
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceUnraveling the magnetoelectric effect using electric field-controlled magnetic anisotropy: a theoretical study
Flaurent
Heully-Alary
a,
Barthélémy
Pradines
a,
Benjamin
Cahier
a,
Nicolas
Suaud
 a,
Talal
Mallah
b and
Nathalie
Guihéry
*a
a,
Talal
Mallah
b and
Nathalie
Guihéry
*a
aUniversité de Toulouse, CNRS UMR5626, LCPQ, 118 route de Narbonne, F-31062 Toulouse, France. E-mail: nathalie.guihery@irsamc.ups-tlse.fr
bInstitut de Chimie Moléculaire et des Matériaux d’Orsay CNRS, Université Paris-Saclay UMR 8182, 17, avenue des Sciences, Orsay, 91400, France
First published on 13th February 2026
Abstract
This article highlights the microscopic origin of the magnetoelectric effect, which could enable crucial control of magnetic properties using electric fields. In particular, it explains the influence of electric fields on the magnetic anisotropy of a Ni(II) complex characterized by significant uniaxial anisotropy and notable rhombicity. Our findings reveal that the electric field's effect on the axial anisotropy parameter primarily arises from atomic displacements, whereas for the rhombic parameter, it is driven by electronic structure changes. While recent observations show that electric fields applied in the direction of the molecular dipole moment induce a greater variation in D than fields applied perpendicular to it due to greater atomic displacement, here we demonstrate a striking exception: applying the electric field in the molecular direction associated with an almost zero dipole moment causes a variation in D seven times greater than that induced in the direction of a large permanent dipole moment (∼10 Debye). This observation reveals a previously unknown magnetoelectric coupling mechanism. We demonstrate that the magnetoelectric effect inducing variations of the zero-field splitting parameters is dictated by the nature of electronic excitations involved in the spin–orbit couplings that produce the magnetic anisotropy, more specifically in the orientation of the orbitals involved in these excitations, rather than by the dipole moment's magnitude. However, as the variation of D is essentially governed by the impact of field-induced geometric changes on the energy of the orbitals involved in these excitations, one may expect larger responses if one of these orbitals points in the direction of the dipole moment. Besides, as this behavior can be rationalized using crystal field theory, it offers a general principle that can help the design of molecules with predictable magnetoelectric responses.
Introduction
Electric fields hold significant promise as efficient external stimuli for tuning the physical properties of magnetic molecules and materials1via the magnetoelectric (ME) effect.2–7 In the context of quantum information, electric fields were initially proposed to perform logical operations on individual atomic spins. This stems from their ability to be more easily focused at the nanoscale than magnetic fields, allowing them to affect electron wave functions and modulate hyperfine interactions.8The strength of the ME response depends on the specific magnetic property under consideration—such as magnetic anisotropy,9g-tensor,10–13 hyperfine coupling14 and exchange interactions.4,15,16 Notably, electric field tuning of local magnetic anisotropy, i.e., zero-field splitting (ZFS) parameters, enables coherent spin control. This has been demonstrated in both atoms embedded in solid-state environments17–21 and in molecular systems.4,5,22
Theoretical calculations have allowed the description of how electric fields influence the spin Hamiltonian parameters of molecular systems, including interactions like magnetic exchange, anisotropy of exchange tensor and the Dzyaloshinskii–Moriya vector for which the impact of the electric field has not yet been measured.23–31 However, ab initio calculations remain scarce. A deep understanding of the microscopic mechanisms underpinning electric field effects on spin degrees of freedom ultimately requires an analysis of low energy spectrum and wave functions. Since spin is inherently insensitive to electric fields, it must couple with orbital degrees of freedom for the ME effect to occur.
We recently reported such a study22 on a series of Mn(II) complexes isostructural to the Ni(II) complex studied here, using pulsed electron paramagnetic resonance (EPR) spectroscopy and coherent control of superposed quantum states. We showed that the ME effect—reflected in changes in the axial ZFS parameter—can be understood through the analysis of spin–orbit coupled wave functions that explain the contributions of the various excited states to ZFS. The primary conclusion was that the electric field modifies the axial ZFS parameter mainly by altering the molecular geometry, rather than directly affecting the electronic structure. Importantly, applying the electric field perpendicular to the bond associated with the largest dipole moment neither influenced the axial magnetic anisotropy nor introduced any detectable rhombic anisotropy, leading to the conclusion that in the case of Mn(II) complexes the electric field has little effect on ZFS if applied along a direction with a weak dipole moment. These findings align with previous results on Mn(II) and Fe(III) ions (both d5 ions with weak spin–orbit coupling) embedded in piezoelectric matrices, where electric fields modulate the local charge environment and amplify ME effects.17,18,20 However, due to the inherently weak ME coupling in such d5 systems that EPR can detect, it is difficult to deconvolute individual mechanisms and fully elucidate the microscopic origin of ME.
The main goal of this study is to elucidate the mechanism by which electric fields influence magnetic anisotropy—one of the key levers to achieve ME coupling in molecular systems—by studying a complex with both a large axial ZFS and non-zero rhombicity. To this end, we investigate a system where the Mn(II) (S = 5/2) magnetic center is replaced by a Ni(II) (S = 1), i.e., [Ni(Me6tren)Cl]+, hereafter referred to as complex 1.32 The high uniaxial ZFS (D ≈ −108 cm−1 at the optimized geometry) results from significant spin–orbit coupling. Jahn–Teller distortion contributes to substantial rhombicity, E, which was strictly zero for the Mn(II) complex that does not exhibit Jahn–Teller distortion. Enhanced spin–orbit coupling effects should lead to stronger electric-field sensitivity of the ZFS parameters.
