time of analysis, these were not consistently accessible for practical implementation. Accordingly, the present study focuses on ligand-based similarity screening methods with stable online availability, as described in the following section.
Computational approaches and resources for ligand-centric target prediction
To provide a systematic overview of the available computational resources for ligand-centric target prediction, we have classified existing tools, servers, and databases according to their methodological approach. Table 1 summarises these resources, distinguishing between ligand-based prediction servers, structure-based reverse docking tools, reference databases, and emerging hybrid/integrative frameworks. For each entry, we indicate the type of method or data used, required inputs, typical outputs, and relevant limitations. Scheme 1 provides a decision flowchart to guide method selection depending on the nature of the query compound and the availability of structural or bioactivity data.| Type | Name | Method/data used | Input(s) | Output(s) | Comments |
|---|---|---|---|---|---|
| Ligand based target prediction server | SwissTargetPrediction | 2D/3D chemical similarity + statistical/ML models | Query small molecule (SMILES/structure) | Ranked list of predicted protein targets | Widely used, good recall; depends on quality & coverage of ligand–target reference data |
| SEA (Similarity Ensemble Approach) | Fingerprint-based similarity/statistical enrichment | Query ligand | Predicted targets (with similarity-based scores) | Common approach: may find off targets, but performance depends on reference set | |
| PPB2 (polypharmacology browser 2) | ML/fingerprint-based similarity | Query ligand | Target predictions (ranked) | Good for diverse small molecules; coverage depends on training data | |
| SuperPred | Ligand similarity/classification-based target prediction | Query ligand | Predicted targets | Conceptually like other ligand-based servers, but dependent on database maintenance & updates | |
| DIA-DB | 3D ligand similarity + shape-based comparison against diabetes-related protein–ligand complexes | Query small molecule (SMILES/structure) | Ranked list of predicted diabetes-related protein targets | Specialised server focused exclusively on antidiabetic targets; not suitable for broad target prediction outside metabolic disease context | |
| Reference database (ligand target binding data) | BindingDB | Curated experimental binding affinities/binding data for many ligand–protein pairs | None (used as resource) | Bioactivity data, known ligand–target associations | Useful as training/reference data; not a predictive server itself |
| Protein structure database for reverse docking | PDTD (potential drug target database) | Collection of proteins with known 3D structures (from PDB) + binding site annotations | None (used as resource) | Repository of 3D protein structures for docking | Key resource for structure based reverse docking; but coverage limited to proteins with available high-quality structures |
| Structure based reverse docking server | TarFisDock | Reverse docking: dock query ligand against many protein structures, compute scores with DOCK algorithm | Query ligand + optionally a target set or full database | Ranked list of potential binding proteins + docking poses | Well established; results depend heavily on protein structure quality and docking limitations (rigid protein, scoring accuracy) |
| Newer hybrid methods (ligand + structure/machine learning) | LigTMap | Combines ligand similarity, docking, and binding similarity analysis for target prediction | Query small molecule | Predicted targets, possible binding modes | Promising hybrid strategy: may balance strengths of ligand and structure-based methods, but still limited by data/structure coverage |
| Integrated/network-based frameworks | — (e.g. STITCH, ChemMapper) | Various (data integration, chemogenomic data, network analysis) | Depends on implementation | Target predictions with network-level context | Availability & maintenance uncertain; can provide broader biological context by integrating chemical, genomic, and network-level data |
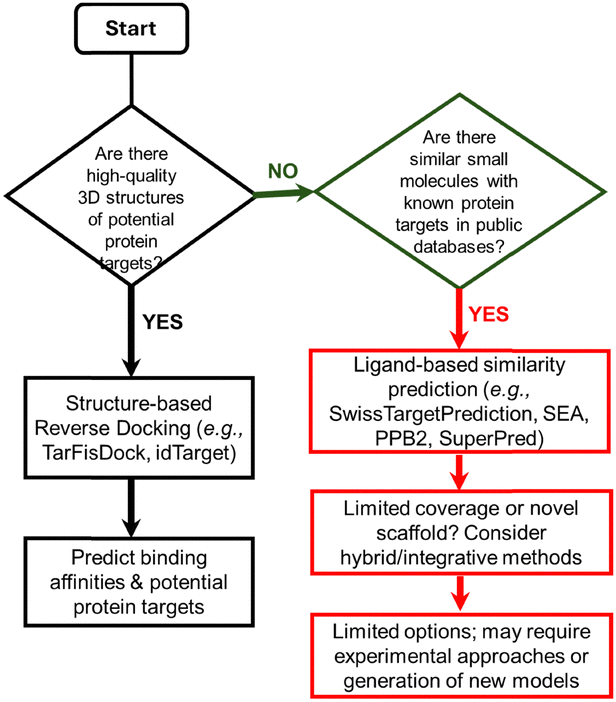 | ||
| Scheme 1 Decision flowchart for selecting a computational approach to identify protein targets of a small molecule. Depending on available information, such as the presence of high-quality 3D protein structures or known similar compounds, users can select structure-based reverse docking, ligand-based similarity prediction, or hybrid/integrative approaches. The flowchart provides guidance for predicting potential protein targets from a query compound. | ||
Rather than re-introducing the broad categories of reverse screening already described above, this section focuses on the methodological basis of ligand-based approaches in greater detail, as these form the basis of the analyses conducted in the present study. Ligand-based shape screening approaches exploit the principle that, in 2D, structurally similar molecules are likely to bind to similar protein targets.13 In 3D, molecules with comparable shapes and aligned chemical features can interact with targets in analogous ways. To perform this type of screening, a server requires access to a library of small molecules annotated with protein-binding data. By comparing the shape and chemical features of a query molecule to those in the database, potential protein targets can be identified.
While predictions of drug–target interactions can be broadly classified as either ligand-based or receptor-based,9 ligand-based approaches are more widely used in practice owing to their speed, efficiency, and applicability across diverse compound classes.7,9 Receptor-based approaches require a high-quality three-dimensional protein structure, which is not always available, whereas ligand-based approaches draw on the structural information of known ligands stored in public databases.
Ligand-based methods are based on the principle that structurally similar compounds tend to exhibit similar biological properties.13 Ligand-based target prediction methods can be further divided into three major classes, based on how molecular similarities are determined: chemical similarity searching, machine learning (ML) approaches, and stacking algorithm methods. Among these, ML approaches have gained significant attention for their predictive performance and scalability with large datasets.8,10,24 ML models are trained on datasets of compounds with known protein targets and employ classification algorithms, such as support vector machines, decision trees, and artificial neural networks, to classify compounds as active or inactive against specific targets. These models can incorporate diverse molecular descriptors and exclude non-informative features, thereby improving the accuracy of protein target identification for novel compounds.
ML-based ligand–target prediction25,26 is typically divided into two main modelling paradigms: conventional quantitative structure–activity relationship (QSAR) models21,27 and proteochemometrics (PCM) modelling.7,28 QSAR models build mathematical relationships between molecular descriptors and biological activity, whereas PCM extends this concept by modelling interactions between multiple ligands and multiple targets, often resulting in improved predictive power.
Chemical similarity searching is the simplest and most computationally efficient method for target prediction but depends on the availability of a reference library containing chemical structures and known binding affinities.29–32 In this approach, each molecule in the library is compared to the user-supplied query compound, and similarity scores are computed. Molecular structures are typically encoded into vectors—using molecular fingerprints—that capture key properties such as the presence of pharmacophores. Three main similarity search strategies are employed: top K hits,33 similarity scores34–36 and statistical similarity scores.37
The top K hits method ranks molecules in the reference library/database based on their similarity to the query molecule using various algorithms and measures. The top K hits are the molecules that have the highest similarity scores compared to the query molecule.