The ME effect in this context is quantified through the variation of the ZFS parameters with the electric field strength ![[F with combining right harpoon above (vector)]](https://www.rsc.org/images/entities/i_char_0046_20d1.gif) , i.e., dD/dF and dE/dF. We also examine the field-orientation dependence of these parameters. Since the molecule possesses a dipole moment, applying the field along the dipole moment orientation is expected to strongly influence its geometry. Finally, following the same strategy as the one we used previously,22 we decouple and quantify the impact of the electric field on the electronic structure and on the geometric structure.
, i.e., dD/dF and dE/dF. We also examine the field-orientation dependence of these parameters. Since the molecule possesses a dipole moment, applying the field along the dipole moment orientation is expected to strongly influence its geometry. Finally, following the same strategy as the one we used previously,22 we decouple and quantify the impact of the electric field on the electronic structure and on the geometric structure.
A further motivation for selecting a Ni(II) complex lies in its integer spin (S = 1) and a negative D parameter, which allows the molecular geometry to give rise to two low-lying states (linear combinations of MS = ±1) that may serve as a qubit at zero magnetic field, forming a so-called “clock transition.” Clock transitions are known to protect spin states—at first order—from magnetic fluctuations that cause decoherence.12,33–37 In such systems, electric fields can be used to tune the superposed quantum states. Understanding the electric field's influence on the rhombic ZFS parameter E is therefore valuable for implementing single-qubit gates in these molecular systems.
Theory and computational information
Methodology for the rationalization of the nature and magnitude of ZFS for [Ni(Me6trenCl)](ClO4) 1 upon electric field application
1 crystallizes in the trigonal space group R3c, imposing a molecular three-fold symmetry axis C3 along the Ni–Cl direction (Fig. 1a) and a C3 symmetry point group.32 However, some of us have already shown through both EPR studies and theoretical calculations that 1 is prone to a Jahn–Teller effect, which induces a distortion in the plane perpendicular to the Ni–Cl direction, leading to a C1 symmetry point group. This effect lifts the degeneracy of the d orbitals (Fig. 1b) and hence of the spin–orbit-free states, responsible for the experimentally observed rhombicity (E ≠ 0). Calculations of the energies of the spin–orbit-free states show an energy difference of 2166 cm−1 between the ground T0 and the first excited T1 triplet states. Each of these two states is mainly spanned by a single determinant: |dxz![[d with combining macron]](https://www.rsc.org/images/entities/char_0064_0304.gif) xzdyzyzdxyxydx2−y2dz2| (see Fig. 1b in C1 symmetry) with a weight of 0.93 in T0 and |dxzxzdyzyzdxydx2−y2x2−y2dz2| (where an electron has been excited from dxy to dx2−y2) with a weight of 0.98 in T1. Therefore, the symmetry lowering due to the Jahn–Teller distortion leads to a situation where the spin is a not-too-bad quantum number. The ZFS spin Hamiltonian is, hence, relevant to describe the energy of the spin states with the axial D and the rhombic E parameters of the D tensor (Fig. 1c). The easy axis of magnetization (Z) is along the pseudo C3 axis and the YZ plane contains the Ni–N3 bond (Fig. 1a).
xzdyzyzdxyxydx2−y2dz2| (see Fig. 1b in C1 symmetry) with a weight of 0.93 in T0 and |dxzxzdyzyzdxydx2−y2x2−y2dz2| (where an electron has been excited from dxy to dx2−y2) with a weight of 0.98 in T1. Therefore, the symmetry lowering due to the Jahn–Teller distortion leads to a situation where the spin is a not-too-bad quantum number. The ZFS spin Hamiltonian is, hence, relevant to describe the energy of the spin states with the axial D and the rhombic E parameters of the D tensor (Fig. 1c). The easy axis of magnetization (Z) is along the pseudo C3 axis and the YZ plane contains the Ni–N3 bond (Fig. 1a).
 | ||
| Fig. 1 (a) Representation of 1 with Ni in green, Cl in pink, N in blue, C in black, and H in white; (b) relative energy of the d orbitals of 1 in the case of a C3 symmetry with the two couples of degenerate orbitals, and the main configuration in the T0 ground state in the presence of a Jahn–Teller distortion inducing a C1 symmetry; note the lift of degeneracy of the d orbitals; (c) the energy difference of the SO states (MS = 0) corresponds to T00, while ϕ+ and ϕ− are the + and − combinations of the MS = 1 (T0+1) and MS = −1 (T0−1) components of the ground state, as a function of the ZFS parameters D and E. | ||
In order to better understand the effect of an electric field, we use the strategy we had implemented in the study of the manganese complex.22 This consists of decoupling and quantifying the impact of the field on the electronic structure (i.e., effect of the electron cloud deformation) from that on the geometric structure. In case (a), the ZFS is calculated by applying the field on the geometry optimized without a field, which allows us to obtain the effect of the field on the electronic structure alone. In case (b), the ZFS is calculated without applying the field but on different geometries optimized under a field. The absence of the field removes the deformation of the electron cloud, allowing us to assess the impact of the field on the geometric structure alone. In case (c), the ZFS is calculated in the presence of the field on the geometries optimized under a field and therefore combines both effects.