Similarity score-based ranking is based on the average similarity score between the query molecule and the molecules in the reference database. A common metric for this comparison is the Tanimoto coefficient (Tc), which is widely used to measure chemical similarity.30 The Tc is calculated by dividing the number of shared features between two molecules by the total number of distinct features in both compounds. As the number of shared features increases, the Tc approaches 1, indicating high similarity, while less similarity results in a lower Tc value. Features in the molecules are typically represented using molecular fingerprints, where a molecular fragment is given a value of 1 in the presence of a feature and 0 in its absence.
The final main search method, statistical similarity scores, involves targeting prediction based on statistical similarity scores. Unlike the first two methods, this approach quantitatively expresses similarities in a statistical framework. The similarities between the query molecule and reference molecules are converted into statistical significance scores, which estimate the likelihood of random associations between molecules. The lower the significance score, the higher the chance that the targets of the reference molecules are shared with the query molecule. This method is particularly useful as it aims to eliminate bias toward targets with a higher number of known ligands, ensuring a more balanced and statistically robust comparison.
An alternative approach is algorithm stacking, which combines the high accuracy of machine learning with the efficiency of chemical similarity searches.7,38,39 This strategy aims to minimize false positives by deprioritizing targets with low similarity to the query compound. In this method, similarity scores are converted into statistical significance values (p-values or e-values), which reflect the likelihood of random associations between the query molecule and the ligands in the database.
Together, these ligand-based similarity approaches provide a versatile toolkit for predicting potential protein targets when structural information on receptors is limited or unavailable. However, the predictive value of such tools is best understood when applied to real compounds with therapeutic relevance. To illustrate their application, we selected hydroxytyrosyl punicate (HT–PA), a recently synthesised phenolipid derived from hydroxytyrosol and punicic acid, as a case study. HT–PA was chosen not only because of its promising biological activities, but also because it represents a class of natural product derivatives with limited mechanistic characterisation. In the following section, we outline the biochemical context of HT–PA and its precursors, before demonstrating how a suite of reverse-screening databases can be employed to explore its potential targets and mechanisms of action.
Our case study: hydroxytyrosyl punicate
Pomegranate (Punica granatum) seed oil, extracted from the seeds of the fruit, is gaining recognition for its diverse array of health-promoting properties. Depending on the pomegranate variety, the seeds contain from 7% to 27% of pomegranate seed oil (PSO),40 rich in bioactive compounds such as punicic acid, flavonoids, and antioxidants. The dominant lipid is punicic acid (PA)41,42 which makes up anywhere between 30% to 80% of the seed oil43 (Scheme 2). PA is of particular interest due to its wide range of biological activities, including but not limited to, antidiabetic, anticarcinogenic and antiproliferative effects.41,43–46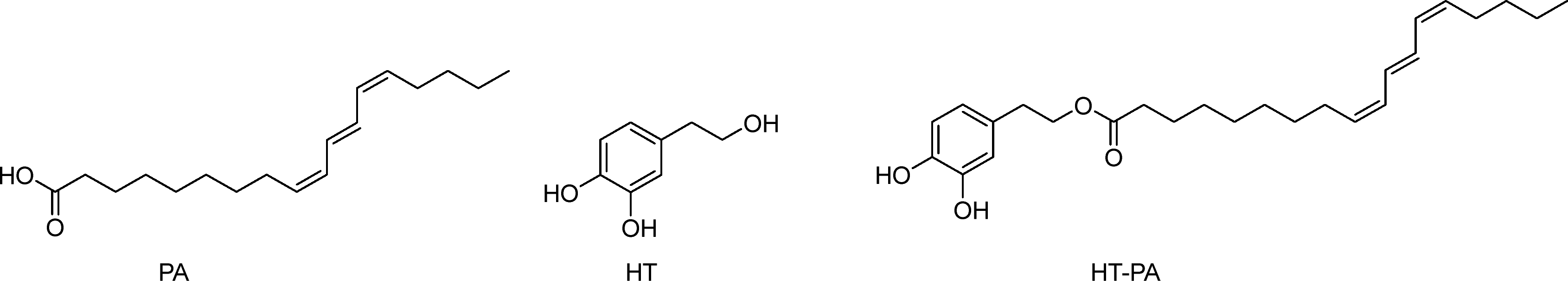 | ||
| Scheme 2 Structures of punicic acid (PA), hydroxytyrosol (HT), and hydroxytyrosyl punicate (HT–PA). | ||
Owing to high levels of PA, PSO has been shown to act as protection against cardiovascular disease, neurotoxicity and osteoporosis.47,48 PSO has also been shown to have an effect in the treatment of type 2 diabetes mellitus (T2DM),45 since PA is thought to be an agonist of peroxisome proliferator-activated receptor gamma (PPARγ), a protein associated with insulin and glucose regulation.49 Similarly, it is effective for the management of mitochondrial dysfunction often associated with diabetes.50 PA has been shown to inhibit the growth of human prostate cancer cells51 and breast cancer cells in vitro.44 Many studies show positive effects on various biological processes like reduced blood pressure, increased antioxidative effects and lowering of blood sugar levels, highlighting the positive biological properties of pomegranates.52
Hydroxytyrosol (HT) is a polyphenolic, highly polar molecule (Scheme 2) which can be readily found in leaves and extra virgin olive oil. The ‘Mediterranean diet’ contains very high levels of HT in the form of foods such as grapes, wheat, and olives. Out of these, the best characterised is that of olive oil, where high olive oil levels in the diet have been linked to lower cancer mortality rates in Mediterranean countries when compared to Western countries.53 It has been suggested that these reductions in mortality rates are in part due to the high levels of HT, monounsaturated fatty acids, and polyphenols in the diet.54–57
Studies on HT have shown that it can have anti-inflammatory, antioxidant and neuroprotective effects.58 Recent work into this compound has focused on developing derivatives to improve metabolism, absorption and excretion processes in the body, with the most notable group being HT esters.55 HT acetate, for example, has been shown to have an improved antioxidant ability over that of HT.55 More esters syntheses have since been carried out between HT and various polyunsaturated fatty acids and the products have shown a variety of biological effects.59–61 HT esters have impacts on cell proliferation59 and have been implicated in reducing myeloma cell survival with no toxicity against human cells.60
A new HT ester has recently been synthesised, hydroxytyrosyl punicate (HT–PA) (Scheme 2). This phenolipid was obtained from the combination of HT and PA demonstrating greater antiproliferative and antitrypanosomal activity compared to its precursors in lung carcinoma A549 cells and against Trypanosoma brucei parasites, respectively.62
Given the limited literature on hydroxytyrosol conjugated with omega-5 polyunsaturated fatty acids (HT–PA), this compound was selected as a representative example to illustrate how computational approaches can be employed to explore potential mechanisms of action through the identification of putative protein targets. To this end, reverse-screening tools across various chemogenomic databases were employed. Identifying HT–PA's targets is essential for understanding how the compound may influence cellular processes, signalling pathways, or disease mechanisms. This knowledge could inform drug development strategies; once the key protein targets are known, the therapeutic potential of HT–PA in treating specific conditions can be explored. Moreover, uncovering these targets may reveal new applications beyond HT–PA's currently known uses. Such insights could also support the advancement of personalised medicine by maximising therapeutic efficacy while minimising adverse effects.
To evaluate the performance and complementarity of different reverse-screening platforms, we next applied a panel of widely used chemogenomic databases and online servers to HT–PA, using this compound as a test case to assess how each tool operates and what types of biological insights can be derived from their predictions.
Databases and online servers
As detailed, six different servers and databases were employed to identify potential protein targets of HT–PA through various ligand-based reverse screening methods. Fluoxetine was used as a control, and the resulting data aligned with expectations in most cases, confirming that these computational approaches are generally reliable for reverse screening and target prediction.Using SwissTargetPrediction, 23 targets were identified. All predicted proteins had identical probability scores, so no ranking could be established. Multiple isoforms of the same protein family were detected such as histone deacetylases, resulting in 16 unique targets. The functions of these potential HT–PA targets are summarized in Fig. 1, which presents pie charts of the most probable target classes for the query molecule based on the top 15, 25, and 50 predicted targets. These findings suggest that HT–PA exhibits a broad spectrum of activity and is not limited to a single protein class, potentially enhancing its pharmacological potential. When fluoxetine was inputted, several serotonin and dopamine receptors were given with a 100% probability, indicative of a reliable method for target prediction.
 | ||
| Fig. 1 Summary of the most probable target classes for the query molecule displayed as a pie chart. Percentages are calculated based on the top 15 (A), 25 (B) and 50 (C) of predicted targets from SwissTargetPrediction. | ||
The similarity ensemble approach (SEA) search results are provided in Table 2. Among the top 10 targets, the TRPV subfamily of ion channels (TRPV1 and TRPV2) appeared multiple times, and cannabinoid receptors were also frequently detected. Some targets corresponded to non-human species, highlighting the need to select biologically relevant proteins for downstream experimental validation.
| 1433G_HUMAN | YWHAG | 14-3-3 protein gamma |
| A0A0C7ACN7_PSEAI | PQSD | 3-Oxoacyl-ACP synthase |
| AA2BR_RAT | ADORA2b | Adenosine receptor A2b |
| ADRB1_MOUSE | ADRB1 | Beta-1 adrenergic receptor |
| AK1BA_HUMAN | AKR1B10 | Aldo–keto reductase family 1 member B10 |
| AK1C4_HUMAN | AKR1C4 | Aldo–keto reductase family 1 member C4 |
| ALF_CANAL | Fructose-bisphosphate aldolase | |
| AMD_HUMAN | PAM | Peptidyl-glycine alpha-amidating monooxygenase |
| ARP19_RAT | ARPP19 | cAMP-regulated phosphoprotein 19 |
| CAH13_MOUSE | CA13 | Carbonic anhydrase 13 |
| CAH5A_HUMAN | CA5A | Carbonic anhydrase 5A, mitochondrial |
| CAH5B_HUMAN | CA5B | Carbonic anhydrase 5B, mitochondrial |
| CAH6_HUMAN | CA6 | Carbonic anhydrase 6 |
| CBS_HUMAN | CBS | Cystathionine beta-synthase |
| CNR1_MOUSE | CNR1 | Cannabinoid receptor 1 |
| CNR1_RAT | CNR1 | Cannabinoid receptor 1 |
| CNR1_HUMAN | CNR1 | Cannabinoid receptor 1 |
| CNR2_MOUSE | CNR2 | Cannabinoid receptor 2 |
| CNR2_HUMAN | CNR2 | Cannabinoid receptor 2 |
| CP74A_ARATH | CYP74A | Allene oxide synthase, chloroplastic |
| DGLA_HUMAN | DAGLA | Sn1-specific diacylglycerol lipase alpha |
| DHB3_HUMAN | HSD17B3 | Testosterone 17-beta-dehydrogenase 3 |
| DPOLB_RAT | POLB | DNA polymerase beta |
| ENPP2_MOUSE | ENPP2 | Ectonucleotide pyrophosphatase/phosphodiesterase family member 2 |
| ERCC1_HUMAN | ERCC1 | DNA excision repair protein ERCC-1 |
| ERG1_RAT | SQLE | Squalene monooxygenase |
| EST1_HUMAN | CES1 | Liver carboxylesterase 1 |
| FAAH1_MOUSE | FAAH | Fatty-acid amide hydrolase 1 |
| FAAH1_HUMAN | FAAH | Fatty-acid amide hydrolase 1 |
| FAAH1_RAT | FAAH | Fatty-acid amide hydrolase 1 |
| FABPH_HUMAN | FABP3 | Fatty acid-binding protein, heart |
| GP174_HUMAN | GPR174 | Probable G-protein coupled receptor 174 |
| GPR34_HUMAN | GPR34 | Probable G-protein coupled receptor 34 |
| GPR34_MOUSE | Gpr34 | Probable G-protein coupled receptor 34 |
| HYES_MOUSE | EPHX2 | Bifunctional epoxide hydrolase 2 |
| HYES_HUMAN | EPHX2 | Bifunctional epoxide hydrolase 2 |
| INHA_MYCTU | INHA | Enoyl-[acyl-carrier-protein] reductase [NADH] |
| KDM4E_HUMAN | KDM4E | Lysine-specific demethylase 4E |
| KPCA_BOVIN | PRKCA | Protein kinase C alpha type |
| KPCA_HUMAN | PRKCA | Protein kinase C alpha type |
| KPCL_MOUSE | PRKCH | Protein kinase C eta type |
| LEF_BACAN | LEF | Lethal factor |
| LKHA4_HUMAN | LTA4H | Leukotriene A-4 hydrolase |
| LOX15_HUMAN | ALOX15 | Arachidonate 15-lipoxygenase |
| LOX15_PIG | ALOX15 | Arachidonate 15-lipoxygenase |
| LOX15_RABIT | ALOX15 | Arachidonate 15-lipoxygenase |
| LOX15_RAT | ALOX15 | Arachidonate 15-lipoxygenase |
| LOX1_SOYBN | LOX1.1 | Seed linoleate 13S-lipoxygenase-1 |
| LOX5_RAT | ALOX5 | Arachidonate 5-lipoxygenase |
| LPAR1_HUMAN | PAR1 | Lysophosphatidic acid receptor 1 |
| LPAR1_MOUSE | LPAR1 | Lysophosphatidic acid receptor 1 |
| LPAR2_HUMAN | LPAR2 | Lysophosphatidic acid receptor 2 |
| LPAR3_HUMAN | PAR3 | Lysophosphatidic acid receptor 3 |
| LPAR4_HUMAN | LPAR4 | Lysophosphatidic acid receptor 4 |
| LPAR4_MOUSE | LPAR4 | Lysophosphatidic acid receptor 4 |
| LPAR6_HUMAN | LPAR6 | Lysophosphatidic acid receptor 6 |
| LPXC_AQUAE | lPXC | UDP-3-O-acyl-N-acetylglucosamine deacetylase |
| LX15B_RAT | ALOX15b | Arachidonate 15-lipoxygenase B |
| M9TGV3_MYCTX | INHA | Enoyl-[acyl-carrier-protein] reductase [NADH] |
| MGLL_RAT | MGLL | Monoglyceride lipase |
| MPIP2_MOUSE | CDC25b | M-phase inducer phosphatase 2 |
| NANA_STREE | Sialidase A | |
| NSMA_RAT | SMPD2 | Sphingomyelin phosphodiesterase 2 |
| NU1M_BOVIN | MT-ND1 | NADH-ubiquinone oxidoreductase chain 1 |
| OXER1_HUMAN | OXER1 | Oxoeicosanoid receptor 1 |
| P2Y10_HUMAN | P2RY10 | Putative P2Y purinoceptor 10 |
| PA24B_HUMAN | PLA2G4B | Cytosolic phospholipase A2 beta |
| PA2A1_NAJMO | Acidic phospholipase A2 CM-I | |
| PA2G5_HUMAN | PLA2G5 | Calcium-dependent phospholipase A2 |
| PA2G5_MOUSE | PLA2G5 | Calcium-dependent phospholipase A2 |
| PA2GA_MOUSE | PLA2G2A | Phospholipase A2, membrane associated |