The electric field is first applied along the Ni–Cl direction, i.e. along Z that corresponds to the easy axis of magnetization and where the component of the dipole moment is maximal. To study the dependence of the ZFS parameters on the field orientation, we then apply the field along Y; the YZ plane contains the N1, Ni and N3 atoms (Fig. 1a). The Ni–N3 bond length is the most different and shortest among the three Ni–N bonds in the XY plane. To rationalize the field dependence of the ZFS parameters, we use a perturbative approach in order to account for the main variations in the contributions of the excited states. The SO Hamiltonian used for the rationalization is  , where i runs over all electrons of the d8 configuration. As already shown,32 the first excited triplet state has the largest (negative) contribution to D and is responsible for its negative sign. This contribution is due to the interaction via SOC between, on the one hand, the two MS = 1 and, on the other hand, the two MS = −1 components of the ground state (T0±1) and the first excited state (T1±1) components. At the second order of perturbation, this contribution is given by eqn (1):
, where i runs over all electrons of the d8 configuration. As already shown,32 the first excited triplet state has the largest (negative) contribution to D and is responsible for its negative sign. This contribution is due to the interaction via SOC between, on the one hand, the two MS = 1 and, on the other hand, the two MS = −1 components of the ground state (T0±1) and the first excited state (T1±1) components. At the second order of perturbation, this contribution is given by eqn (1):
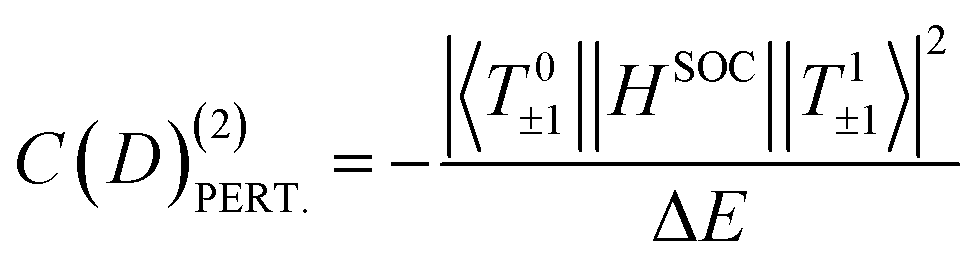 | (1) |
Of course, other excited triplet states, coupled through the lixsix + liysiy part of the SOC operator, can bring positive contributions to D by couplings their MS = ±1 components to the MS = 0 (T00) component of the ground state. The open-shell singlet S1 based on the same electronic configuration as T1 also couples through lizsiz to T00, providing a positive contribution. These contributions must be taken into account in order to recover the correct value of D. However, the electric field has almost no impact on those terms (see Table S1); they are therefore not considered in the rationalization of the electric field effect. While eqn (1) is used to rationalize the results, more precise values of the contributions are provided by a procedure38,39 based on the effective Hamiltonian theory,40,41 which is implemented in the standard code ORCA (see the Computational information section).42,43 We refer interested readers to the four previously cited articles.
If the X and Y axes of the molecular coordinates are those of the D tensor, the rhombic parameter E comes from the effective coupling between the MS = 1 and −1 components of the ground triplet state (T01 and T0−1), which leads to two spin–orbit non-degenerate states that are linear combinations of these components. This effective coupling is due to the SOCs between these components and the excited states. The contributions of the excited states to the rhombic parameter E are also provided by the code ORCA. Unfortunately, as this quantity is particularly small for 1, the trends in E as a function of the field are not always reproduced by the sum of the contributions of each excited state, which in some cases is even opposite to the variation in E. Therefore, we do not use these contributions for rationalization purposes. Instead, we use the analysis of the wave functions of the two excited states that contribute most to the rhombic parameter E and give information on the difference in electron density in the X and Y directions, and therefore on the magnitude of E.
The field is applied in two opposite directions referred to as positive +F and negative −F with a maximal amplitude of |F| = 1.028 × 109 V m−1. The positive direction for the field in the Z direction induces an elongation of the Ni2+–Cl− bond, i.e., the positive region of the electric potential is above Cl− in Fig. 1. The positive direction for the field in the Y direction induces an elongation of the Ni2+–N3 bond, i.e., the positive region of the electric potential is to the right of N3 in Fig. 1a. One may note that, as is the case in other ab initio calculations of the impact of the electric field on electronic and/or magnetic properties,22,29,31 the values of the field (that would be achievable by STM) are much greater than those used experimentally (in EPR experiments, for instance). As shown later, (i) geometric distortions are particularly small, so for the sake of precision, it is safer to apply strong fields, and (ii) the variations of the ZFS parameters with the field are linear and the slopes would be unchanged for lower values of the field.
Computational information
Wave function-based calculations have been performed using the ORCA package.42,43 In the first place, complete active space self-consistent field (CASSCF) calculations have been performed to account for non-dynamic correlations. The CAS(8,5) active space contains 8 electrons in the 5 orbitals mainly carried by the 3d orbitals of the Ni(II) ion, and the set of orbitals has been optimized using an average procedure over all triplet and all singlet states of the configuration. To account for dynamic correlations at the 2nd order of perturbation, NEVPT244 calculations have been carried out for all states. The spin–orbit state-interaction method45 was then used to account for spin–orbit couplings (SOCs) between the MS components of all spin states using the NEVPT2 correlated energies as diagonal elements of the SO matrix. The QZVPP extended basis sets46 have been used for Ni atoms (14s10p5d4f2g), SVP for H (2s1p) and TZVP for all other atoms (6s3p2d1f for N, 8s4p2d1f for Cl and 6s3p2d1f for C). This approach has been successfully employed in numerous studies investigating the magnetic anisotropy of transition metal47–57 and lanthanide and actinide58–65 complexes.As the field may change the geometric structure, geometry optimizations were performed for different values and orientations of the field.42 We used the Hay–Wadt LanL2TZ(f) basis set66,67 for Ni and its corresponding relativistic effective core, a Pople triple-ζ basis set (6-311G) for N68 and Cl69 atoms, a Pople double-ζ plus polarization basis set (6-31G*)70 for C atoms, and a Pople double-ζ basis set (6-31G) for H atoms70 and the B3LYP functional.71 As magnetic anisotropy parameters may be very sensitive to the structure of the coordination sphere of the metal,56,72 convergence thresholds for geometry optimization were set as VERYTIGHT. It should be noted that only geometry optimizations were performed at the DFT level; all values of magnetic anisotropy parameters were obtained at the CAS + NEVPT2 + SOSI level.