| PA2GA_RABIT | PLA2G2A | Phospholipase A2, membrane associated |
| PA2GA_RAT | PLA2G2A | Phospholipase A2, membrane associated |
| PA_I000X | PA | Polymerase acidic protein |
| PLCG1_BOVIN | PLCG1 | 1-Phosphatidylinositol 4,5-bisphosphate phosphodiesterase gamma-1 |
| POLH_HUMAN | POLH | DNA polymerase eta |
| POLI_MOUSE | POLI | DNA polymerase iota |
| POLK_HUMAN | DNA polymerase kappa | |
| PPO2_AGABI | PPO2 | Polyphenol oxidase 2 |
| Q39829_SOYBN | Lipoxygenase | |
| Q6UCJ9_TOXGO | ENR | Enoyl-acyl carrier reductase |
| Q7ZJM1_9HIV1 | POL | Integrase |
| Q95214_RABIT | ACAT | Acyl-CoA:cholesterol acyltransferase |
| RPOB_ECOLI | RPOB | DNA-directed RNA polymerase subunit beta |
| S6A11_MOUSE | Sodium- and chloride-dependent GABA transporter 3 | |
| S6A13_MOUSE | Sodium- and chloride-dependent GABA transporter 2 | |
| THA_HUMAN | THRA | Thyroid hormone receptor alpha |
| THB_HUMAN | THRB | Thyroid hormone receptor beta |
| TLR2_HUMAN | TLR2 | Toll-like receptor 2 |
| TRPA1_RAT | TRPA1 | Transient receptor potential cation channel subfamily A member 1 |
| TRPV2_RAT | TRPV2 | Transient receptor potential cation channel subfamily V member 2 |
| TRPV1_RAT | TRPV1 | Transient receptor potential cation channel subfamily V member 1 |
| TRPV1_HUMAN | TRPV1 | Transient receptor potential cation channel subfamily V member 1 |
| TTHY_HUMAN | TTR | Transthyretin |
| TYTR_CRIFA | TPR | Trypanothione reductase |
| XPF_HUMAN | ERCC4 | DNA repair endonuclease XPF |
SuperPred identified 91 predicted targets, nine of which scored above 90% in probability. However, three, including the highest-ranked protein with a 98.74% probability score, had model accuracy scores below 80%, indicating lower reliability. The protein with the highest combined probability and accuracy was a glycine transporter (98.6% probability, 99.17% model accuracy), while the lowest recorded probability score was 50.53%. When fluoxetine was run through this server, as it is such a well-documented ligand, a table of known strong binders was given, including serotonin, adrenaline and norepinephrine transporters, and thus validates this tool as the approach correctly identified known protein targets.
BindingDB returned only a single potential target: ferricytochrome c peroxidase. This enzyme catalyzes the oxidation of organic substrates via hydrogen peroxide and is localized to the mitochondrial intermembrane space, where it contributes to apoptosis through cardiolipin oxidation. When fluoxetine was submitted, serotonin and dopamine transporters were identified with a Tanimoto coefficient of 1.00, indicating a perfect match and validating this search method.
Using DIA-DB, 18 potential targets were identified. The highest-scoring protein lacked a name and PDB entry and was therefore excluded. The next highest-ranking target was aldose reductase, and among the top 10, peroxisome proliferator-activated receptors (PPARs) were identified three times, corresponding to the α, δ, and γ isoforms. This finding is particularly significant because PA is a known PPARγ agonist, suggesting that HT–PA may share protein targets and biological functions with its parent compound. Fluoxetine was inputted via a SMILE code, but no results were generated. Given that this server is exclusively focused on diabetes-related matters, and fluoxetine does not pertain to diabetes, the absence of data was deemed acceptable and incorporated into this study.
Polypharmacology browser 2 (PPB2) yielded 20 targets across its best-performing methods. Several proteins were identified multiple times, including arachidonate 5-lipoxygenase (ALOX5), cannabinoid CB2 receptor, and PPARγ, suggesting these proteins are strong candidates for HT–PA. Predicted target classes are displayed in Fig. 2, which shows compound–protein associations derived from multiple similarity and machine learning methods. Among these, the best-performing methods were ECfp4 NN(ECfp4) + NB(ECfp4) and Xfp NN(Xfp) + NB(ECfp4), highlighting the complementary nature of different computational strategies. When fluoxetine was inputted, serotonin and norepinephrine transporters were given as targets, with the nearest neighbours having a Tc value of 1.00, once again indicating a perfect match and validating this search method.
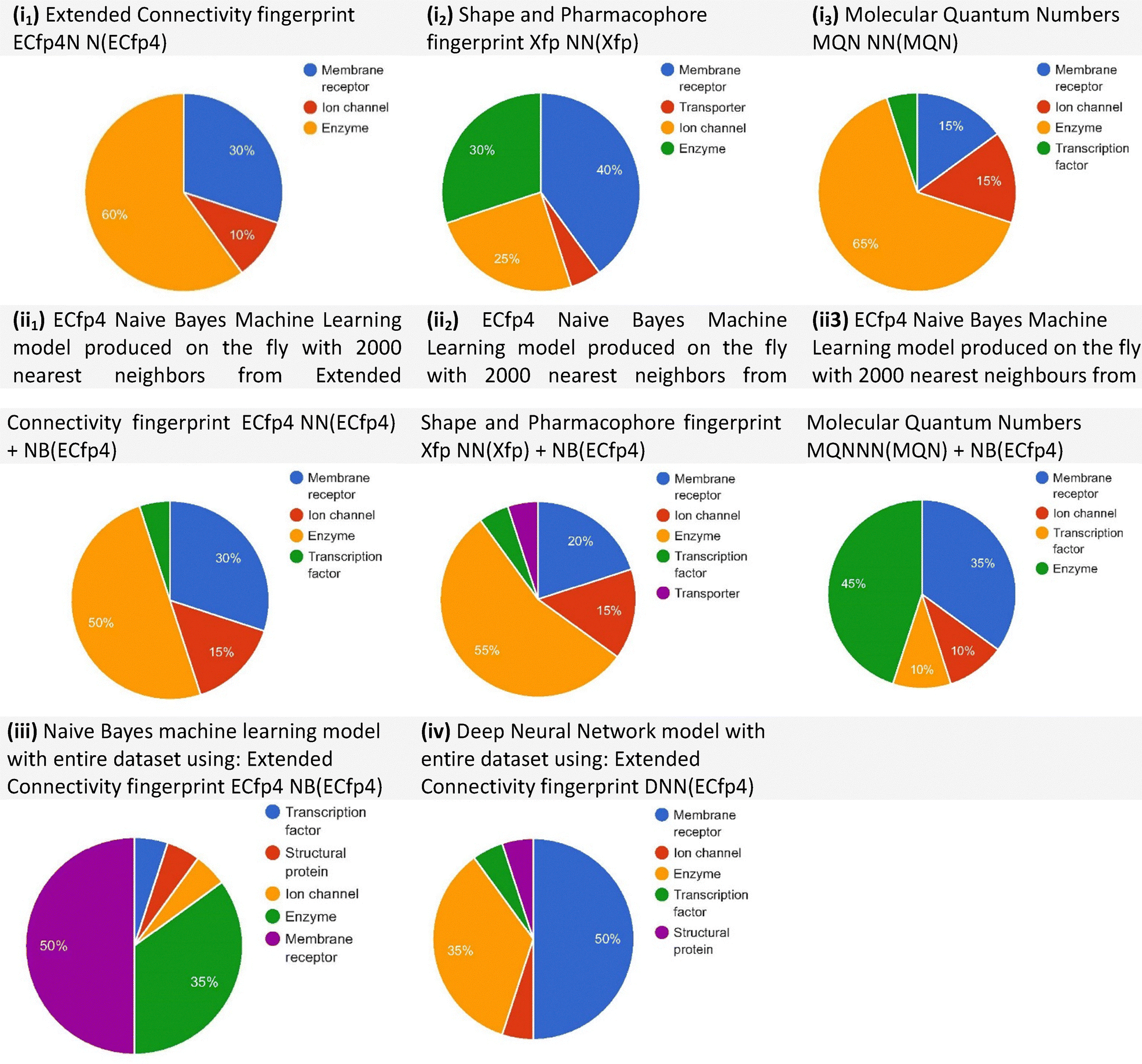 | ||
| Fig. 2 Predicted target classes displayed as pie charts based on compound–protein target associations in ChEMBL22, using different methods from the polypharmacology browser 2 (PPB2). The methods include: (i) nearest-neighbour search using: extended connectivity fingerprint (ECfp4 NN(ECfp4)), shape and pharmacophore fingerprint (Xfp NN(Xfp)), and molecular quantum numbers (MQN NN(MQN)); (ii) Naive Bayes (NB) models generated on the fly with the 2000 nearest neighbours, combining: ECfp4 NN(ECfp4) + NB(ECfp4), Xfp NN(Xfp) + NB(ECfp4), and MQN NN(MQN) + NB(ECfp4); (iii) Naive Bayes model trained on the entire dataset using ECfp4 (NB(ECfp4)); (iv) deep neural network (DNN) model trained on the entire dataset using ECfp4 (DNN(ECfp4)). Among these, the best-performing methods were ECfp4 NN(ECfp4) + NB(ECfp4) and Xfp NN(Xfp) + NB(ECfp4). | ||
Across all servers, nearly 300 proteins were identified. Given the size of this list, an exhaustive analysis of every target was impractical. Differences in scoring, probability values, and ranking across platforms further complicated direct comparisons. Consequently, subsequent analyses focused on proteins consistently identified across multiple databases, as shown in Table 3. Recurrent identification across servers strengthens confidence in these proteins as potential HT–PA targets. Three proteins were prioritized for more detailed discussion based on biological relevance: ALOX5, PPARγ, and TRP channels. ALOX5, identified by three servers, is a cytosolic enzyme that catalyses the conversion of arachidonic acid into leukotrienes, which are potent mediators of inflammation. HT–PA may exert anti-inflammatory effects via modulation of ALOX5 activity, complementing findings from studies of polyphenol-rich compounds, which inhibit lipoxygenases and cyclooxygenases. PPARγ is a nuclear hormone receptor regulating lipid, glucose, and lipoprotein metabolism. It is a well-characterized target of PA, and its identification by DIA-DB and PPB2 suggests that HT–PA may retain similar metabolic and neuroprotective activities. PPARγ is predominantly expressed in adipose tissue, regulating genes involved in lipid metabolism and insulin signalling, and contributes to central nervous system processes such as inflammation and energy homeostasis. Activation of PPARγ by HT–PA could therefore offer therapeutic benefits in metabolic disorders and neurodegenerative diseases. TRP channels are membrane proteins involved in nociception and pain perception. Their repeated identification across SEA and SwissTargetPrediction indicates that HT–PA could modulate these ion channels, suggesting potential analgesic applications. Several TRP subfamilies were highlighted among the predicted targets, as detailed in Tables 2 and 4.
| Protein | Server | Function |
|---|---|---|
| Arachidonate 5-lipoxygenase (ALOX5, 5-LOX) | STP | Catalyses the oxygenation of arachidonate, an intermediate in the formation of leukotrienes, key regulators of inflammation.63 |
| SEA | ||
| PPB2 | Plays a role in glucose homeostasis and cancer cell proliferation. | |
| Arachidonate 15-lipoxygenase (ALOX15, 15-LOX-1) | SEA | Enzyme catalysing stereospecific peroxidation of fatty acids, with effects linked to cell differentiation, inflammation, carcinogenesis and atherogenesis.64 |
| PPB2 | ||
| Protein kinase C alpha (PKCα) | SEA | Plays both a positive and/or negative role in cell proliferation, differentiation, motility, apoptosis and inflammation.65 |
| PPB2 | ||
| Peroxisome proliferator-activated receptor (PPARγ) | DIA-DB | Transcription factor with a role in energy metabolism, cell differentiation, apoptosis and inflammation66 |
| PPB2 | Has action as an anticancer agent, by slowing the growth and differentiation of cancer cells.67 | |
| Putative P2Y purinoreceptor 10 (P2RY10) | SEA | Suspected to have a role in the regulation of migration of T cells.68 P2 purinoreceptors might have a role in various conditions like cancer, diabetes, renal failure and thrombosis69 |
| PPB2 | ||
| Sphingosine 1-phosphate receptor 3 (S1PR3) | STP | Overexpressed in many forms of cancers, potentially increasing tumour growth70 |
| SuperPred | ||
| Transient receptor potential cation channel subfamily A member 1 (TRPA1) | SEA | Excitatory ion channel, acting as a sensor for pain, temperature, and water regulation71 |
| PPB2 |
| Protein | Server (subfamily) | Function |
|---|---|---|
| Cannabinoid receptors 1 and 2 (CRN1, CRN2) | SEA (CRN1) | Receptor 1 has a role in synaptic signalling and mediation of GABA72 |
| PPB2 (CRN2) | Receptor 2 is expressed predominantly in the immune system and control cytokine release72 | |
| Pyruvate dehydrogenase kinase isoforms 1 and 2 (PDK1, PDK2) | STP (PDK1) | PDK1 and PDK2 have similar roles in cell metabolism and energy production in mitochondria73 |
| DIA-DB (PDK2) | ||
| Transient receptor potential cation channel subfamilies M, V (TRVM8, TRVP1, TRVP2) | STP (TRVM8) | Ion channels which control the entry of Ca2+ into the cell for various cellular pathways74 |
| SEA (TRPV1/2) | ||
| Adenosine receptor A2a and A2b (ADORA2a, ADORA2b) | STP (ADORA2a) | Control the inhibition or stimulation of adenylyl cyclase, regulating cytoprotective effects75 |
| SEA (ADORA2b) | ||
| Dipeptidyl peptidase 2, 4, 8 and 9 (DPP2, DPP4, DPP8, DPP9) | DIA-DB (DPP4) | Major role in glucose metabolism, with many inhibitors being developed for this protein to treat type 2 diabetes76 |
| SuperPred (DPP2/8/9) | ||
| Protein-tyrosine phosphatase 2C, 1B and 9 (PRPN1, PRPN2, PRPN9) | SuperPred (PRPN1/2) | Regulate phosphorylation of various signalling molecules in signal transduction cascades77 |
| DIA-DB (PRPN9) |
Limitations and methodological considerations
Several limitations must be acknowledged when interpreting these results. Database accessibility and updates are critical, as online servers are continuously updated with new compounds, protein structures, and experimental data. Repeating calculations in the future may yield different predictions, affecting reproducibility and comparability across studies.The study relied exclusively on ligand-based shape similarity screening, which, while efficient, may overlook targets with flexible or atypical binding sites not represented in current ligand libraries. Scoring and ranking limitations are also present, as probability and Tanimoto similarity values differ between platforms, and some servers provide unranked predictions (Fig. 1 and Table 2). Species differences further underscore the importance of selecting biologically relevant organisms for downstream validation. Finally, computational predictions alone cannot fully confirm HT–PA's interactions; experimental studies including biochemical assays, cellular models, and in vivo investigations are essential.
Despite these limitations, integrating multiple servers provides a more comprehensive view of potential targets. Combining ligand-based approaches with other computational methods, such as structure-based docking, protein–protein interaction analysis, and molecular dynamics simulations, may uncover targets not detectable through ligand-based screening alone. Table 4 illustrates proteins consistently identified across servers, demonstrating how cross-validation improves confidence in predicted targets.