Results and discussion
Impact of the electric field on the axial ZFS parameter D
We begin by studying the impact of the electric field on the variation in the ground state energy illustrated in Fig. S1 and on the dipole moment presented in Table 1. The application of the field along the Z direction from −F to +F leads globally to a linear variation of the energy of the ground state dominated by the electronic structure variation (case (a)) as expected for a Stark effect (Fig. S1). This linear behavior results from the fact that μZ is the largest component of the dipole moment so that the variation of the field does not cause a change of its sign (Table 1). The situation is different when the field is applied along the Y direction. Here, the energy of the ground state has a global quadratic variation also dominated by the electronic structure variation (case (a); Fig. S1). The quadratic behavior stems from the change of the sign of μY when the field is varied (Table 1), since μY is almost zero (−0.054 Debye) for F = 0. It is worth noting that the overall variation of the dipole moment (Δμi) has the same magnitude when F is parallel or perpendicular to Z (i.e. along the Y direction) even though μZ is almost one order of magnitude larger than μY for all field values.| (a) | (b) | (c) | |
|---|---|---|---|
| μ Z at −F | 7.19 | 8.04 | 7.13 |
| μ Z at +F | 9.02 | 8.17 | 9.08 |
| ΔμZ | 1.83 | 0.13 | 1.95 |
| μ Y at −F | −0.97 | −0.12 | −1.03 |
| μ Y at +F | 0.86 | 0.01 | 0.93 |
| ΔμY | 1.83 | 0.13 | 1.96 |
The variation of D with the field is linear in all three cases, whether the field is parallel or perpendicular to Z, as expected for non-centrosymmetric species (Fig. 2). The additivity of the slopes (Table 2) shows that our method of decomposition of the impact of the field on the electronic structure and geometry is reliable. Comparison of the results obtained for the three cases shows that the overall variation of D (case (c)) is dominated by the impact of the field on the geometry (case (b)) for both directions of the field.
 | ||
| Fig. 2 Axial parameter D as a function of the field applied in the Z (left) and Y (right) directions. Case (a): electronic effect only (blue dot-dashed line), case (b): geometric effect only (green dashed line) and case (c): both electronic and geometric effects (red solid line). | ||
| Hz/(V m−1) | Case (a) | Case (b) | Case (c) | Sum of cases (a) and (b) |
|---|---|---|---|---|
| dD/dF (F//Z) | −13.52 | +26.66 | +13.15 | +13.14 |
| dD/dF (F//Y) | 25.32 | −110.75 | −85.45 | −85.43 |
When F//Z increases, |D| increases with the change in electronic structure (case (a)), while it decreases in both cases (b) and (c). The situation is opposite when the field is perpendicular to Z (F//Y), i.e., |D| decreases for a change in the electronic structure (case (a)), while it increases in both cases (b) and (c). More importantly, the variation of |D| is almost 7 times larger when F is along the Y axis than when it is along the Z axis, even though the dipole moment is one order of magnitude larger along the Z axis than along the Y axis.
To rationalize these results, we analyze the contribution C(D) to D from the most contributing excited states. The largest contribution is provided by the first excited triplet state T1 (Tables S1 and S2) and this contribution is also the most sensitive to the field, i.e., the variation ΔC(D) with the field has by far the highest value. Therefore, only the properties of T1 are reported in Table 3. It is worth noting that the variations with the field (Tables S1 and S2) of the contributions calculated perturbatively C(D)(2)PERT. (eqn (1)) are in good agreement with those calculated ab initio, showing that one may use the variation of the energy differences between the spin–orbit free states to rationalize the impact of the field on the parameter D. In the following, we consider the variation of the absolute value of D (|D|) when the field is varied from −F to +F.
| Orientation of F for cases (a), (b) and (c) | ΔC(D) | Δ(ΔE) | Δ(Δε(dx2−y2 − dxy)) | |
|---|---|---|---|---|
| F//Z | (a) | −0.88 | −1.60 | 6.58 |
| (b) | 2.70 | 36.2 | 258.98 | |
| (c) | 1.83 | 34.7 | 267.76 | |
| F//Y | (a) | 1.73 | 24.40 | 274.34 |
| (b) | −8.84 | −122.40 | −561.86 | |
| (c) | −7.11 | −98.20 | −287.51 | |
The energy difference ΔE between the ground state and T1 is mainly governed by the energy difference Δε(dx2−y2 − dxy) between the dx2−y2 and dxy orbitals because T1 is obtained following an excitation from dxy to dx2−y2 (Fig. 1). Table 3 also shows the variation Δ(Δε(dx2−y2 − dxy)) of these energy differences between −F and +F. They can be directly linked to the change in the ligand field under the effect of the electric field. Indeed, for +F, the Ni–Cl bond length increases (Table 4) in order to maximize the dipole moment and thus the interaction with the electric field. Concomitantly, all the other distances decrease, inducing a stronger ligand field in the X and Y directions, which affects the energy of the dx2−y2 orbital directly pointing towards the N3 atoms (see Fig. 3) more than that of dxy. The energy difference between these two orbitals therefore increases with the field, resulting in a greater energy difference between the ground state and the first excited state, responsible for the decrease in |D|, according to eqn (1).
| d(Ni–X) | Δ(d), F//Z | Δ(d), F//Y |
|---|---|---|
| Cl | 131.8 × 10−4 | 2.4 × 10−4 |
| N1 (apical) | −28.9 × 10−4 | 1.7 × 10−4 |
| N2 | −69.6 × 10−4 | −27.3 × 10−4 |
| N3 | −48.9 × 10−4 | 11.4 × 10−4 |
| N4 | −66.5 × 10−4 | −4.1 × 10−4 |
 | ||
| Fig. 3 Picture of the dxy (left) and dx2−y2 (right) orbitals optimized at the CASSCF level in the absence of a field. | ||
The application of the field along the Y axis leads to an increase of the Ni–N3 bond length and to a decrease of the two other Ni–N distances in the XY plane (Table 3). These geometric changes result in a weaker ligand field in the Y direction and a stronger ligand field between the X and Y axes. The tails of the dx2−y2 and dxy orbitals on the ligands (Fig. 3) illustrate the delocalization of dx2−y2 mainly on N3, while dxy delocalizes on N2 and N4. Consequently, the dx2−y2 orbital is stabilized, while dxy is destabilized, leading to a decrease of the energy difference Δε between the two orbitals and therefore a decrease of the energy difference ΔE between the ground and the first excited state. According to eqn (1), this rationalizes the increase of |D| (Table 3).
The principal conclusion drawn from the above studies and their analysis is that the electric field exerts a stronger influence on D when applied along the Y direction, which is perpendicular to the largest dipole moment. This behavior, which is in contrast to previously reported experimental observations and theoretical predictions,17,18,20,22 stems from the nature of the excitation responsible for the strong contribution to D. Indeed, the energy of the excitation from dxy to dx2−y2 is much more sensitive to a perturbation applied along the Y than along the Z axis, rationalizing the stronger response when the field is perpendicular to the largest dipole moment.
Impact of the electric field on the rhombic term E
The variation of E with the field is linear in all three cases, whether the field is parallel or perpendicular to the Z axis (Fig. 4); however, due to a lack of precision in calculating this particularly small quantity, the coefficient of trend line determination for case (b) and F//Z is lower (0.88) than that of all others (>0.99). Here again the additivity of the slopes (Table 5) demonstrates the accuracy of our method of decomposition. Comparison of the results obtained for the three cases shows that the overall variation of E (case (c)) is dominated by the impact of the field on the electronic structure (case (a)) for both directions of the field. | ||
| Fig. 4 Rhombic parameter E as a function of the field applied in the Z (left) and Y (right) directions. Case (a): electronic effect only (blue dot-dashed line), case (b): geometric effect only (green dashed line) and case (c): both electronic and geometric effects (red solid line). | ||
| Hz/(V m−1) | Case (a) | Case (b) | Case (c) | Sum of cases (a) and (b) |
|---|---|---|---|---|
| dE/dF (F//Z) | −2.53 | −0.36 | −2.88 | −2.89 |
| dE/dF (F//Y) | −2.39 | 1.20 | −1.19 | −1.19 |
When F//Z increases, E decreases in cases (a) and (c), while it remains almost constant in case (b). The situation is similar for cases (a) and (c) when the field is perpendicular to Z (F//Y), but it now increases in case (b). It can also be noted that the strongest response to the field is now obtained for the direction F//Z.
Unfortunately, it was not possible to use the contributions to E calculated ab initio to rationalize the obtained results. Indeed, the sum of the contributions increases with the field, while the overall computed values of E decrease. Nevertheless, it is possible to analyze the wave functions of the third and fourth triplet states T2 and T3 that contribute mostly to E, with contributions of opposite sign. The wave functions of these states have large coefficients on the determinants with three electrons in the pair of orbitals dxz and dyz, i.e. resulting from single excitations (from T0) from the dxz or dyz orbital to either dx2−y2 or dz2 (see Fig. 1c). Since dx2−y2, dxy and dz2 are symmetric in the X and Y directions, only six determinants with MS = 1 distinguish the X and Y axes: three have a single occupation in dxz and the other three have a single occupation in dyz. In the case of purely axial anisotropy, i.e. E = 0, we can expect the weights of the first three determinants to be equal to those of the last three ones in T2 and T3, ensuring equal electron density in the X and Y directions and therefore strictly offsetting contributions. The weights w(dxz) and w(dyz) of determinants having a single occupation in dxz or dyz, respectively, their differences Δw(X − Y) and the variations of their difference Δ(Δw(Y − X)) between −F and +F are reported in Table S3. Only the variations of their differences are reported in Table 6.
| F//Z | F//Y | |
|---|---|---|
| Case (a) | −0.528 | −0.174 |
| Case (b) | −0.088 | +0.002 |
| Case (c) | −0.621 | −0.170 |
The strongest response of the rhombic parameter E to the electric field is observed when it is applied in the Z direction. This result is here as well determined by the nature of the excitations providing the main contributions to E. Indeed, in the two states T2 and T3, the orbitals, namely dxz or dyz, involved in the excitation have a component along the Z axis. Hence, the corresponding excitation energies will be more sensitive to a perturbation applied in the Z than along the Y direction.
Conclusions
Through an accurate decomposition of the impact of the electric field on the electronic and geometric structures, it has been possible to provide an in-depth analysis of the effect of the electric field on ZFS parameters. The main results can be summarized as follows:(i) The variation in D caused by the impact of the field on the electronic structure alone (case (a)) is opposite to that induced by geometric changes (case (b)). When both electronic and geometric changes are accounted for in the calculation (case (c)), a similar behavior to that in case (b) is observed, showing that the impact of the field on the axial parameter in mainly governed by the field-induced geometric changes. It is true for both directions of the application of the field: for F//Z, |D| decreases, while it increases for F//Y. As the geometric changes dominate the response of D to the field, an analysis of the ligand field changes induced by the field can be used to rationalize both results.
(ii) Concerning the rhombicity E, we observe that the overall (case (c)) response to the electric field is dominated by the electronic structure changes (case (a)). To rationalize these results, we analyzed the wave functions of the excited states contributing most to E.