This study provides several broader lessons for reverse screening. Combining multiple servers increases reliability and helps identify overlapping, high-confidence targets such as PPARγ, ALOX5, and TRP channels (Table 4). Machine learning and similarity-based descriptors complement experimental databases, providing unique insights into protein interactions, as visualized in Fig. 2. Awareness of database updates, scoring limitations, and species specificity is critical for interpreting results accurately. Cross-validation against well-characterized compounds, exemplified by fluoxetine, enhances confidence in predictions. Collectively, these findings underscore the value of using complementary computational approaches alongside experimental validation. While exemplified here with HT–PA, these insights are broadly relevant to natural products, drug candidates, and phenolipid derivatives, offering practical lessons for assessing confidence and potential biases in reverse screening workflows.
Conclusions
Reverse docking has emerged as a valuable computational strategy for identifying potential protein targets of small molecules, facilitating drug repurposing, polypharmacology studies, and toxicity prediction. However, despite its advantages, several challenges limit its broader applicability and reliability. One major limitation is the availability and accessibility of target databases. While some tools come with built-in datasets, others rely on user-defined collections, which can introduce biases depending on the curation process. Additionally, many reverse docking databases are not openly accessible, restricting researchers’ ability to validate findings across different platforms.Another significant limitation is the reliability of protein structure databases. In shape- and pharmacophore-based screening, the accuracy of predictions depends on the quality of reference target–ligand interactions. These methods tend to identify well-characterized proteins but perform less reliably for novel targets, reinforcing a bias toward established pathways. Moreover, most scoring functions were originally developed for forward docking and are not fully optimized for large-scale reverse screening, introducing systematic bias into binding affinity estimates. The absence of standardized negative datasets further complicates performance assessment, as it remains difficult to distinguish true targets from false positives. Receptor flexibility also remains a persistent challenge, since most reverse docking approaches treat proteins as rigid structures and fail to capture the conformational dynamics that influence ligand binding. Ensemble and flexible docking algorithms are under active development, but their high computational demands currently limit their widespread application.
Despite these challenges, reverse docking continues to evolve. Improvements in score normalization, incorporation of receptor dynamics, and integration with complementary computational methods, including machine learning, are expanding its potential. As structural databases grow, and computational resources become more powerful, reverse docking may become a more robust tool for target identification and drug discovery. Addressing the key issues of database accessibility, receptor flexibility, and inter-target score normalization will be crucial to realizing its full utility.
While hydroxytyrosyl punicate (HT–PA) serves as an illustrative case study, the following conclusions draw broader lessons from the comparative use of reverse screening approaches and databases. HT–PA is a novel phenolipid compound formed by combining hydroxytyrosol (HT) with punicic acid (PA), a fatty acid highly concentrated in pomegranate seed oil. This combination is expected to enhance their therapeutic effectiveness against specific disorders, exemplifying a circular economy approach in drug discovery. Using ligand-based reverse similarity screening, numerous potential targets of HT–PA were identified across multiple computational platforms. These findings suggest that HT–PA may act on diverse targets, which could be beneficial in a therapeutic context but also raises the need to carefully evaluate its specificity and safety profile. In this study, all targets were identified through ligand-based shape similarity screening. While effective, this approach is limited to identifying targets structurally related to known ligands and may overlook proteins with flexible or atypical binding sites capable of accommodating broader chemical diversity. To expand on these insights, additional computational methods such as structure-based docking or protein–protein interaction network analysis could uncover novel targets beyond those detected with ligand-based approaches. Experimental validation through in vitro and in vivo studies, including biochemical assays, cellular models, and animal models, will be critical to confirm HT–PA's interactions with its predicted targets. Advanced computational techniques that account for protein conformational flexibility, such as molecular dynamics simulations, are also being explored and may identify additional targets that static models miss.
Evaluating HT–PA's binding specificity will be equally important. Binding affinity studies and structure–activity relationship (SAR) analyses could clarify its selectivity profile and highlight potential off-target effects, guiding the rational design of derivatives with improved therapeutic windows. By integrating multiple computational and experimental strategies, a deeper understanding of HT–PA's mechanisms of action and pharmacological potential can be achieved. This comprehensive approach will not only enhance the safety and efficacy of HT–PA but also enable the development of more targeted therapies with improved clinical outcomes. Importantly, the potential for HT–PA to align with the principles of the circular economy, through the sustainable use of natural compounds, offers an additional opportunity to promote more environmentally responsible drug discovery.
Beyond the specific case of HT–PA, this study highlights broader insights into the current state of reverse screening approaches. Comparing multiple servers and ligand-based methods reveals their respective strengths, limitations, and potential biases, providing a practical framework for researchers seeking to identify protein targets of small molecules. Ligand-based methods are efficient and widely accessible but may overlook targets with flexible or atypical binding sites. Integrating complementary approaches, such as structure-based docking, network analysis, or machine learning, can improve coverage and predictive accuracy. Taken together, these findings provide practical guidance for applying reverse screening methods to natural products and small molecules, and underscore the value of integrating computational predictions with experimental validation.
Author contributions
The work was designed by C. D. experimental work was carried out by J. S. and C. D. the manuscript was written by J. S. and C. D. and with contributions from all authors. Figures and schemes were generated with contributions from J. S., M. C., and C. D. All authors have given approval to the final version of the manuscript.Conflicts of interest
There are no conflicts to declare.List of abbreviations
| PA | Punicic acid |
| HT | Hydroxytyrosol |
| HT–PA | Hydroxytyrosol punicate |
| ALOX5 | Arachidonate 5-lipoxygenase |
| TRPV1 | Transient receptor potential cation channel |
| PPARγ | Peroxisome proliferator-activated receptor |
| ML | Machine learning |
| QSAR models | Conventional quantitative structure–activity relationship |
| PCM | Proteochemometrics |
| PPB2 | Polypharmacology browser 2 |
| PSO | Pomegranate seed oil |
| T2DM | Type 2 diabetes mellitus |
| HTS | High-throughput screening |
| T c | Tanimoto coefficient |
| SMILE code | Simplified molecular input line entry system |
| FP2 | FingerPrint2D |
| ATC | Anatomical therapeutic chemical |
| SEA | Similarity ensemble approach |
| PPB2 | Polypharmacology browser 2 |
| ECfp4 | Extended-connectivity fingerprint 4 |
| MQN | Molecular quantum numbers |
| Xfp | Extended fingerprints |
| CBD | City block distances |
| NN | Nearest neighbour |
| NB | Naïve Bayes |
| DNN | Deep neural network |
Data availability
The data supporting the findings of this study are available from the corresponding author upon reasonable request. Additionally, the analyses used publicly available, third-party open-access databases, which can be accessed via the following links: SwissTargetPrediction: https://www.expasy.org/resources/swisstargetprediction. Similarity ensemble approach (SEA): https://sea.bkslab.org/. SuperPred: https://prediction.charite.de/. BindingDB: https://www.bindingdb.org/rwd/bind/index.jsp. DIA-DB: https://bio-hpc.ucam.edu/dia-db/index.php. Polypharmacology browser 2 (PPB2): https://ppb2.gdb.tools/.Acknowledgements
C. D. thanks HECBioSim, the UK High End Computing Consortium for Biomolecular Simulation (https://hecbiosim.ac.uk), which is supported by the EPSRC (EP/L000253/1). For the purpose of open access, C. D. has applied a Creative Commons Attribution (CC-BY) license to any Author Accepted Manuscript version arising.References
- J. Abramson, J. Adler, J. Dunger, R. Evans, T. Green, A. Pritzel, O. Ronneberger, L. Willmore, A. J. Ballard, J. Bambrick, S. W. Bodenstein, D. A. Evans, C. C. Hung, M. O'Neill, D. Reiman, K. Tunyasuvunakool, Z. Wu, A. Žemgulytė, E. Arvaniti, C. Beattie, O. Bertolli, A. Bridgland, A. Cherepanov, M. Congreve, A. I. Cowen-Rivers, A. Cowie, M. Figurnov, F. B. Fuchs, H. Gladman, R. Jain, Y. A. Khan, C. M. R. Low, K. Perlin, A. Potapenko, P. Savy, S. Singh, A. Stecula, A. Thillaisundaram, C. Tong, S. Yakneen, E. D. Zhong, M. Zielinski, A. Žídek, V. Bapst, P. Kohli, M. Jaderberg, D. Hassabis and J. M. Jumper, Nature, 2024, 630, 493–500 CrossRef CAS PubMed.