The most spectacular and interesting result is the variation of the axial parameter D with the field that is considerably ∼100 times greater than that observed in our previous study on [Mn(Me6tren)X]+. More importantly, d|D|/dF is ∼7 times larger when the field is applied along the direction where the dipole moment component Y is very small than along the Z direction where it is close to 10 Debye. The variation of the parameter E is less important but still large in particular for F//Z. As E quantifies the anisotropy between X and Y, one might have expected that applying the field along the Y axis would generate greater effects than along the Z axis. Actually, these a priori non-intuitive results are rationalized by the nature of the excitations involved in the SOCs that generate the magnetic anisotropy. Indeed, in the case of the axial parameter D, the first excited T1 state, which provides the most significant and sensitive contribution to the field, results from the excitation from dxy to dx2−y2. The energy difference between these two orbitals is less affected by the application of the field along the Z than the Y axis, which clearly discriminates the two orbitals as dx2−y2 directly points toward the Y axis. The same conclusion is reached when rationalizing the response to the field of the parameter E. The most important contributions to E come from the T2 and T3 excited states, which are coupled to the ground state by excitations from dxz or dyz to the dz2 or dx2−y2 orbital, involving mainly d orbitals with a Z component. It is therefore natural for these excitations to be more sensitive to the field along the Z direction than along the Y direction.
Although our study of the [MnMe6trenCl]+ complex22 concluded that the strongest electric-field response of D occurs when the field is applied collinearly with the dipole moment, these new results do not contradict that conclusion. In the Mn(II) system, the d5 electronic configuration gives rise to numerous excitations involving all d orbitals, each contributing weakly and often with opposite signs to the axial parameter D. Therefore, no clear conclusion regarding the role of excitation character could be drawn. Instead, geometric distortions—enhanced when the field is applied along the dipole moment—dominate the electric-field dependence of D. In contrast, the present results demonstrate that the dominant factor can be the nature of the orbitals involved in the relevant electronic excitations. The conclusions of both studies suggest a general design principle for maximizing electric-field responses: when one of the two orbitals involved in a key excitation is aligned with the dominant dipole component, the excitation energy becomes particularly sensitive to the applied field, leading to an enhanced response of the ZFS parameters.
The main message of this work is that the magnetoelectric effect—expressed here as a change in the ZFS parameters, i.e. the slopes dD/dF and dE/dF—can be very large. The modulation of the magnetic properties by the electric field is not necessarily large only when the field is oriented in the direction of the dipole moment. For both D and E, the nature of the excitations that couple the ground state to the various excited states responsible for the strongest contributions is the determining factor. Finally, we have shown that most of the rationalization of these effects requires analysis of the impact of the field on the orbitals involved in these excitations and can be related to ligand field theory. These conclusions, even though they were highlighted in a specific case, are general in nature, which is promising for the design of new complexes whose magnetic properties can be controlled and tuned by the electric field. Finally, the predicted large change in the ZFS parameters (dD/dF and dE/dF) can be useful for technological applications in the sense that weak electric fields may induce detectable magnetoelectric effects.
Conflicts of interest
There are no conflicts to declare.Data availability
The data that support the findings of this study are available from the authors upon request.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d5cp03260a.
Acknowledgements
N. G., N. S. and T. M. thank the ANR for financial support through project ANR-20-CE29-0010. This work also received government funding managed by the French National Research Agency under France 2030, reference ANR-23-PETQ-0007.References
- R. Ramesh and S. Manipatruni, Proc. R. Soc. A, 2021, 477, 20200942 CrossRef.
- J. Long, M. S. Ivanov, V. A. Khomchenko, E. Mamontova, J.-M. Thibaud, J. Rouquette, M. Beaudhuin, D. Granier, R. A. S. Ferreira, L. D. Carlos, B. Donnadieu, M. S. C. Henriques, J. A. Paixão, Y. Guari and J. Larionova, Science, 2020, 367, 671–676 CrossRef CAS PubMed.
- Y.-X. Wang, D. Su, Y. Ma, Y. Sun and P. Cheng, Nat. Commun., 2023, 14, 7901 CrossRef CAS PubMed.
- J. Liu, J. Mrozek, W. K. Myers, G. A. Timco, R. E. P. Winpenny, B. Kintzel, W. Plass and A. Ardavan, Phys. Rev. Lett., 2019, 122, 037202 CrossRef CAS PubMed.
- A. K. Boudalis, J. Robert and P. Turek, Chem. – Eur. J., 2018, 24, 14896–14900 CrossRef CAS PubMed.
- M. Lewkowitz, J. Adams, N. S. Sullivan, P. Wang, M. Shatruk, V. Zapf and A. S. Arvij, Sci. Rep., 2023, 13, 2769 CrossRef CAS PubMed.
- N. Guihery, G. Durand, M.-B. Lepetit and J.-P. Malrieu, Chem. Phys., 1994, 183, 61–72 CrossRef CAS.
- B. E. Kane, Nature, 1998, 393, 133–137 CrossRef CAS.
- A. S. Zyazin, J. W. G. van den Berg, E. A. Osorio, H. S. J. van der Zant, N. P. Konstantinidis, M. Leijnse, M. R. Wegewijs, F. May, W. Hofstetter, C. Danieli and A. Cornia, Nano Lett., 2010, 10, 3307–3311 CrossRef CAS PubMed.
- B. Kintzel, M. Fittipaldi, M. Böhme, A. Cini, L. Tesi, A. Buchholz, R. Sessoli and W. Plass, Angew. Chem., Int. Ed., 2021, 60, 8832–8838 CrossRef CAS PubMed.
- Z. Liu, Y.-X. Wang, Y.-H. Fang, S.-X. Qin, Z.-M. Wang, S.-D. Jiang and S. Gao, Natl. Sci. Rev., 2020, 7, 1557–1563 CrossRef CAS PubMed.
- J. Liu, J. Mrozek, A. Ullah, Y. Duan, J. J. Baldoví, E. Coronado, A. Gaita-Ariño and A. Ardavan, Nat. Phys., 2021, 17, 1205–1209 Search PubMed.
- L. Tacconi, A. Cini, A. Raza, L. Tesi, P. Bartolini, A. Taschin, J. van Slageren, M. Briganti, L. Sorace, M. Fittipaldi and M. Perfetti, J. Am. Chem. Soc., 2025, 147, 33040–33051 CrossRef CAS PubMed.
- S. Thiele, F. Balestro, R. Ballou, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Science, 2014, 344, 1135–1138 CrossRef CAS PubMed.
- M. Fittipaldi, A. Cini, G. Annino, A. Vindigni, A. Caneschi and R. Sessoli, Nat. Mater., 2019, 18, 329–334 CrossRef CAS PubMed.
- A. Cini, M. Böhme, B. Kintzel, M. Perfetti, W. Plass, R. Sessoli and M. Fittipaldi, Nat. Commun., 2025, 16, 6564 CrossRef CAS PubMed.
- R. E. George, J. P. Edwards and A. Ardavan, Phys. Rev. Lett., 2013, 110, 027601 CrossRef PubMed.
- J. Liu, V. V. Laguta, K. Inzani, W. Huang, S. Das, R. Chatterjee, E. Sheridan, S. M. Griffin, A. Ardavan and R. Ramesh, Sci. Adv., 2021, 7, eabf8103 CrossRef CAS PubMed.
- Y.-H. Fang, Z. Liu, S. Zhou, P.-X. Fu, Y.-X. Wang, Z.-Y. Wang, Z.-M. Wang, S. Gao and S.-D. Jiang, J. Am. Chem. Soc., 2022, 144, 8605–8612 CrossRef CAS PubMed.
- Y.-C. Liu, J.-X. Chen, P.-X. Fu, Y.-Q. Liao, Y.-H. Wang, Y.-X. Wang, Z. Liu, S. Gao and S.-D. Jiang, J. Am. Chem. Soc., 2024, 146, 19397–19404 CrossRef CAS PubMed.
- Y.-C. Liu, J.-X. Chen, J.-F. Bu, P.-X. Fu, Y.-X. Wang, Z. Liu, S. Gao and S.-D. Jiang, J. Am. Chem. Soc., 2025, 147, 10423–10429 CrossRef CAS PubMed.
- M. V. Vaganov, N. Suaud, F. Lambert, B. Cahier, C. Herrero, R. Guillot, A.-L. Barra, N. Guihéry, T. Mallah, A. Ardavan and J. Liu, Nat. Chem., 2025, 17, 1903–1909 CrossRef CAS PubMed.
- B. Pradines, B. Cahier, N. Suaud and N. Guihéry, J. Chem. Phys., 2022, 157, 204308 CrossRef CAS PubMed.
- F. Meier, J. Levy and D. Loss, Phys. Rev. Lett., 2003, 90, 047901 CrossRef PubMed.
- F. Troiani, Phys. Rev. B, 2019, 100, 155424 CrossRef.
- A. Sarkar and G. Rajaraman, Chem. Sci., 2020, 11, 10324–10330 RSC.
- F. Heully-Alary, B. Pradines, N. Suaud and N. Guihéry, J. Chem. Phys., 2024, 161, 054310 CrossRef PubMed.
- Y. Lu, Y. Wang, L. Zhu, L. Yang and L. Wang, Phys. Rev. B, 2022, 106, 064405 CrossRef.
- T. Sharma, A. Singh and G. Rajaraman, Chem. – Eur. J., 2025, 31, e202402868 CrossRef PubMed.
- M. Trif, F. Troiani, D. Stepanenko and D. Loss, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 82, 045429 CrossRef.
- W. T. Morrillo, H. I. J. Cumming, A. Mattioni, J. K. Staab and N. F. Chilton, J. Am. Chem. Soc., 2024, 146, 25841–25851 CrossRef PubMed.
- R. Ruamps, R. Maurice, L. Batchelor, M. Boggio-Pasqua, R. Guillot, A. L. Barra, J. Liu, E.-E. Bendeif, S. Pillet, S. Hill, T. Mallah and N. Guihéry, J. Am. Chem. Soc., 2013, 135, 3017–3026 CrossRef PubMed.
- M. Shiddiq, D. Komijani, Y. Duan, A. Gaita-Ariño, E. Coronado and S. Hill, Nature, 2016, 531, 348–351 CrossRef PubMed.
- S. Giménez-Santamarina, S. Cardona-Serra, J. M. Clemente-Juan, A. Gaita-Ariño and E. Coronado, Chem. Sci., 2020, 11, 10718–10728 RSC.
- I. Tlemsani, F. Lambert, N. Suaud, C. Herrero, R. Guillot, A.-L. Barra, S. Gambarelli and T. Mallah, J. Am. Chem. Soc., 2025, 147, 4685–4688 CrossRef PubMed.
- G. Chen, B. C. Sheehan, I. Nikolov, J. W. Logan, C. A. Collett, G. Joshi, G. A. Timco, J. E. Denhardt, K. R. Kittilstved, R. E. P. Winpenny and J. R. Friedman, arXiv, 2025, preprint, arXiv:arXiv:2507.13714 DOI:10.48550/arXiv.2507.13714.
- G. Wolfowicz, A. M. Tyryshkin, R. E. George, H. Riemann, N. V. Abrosimov, P. Becker, H.-J. Pohl, M. L. W. Thewalt, S. A. Lyon and J. J. L. Morton, Nat. Nanotechnol., 2013, 8, 561–564 Search PubMed.
- R. Maurice, R. Bastardis, C. de Graaf, N. Suaud, T. Mallah and N. Guihéry, J. Chem. Theory Comput., 2009, 5, 2977–2984 CrossRef PubMed.