- D. D. Wang, W. Wu and R. Wang, J. Cheminf., 2024, 16, 2 Search PubMed.
- Z. Qiao, W. Nie, A. Vahdat, T. F. Miller and A. Anandkumar, Nat. Mach. Intell., 2024, 6, 195–208 CrossRef.
- R. Chowdhury, N. Bouatta, S. Biswas, C. Floristean, A. Kharkar, K. Roy, C. Rochereau, G. Ahdritz, J. Zhang, G. M. Church, P. K. Sorger and M. AlQuraishi, Nat. Biotechnol., 2022, 40, 1617–1623 CrossRef CAS PubMed.
- Q. Luo, S. Wang, H. Y. Li, L. Zheng, Y. Mu and J. Guo, Protein Sci., 2024, 33, e5167 CrossRef CAS PubMed.
- H. Huang, G. Zhang, Y. Zhou, C. Lin, S. Chen, Y. Lin, S. Mai and Z. Huang, Front. Chem., 2018, 6, 1–28 Search PubMed.
- S.-Q. Yang, Q. Ye, J.-J. Ding, Y. Ming-Zhu, A.-P. Lu, X. Chen, T.-J. Hou and D.-S. Cao, Wiley Interdiscip. Rev.:Comput. Mol. Sci., 2021, 11, e1504 CAS.
- J. Vamathevan, D. Clark, P. Czodrowski, I. Dunham, E. Ferran, G. Lee, B. Li, A. Madabhushi, P. Shah, M. Spitzer and S. Zhao, Nat. Rev. Drug Discovery, 2019, 18, 463–477 CrossRef CAS PubMed.
- M. Awale and J.-L. Reymond, Systems Chemical Biology: Methods and Protocols, 2019, pp. 255–272 Search PubMed.
- S. Dara, S. Dhamercherla, S. S. Jadav, C. M. Babu and M. J. Ahsan, Artif. Intell. Rev., 2022, 55, 1947–1999 Search PubMed.
- A. Daina and V. Zoete, Commun. Chem., 2024, 7, 105 CrossRef CAS PubMed.
- J. C. Wang, P. Y. Chu, C. M. Chen and J. H. Lin, Nucleic Acids Res., 2012, 40, W393–W399 CrossRef CAS PubMed.
- Y. C. Martin, J. L. Kofron and L. M. Traphagen, J. Med. Chem., 2002, 45, 4350–4358 CrossRef CAS PubMed.
- K. Y. Ji, C. Liu, Z. Q. Liu, Y. F. Deng, T. J. Hou and D. S. Cao, Briefings Bioinf., 2023, 24, 1–15 CAS.
- A. Daina, O. Michielin and V. Zoete, Nucleic Acids Res., 2019, 47, W357–W364 CrossRef CAS PubMed.
- F. Shaikh, H. K. Tai, N. Desai and S. W. I. Siu, J. Cheminf., 2021, 13, 44 CAS.
- X. Liu, S. Ouyang, B. Yu, Y. Liu, K. Huang, J. Gong, S. Zheng, Z. Li, H. Li and H. Jiang, Nucleic Acids Res., 2010, 38, W609–W614 CrossRef CAS PubMed.
- B. Bhattacharjee and J. Chatterjee, Asian Pac. J. Cancer Prev., 2013, 14, 3735–3742 CrossRef PubMed.
- S. M. Ge, D. L. Zhan, S. H. Zhang, L. Q. Song and W. W. Han, Am. J. Transl. Res., 2016, 8, 5187–5198 CAS.
- H. Li, Z. Gao, L. Kang, H. Zhang, K. Yang, K. Yu, X. Luo, W. Zhu, K. Chen, J. Shen, X. Wang and H. Jiang, Nucleic Acids Res., 2006, 34, W219–W224 CrossRef CAS PubMed.
- X. Chen, C. Y. Ung and Y. Chen, Nat. Prod. Rep., 2003, 20, 432–444 Search PubMed.
- F. Wang, F.-X. Wu, C.-Z. Li, C.-Y. Jia, S.-W. Su, G.-F. Hao and G.-F. Yang, J. Cheminf., 2019, 11, 73 Search PubMed.
- X. Xu, M. Huang and X. Zou, Biophys. Rep., 2018, 4, 1–16 CrossRef CAS PubMed.
- J. Li, J. Zhang, R. Guo, J. Dai, Z. Niu, Y. Wang, T. Wang, X. Jiang and W. Hu, Eur. J. Med. Chem., 2025, 285, 117269 Search PubMed.
- M. K. Khan, M. Raza, M. Shahbaz, I. Hussain, M. F. Khan, Z. Xie, S. S. A. Shah, A. K. Tareen, Z. Bashir and K. Khan, Front. Chem., 2024, 12, 1408740 CrossRef CAS PubMed.
- C. Hasselgren and T. I. Oprea, Annu. Rev. Pharmacol. Toxicol., 2024, 64, 527–550 CrossRef CAS PubMed.
- M. Astigarraga, A. Sánchez-Ruiz and G. Colmenarejo, Artif. Intell. Life Sci., 2025, 7, 100127 Search PubMed.
- S. D’Souza, K. V. Prema and S. Balaji, Drug Discovery Today, 2020, 25, 748–756 CrossRef PubMed.
- P. Willett, Annu. Rev. Inf. Sci. Technol., 2009, 43, 3–71 Search PubMed.
- G. Maggiora, M. Vogt, D. Stumpfe and J. Bajorath, J. Med. Chem., 2014, 57, 3186–3204 Search PubMed.
- P. Willett, J. M. Barnard and G. M. Downs, J. Chem. Inf. Comput. Sci., 1998, 38, 983–996 Search PubMed.
- N. Mathai and J. Kirchmair, Int. J. Mol. Sci., 2020, 21, 3585 CrossRef CAS PubMed.
- A. Peón, C. C. Dang and P. J. Ballester, Front. Chem., 2016, 4, 15 Search PubMed.
- X. Liu, Y. Xu, S. Li, Y. Wang, J. Peng, C. Luo, X. Luo, M. Zheng, K. Chen and H. Jiang, J. Cheminf., 2014, 6, 33 Search PubMed.
- S. Struckmann, M. Ernst, S. Fischer, N. Mah, G. Fuellen and S. Möller, Briefings Bioinf., 2021, 22, 1–8 Search PubMed.
- P. Willett, Methods Mol. Biol., 2011, 672, 133–158 CrossRef CAS PubMed.
- P. Baldi and R. Nasr, J. Chem. Inf. Model., 2010, 50, 1205–1222 Search PubMed.
- Y.-F. Shi, Z.-X. Yang, S. Ma, P.-L. Kang, C. Shang, P. Hu and Z.-P. Liu, Engineering, 2023, 27, 70–83 CrossRef CAS.
- K. López-Pérez, J. F. Avellaneda-Tamayo, L. Chen, E. López-López, K. E. Juárez-Mercado, J. L. Medina-Franco and R. A. Miranda-Quintana, Artif. Intell. Chem., 2024, 2, 100077 Search PubMed.