- J. P. Malrieu, R. Caballol, C. J. Calzado, C. de Graaf and N. Guihéry, Chem. Rev., 2014, 114, 429–492 CrossRef PubMed.
- C. Bloch, Nucl. Phys., 1958, 6, 329–347 CrossRef.
- J. des Cloizeaux, Nucl. Phys., 1960, 20, 321–346 CrossRef.
- F. Neese, F. Wennmohs, U. Becker and C. Riplinger, J. Chem. Phys., 2020, 152, 224108 CrossRef PubMed.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2022, 12, e1606 Search PubMed.
- C. Angeli, R. Cimiraglia, S. Evangelisti, T. Leininger and J.-P. Malrieu, J. Chem. Phys., 2001, 114, 10252–10264 CrossRef.
- P. Å. Malmqvist, B. O. Roos and B. Schimmelpfennig, Chem. Phys. Lett., 2002, 357, 230–240 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- Pentagonal Bipyramidal First-Row Transition Metal Complexes with Macrocyclic Ligand Containing Two Pyridine-N-Oxide Pendant Arms, https://colab.ws/articles/10.1021%2Facs.inorgchem.5c03514, (accessed December 17, 2025).
- M. Malik, A. Bieńko, R. Boča, J. Titiš, C. Rajnák, A. Świtlicka, D. C. Bieńko, M. Ozerov and A. Ozarowski, Dalton Trans., 2025, 54, 16955–16965 RSC.
- K. Bretosh, V. Béreau, F. Heully-Alary, N. Suaud, C. Duhayon, E. Duverger-Nédellec, N. Guihéry and J.-P. Sutter, Inorg. Chem. Front., 2025, 12, 3456–3468 RSC.
- A. Colin, Y. Wang, F. Lambert, N. Bridonneau, N. Suaud, R. Guillot, E. Rivière, Z. Halime, N. Guihéry, S. Ohkoshi and T. Mallah, Magnetochemistry, 2024, 10, 102 CrossRef.
- A. Mukhopadhyaya and Md. E. Ali, J. Phys. Chem. A, 2024, 128, 2339–2348 CrossRef PubMed.
- D.-C. Sergentu, B. L. Guennic and R. Maurice, Phys. Chem. Chem. Phys., 2024, 26, 6844–6861 RSC.
- J. Titiš, C. Rajnák and R. Boča, J. Phys. Chem. A, 2023, 127, 6412–6424 CrossRef.
- S. K. Gupta, S. V. Rao, S. Demeshko, S. Dechert, E. Bill, M. Atanasov, F. Neese and F. Meyer, Chem. Sci., 2023, 14, 6355–6374 RSC.
- M. Atanasov, N. Spiller and F. Neese, Phys. Chem. Chem. Phys., 2022, 24, 20760–20775 RSC.
- S. R. Chowdhury and S. Mishra, Phys. Chem. Chem. Phys., 2017, 19, 16914–16922 RSC.
- R. Maurice, K. Sivalingam, D. Ganyushin, N. Guihéry, C. de Graaf and F. Neese, Inorg. Chem., 2011, 50, 6229–6236 CrossRef PubMed.
- Y. Ai, Z.-W. Li, Z.-B. Guan and H. Jiang, J. Chem. Theory Comput., 2025, 21, 9631–9640 CrossRef PubMed.
- Md. A. Islam, C. Berthon, J. Jung and H. Bolvin, Inorg. Chem., 2023, 62, 17254–17264 CrossRef.
- A. B. Canaj, M. K. Singh, C. Wilson, G. Rajaraman and M. Murrie, Chem. Commun., 2018, 54, 8273–8276 RSC.
- M. Perfetti, M. Gysler, Y. Rechkemmer-Patalen, P. Zhang, H. Taştan, F. Fischer, J. Netz, W. Frey, L. W. Zimmermann, T. Schleid, M. Hakl, M. Orlita, L. Ungur, L. Chibotaru, T. Brock-Nannestad, S. Piligkos and J. van Slageren, Chem. Sci., 2019, 10, 2101–2110 RSC.
- M. Vonci, K. Mason, E. A. Suturina, A. T. Frawley, S. G. Worswick, I. Kuprov, D. Parker, E. J. L. McInnes and N. F. Chilton, J. Am. Chem. Soc., 2017, 139, 14166–14172 CrossRef PubMed.
- B. Hong, A. Näder, T. Sawallisch, T. Bode, S. Fichter, R. Gericke, P. Kaden, M. Patzschke, T. Stumpf, M. Schmidt and J. März, Inorg. Chem., 2024, 63, 17488–17501 CrossRef PubMed.
- S. Dey, G. Rajaraman and H. Bolvin, Chem. – Eur. J., 2022, 28, e202201883 CrossRef PubMed.
- F. Gendron and J. Autschbach, J. Phys. Chem. Lett., 2017, 8, 673–678 CrossRef PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef.
- L. E. Roy, P. J. Hay and R. L. Martin, J. Chem. Theory Comput., 2008, 4, 1029–1031 CrossRef PubMed.
- R. Krishnan, J. S. Binkley, R. Seeger and J. A. Pople, J. Chem. Phys., 1980, 72, 650–654 CrossRef.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef.
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef.
- D. G. A. Smith, L. A. Burns, K. Patkowski and C. D. Sherrill, J. Phys. Chem. Lett., 2016, 7, 2197–2203 CrossRef PubMed.
- P. A. Brayshaw, J.-C. G. Buenzli, P. Froidevaux, J. M. Harrowfield, Y. Kim and A. N. Sobolev, Inorg. Chem., 1995, 34, 2068–2076 CrossRef.
| This journal is © the Owner Societies 2026 |