- A. Paul and M. Radhakrishnan, Trends Food Sci. Technol., 2020, 105, 273–283 CrossRef CAS.
- M. Mete, U. U. Unsal, I. Aydemir, K. P. Sönmez and I. M. Tuglu, Anti-Cancer Agents Med. Chem., 2019, 19, 1120–1131 CrossRef CAS PubMed.
- G. F. P. Tiwari, R. B. Singh, F. Mehdi, W. Al-Awaida, N. Hadi, A. Mehdi, M. H. Shahrajabian, M. Khatibi, S. Omidvar and S. S. Zadeb, in Funcfional Foods and Nutraceuficals in Metabolic and Non-Communicable Diseases, ed. R. B. S. S. W. A. Isaza, Academic Press, 2022, pp. 121–135 Search PubMed.
- P. Aruna, D. Venkataramanamma, A. K. Singh and R. P. Singh, Compr. Rev. Food Sci. Food Saf., 2016, 15, 16–27 CrossRef CAS PubMed.
- M. E. Grossmann, N. K. Mizuno, T. Schuster and M. P. Cleary, Int. J. Oncol., 2010, 36, 421–426 CAS.
- Y. Khajebishak, L. Payahoo, M. Alivand and B. Alipour, J. Cell. Physiol., 2019, 234, 2112–2120 CrossRef CAS PubMed.
- M. A. Shabbir, M. R. Khan, M. Saeed, I. Pasha, A. A. Khalil and N. Siraj, Lipids Health Dis., 2017, 16, 99 CrossRef PubMed.
- M. T. Boroushaki, H. Mollazadeh and A. R. Afshari, Int. J. Pharm. Sci. Res., 2016, 7, 430 Search PubMed.
- M. Pirzadeh, N. Caporaso, A. Rauf, M. A. Shariati, Z. Yessimbekov, M. U. Khan, M. Imran and M. S. Mubarak, Crit. Rev. Food Sci. Nutr., 2021, 61, 982–999 CrossRef CAS PubMed.
- C. M. Guerra-Vázquez, M. Martínez-Ávila, D. Guajardo-Flores and M. Antunes-Ricardo, Foods, 2022, 11, 252 CrossRef PubMed.
- S. S. Anusree, V. M. Nisha, A. Priyanka and K. G. Raghu, Mol. Cell. Endocrinol., 2015, 413, 120–128 CrossRef CAS PubMed.
- E. P. Lansky, G. Harrison, P. Froom and W. G. Jiang, Invest. New Drugs, 2005, 23, 121–122 CrossRef CAS PubMed.
- P. Kandylis and E. Kokkinomagoulos, Foods, 2020, 9, 122 CrossRef CAS PubMed.
- S. Granados-Principal, J. L. Quiles, C. L. Ramirez-Tortosa, P. Sanchez-Rovira and M. C. Ramirez-Tortosa, Nutr. Rev., 2010, 68, 191–206 CrossRef PubMed.
- J. L. Quiles, M. C. Ramírez-Tortosa and P. Yaqoob, Olive oil and health, Cabi, 2006 Search PubMed.
- M. Robles-Almazan, M. Pulido-Moran, J. Moreno-Fernandez, C. Ramirez-Tortosa, C. Rodriguez-Garcia, J. L. Quiles and M. Ramirez-Tortosa, Food Res. Int., 2018, 105, 654–667 CrossRef CAS PubMed.
- R. Fabiani, A. De Bartolomeo, P. Rosignoli, M. Servili, G. F. Montedoro and G. Morozzi, Eur. J. Cancer Prev., 2002, 11, 351–358 CrossRef CAS PubMed.
- J. Han, T. P. Talorete, P. Yamada and H. Isoda, Cytotechnology, 2009, 59, 45–53 CrossRef CAS PubMed.
- M. Bertelli, A. K. Kiani, S. Paolacci, E. Manara, D. Kurti, K. Dhuli, V. Bushati, J. Miertus, D. Pangallo, M. Baglivo, T. Beccari and S. Michelini, J. Biotechnol., 2020, 309, 29–33 CrossRef CAS.
- R. Bernini, F. Crisante, N. Merendino, R. Molinari, M. C. Soldatelli and F. Velotti, Eur. J. Med. Chem., 2011, 46, 439–446 CrossRef CAS PubMed.
- K. Todoerti, M. E. Gallo Cantafio, M. Oliverio, G. Juli, C. Rocca, R. Citraro, P. Tassone, A. Procopio, G. De Sarro, A. Neri, G. Viglietto and N. Amodio, Int. J. Mol. Sci., 2021, 22, 11639 CrossRef CAS.
- E. Belmonte-Reche, M. Martínez-García, P. Peñalver, V. Gómez-Pérez, R. Lucas, F. Gamarro, J. M. Pérez-Victoria and J. C. Morales, Eur. J. Med. Chem., 2016, 119, 132–140 CrossRef CAS PubMed.
- O. Cruz-López, E. Díaz-de-Cerio, B. Rubio-Ruiz, J. M. Espejo-Román, P. Peñalver, J. C. Morales, M. F. Caboni, A. Conejo-García and V. Verardo, J. Funct. Foods, 2024, 117, 106249 CrossRef.
- O. Rådmark, O. Werz, D. Steinhilber and B. Samuelsson, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2015, 1851, 331–339 CrossRef PubMed.
- H. Kuhn, M. Walther and R. J. Kuban, Prostaglandins Other Lipid Mediators, 2002, 68–69, 263–290 CrossRef CAS PubMed.
- S. Nakashima, J. Biochem., 2002, 132, 669–675 CrossRef CAS PubMed.
- K. L. Houseknecht, B. M. Cole and P. J. Steele, Domest. Anim. Endocrinol., 2002, 22, 1–23 CrossRef CAS PubMed.
- H. P. Koeffler, Clin. Cancer Res., 2003, 9, 1–9 CAS.
- M. Gurusamy, D. Tischner, J. Shao, S. Klatt, S. Zukunft, R. Bonnavion, S. Günther, K. Siebenbrodt, R.-I. Kestner, T. Kuhlmann, I. Fleming, S. Offermanns and N. Wettschureck, Nat. Commun., 2021, 12, 6798 CrossRef CAS PubMed.
- M. P. Abbracchio and G. Burnstock, Pharmacol. Ther., 1994, 64, 445–475 CrossRef CAS.
- Q. Li, Y. Li, C. Lei, Y. Tan and G. Yi, Clin. Chim. Acta, 2021, 519, 32–39 CrossRef CAS PubMed.
- S. Bevan, T. Quallo and D. A. Andersson, Handbook of Experimental Pharmacology, Springer, Berlin Heidelberg, 2014, pp. 207–245 DOI:10.1007/978-3-642-54215-2_9.
- R. G. Pertwee, Int. J. Obes., 2006, 30, S13–S18 CrossRef CAS PubMed.
- G. Sutendra and E. D. Michelakis, Front. Oncol., 2013, 3, 38 Search PubMed.
- B. Nilius, G. Owsianik, T. Voets and J. A. Peters, Physiol. Rev., 2007, 87, 165–217 CrossRef CAS PubMed.
- K. A. Jacobson and Z.-G. Gao, Nat. Rev. Drug Discovery, 2006, 5, 247–264 CrossRef CAS.
- H.-U. Demuth, C. H. S. McIntosh and R. A. Pederson, Biochim. Biophys. Acta, Proteins Proteomics, 2005, 1751, 33–44 CrossRef CAS PubMed.
- N. K. Tonks, Nat. Rev. Mol. Cell Biol., 2006, 7, 833–846 CrossRef CAS PubMed.
| This journal is © the Owner Societies 2026 |