Advantages of discrete variable representation in variational quantum eigensolvers for vibrational energy calculations
Received
27th July 2025
, Accepted 20th February 2026
First published on 17th March 2026
Abstract
While quantum computing algorithms have been widely applied for electronic structure calculations, applications to molecular dynamics remain scarce. Complex and varied landscapes of molecular potential energy surfaces give rise to vibrational states with a wide range of properties, making it difficult to construct a general representation of ro-vibrational states by a quantum computer with a limited number of qubits and gates. Another challenge is the exponential growth of the computational complexity – for example, the number of terms required to expand a general Hamiltonian in Pauli strings increases exponentially with the number of qubits. Here, we show that discrete variable representation (DVR) can be leveraged to represent molecular Hamiltonians by the polynomial (in the number of qubits) number of quantum circuits. We then demonstrate that DVR Hamiltonians lead to very efficient quantum ansatze for vibrational states. For this purpose, we develop a compositional quantum ansatz search that adapts gate sequences in variational quantum eigensolvers (VQE) to a specific molecular state. We apply VQE to compute the vibrational energy levels of Cr2 in seven electronic states, as well as those of van der Waals complexes Ar–HCl and Mg–NH. Our numerical results show that an accuracy of 1 cm−1 can be achieved by very shallow quantum circuits with 2 to 9 entangling gates.
I. Introduction
Accurate calculation of molecular properties is considered a promising application of quantum computing. The eigenstates of molecular Hamiltonians can be obtained on quantum computers by variational quantum eigensolvers (VQEs)1,2 that employ sequences of gates operating on qubits (quantum circuits) to prepare quantum states tailored for specific problems. VQEs have been applied for solving the electronic structure problem for molecules3–11 and lattice models.1,7,12 However, applications of VQEs to computations of ro-vibrational energies and states have been limited.13–20 Ref. 16 and 17 demonstrated a general approach for computing ro-vibrational energy levels of polyatomic molecules inspired by previous work on the electronic structure. However, the methods used in ref. 16 and 17 require extended quantum circuits including a large number of entangling gates. This makes the applications of VQEs to vibrational energy calculations challenging for quantum computers limited by errors and noise. The errors grow with the circuit size and make large quantum circuits impractical for implementation in current quantum devices. This challenge is compounded by transpilation of quantum circuits into hardware-specific gate sequences, which often further extends the size of the quantum circuits.
A central goal for quantum computing of molecular dynamics can thus be formulated to develop a general approach that (i) is applicable to a broad range of molecules with widely varying ro-vibrational states, from deeply bound to van der Waals states; (ii) yields high accuracy with shallow quantum circuits; and (iii) exhibits at most a polynomial scaling with the dimensionality of the molecular configuration space or, alternatively, in the number of qubits if binary basis encoding is used. Within the VQE framework, requirement (i) demands an ansatz for quantum circuits that can represent molecular states across widely varying landscapes of potential energy surfaces; requirement (ii) is necessary due to the noise and hardware limitations of current quantum computers; and requirement (iii) is essential for realizing the quantum advantage over classical methods.
Here, we explore an approach for computing the vibrational energy levels of molecules with VQEs based on discrete variable representation (DVR) of molecular Hamiltonians.21,22 We first derive the theoretical bounds on the quantum measurement complexity with VQE based on Fourier grid DVR. Our analysis shows that the structure of the DVR matrices can be exploited to reduce the expansion of the molecular Hamiltonians in measurement operators to the desired polynomial scaling in the number of qubits. In the second part of this work, we explore an automated construction of the quantum circuits for the vibrational energy computations. Our goal is to build quantum circuits, yielding accurate VQE results, without any constraints on the quantum ansatz. Our results demonstrate that DVR Hamiltonians lead to very efficient (small number of qubits and gates) quantum circuits for representing vibrational states of molecules by states of a quantum computer. To probe the minimal gate count for accurate vibrational VQE computations, we develop a compositional search algorithm that incrementally grows quantum circuits to identify optimal sequences in the space of gate permutations.
To illustrate the generality of this approach and the efficiency of the resulting quantum circuit representations of vibrational states, we consider Cr2 in seven different electronic states23 and van der Waals complexes Ar–HCl(1Σ) and Mg–NH(3Σ). These molecular systems exhibit vibrational states with widely different energies (from −55 to −15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cm−1 from the dissociation threshold) and spatial variations of wave functions and energy level patterns. Our compositional search yields quantum circuits that produce VQE results with accuracy <1 cm−1 for ground and excited vibrational energy levels, illustrating the ability of VQEs to compute the rotational constants and vibrational anharmonicity, with between 2 and 9 entangling gates, for diatomic and triatomic molecules. For reference, previous VQE calculations of vibrational energy levels required extended quantum circuits with >200 (for CO, COH and O3 molecules17) or between 44 and 140292 (for CO2, H2CO and HCOOH molecules16) entangling gates.
000 cm−1 from the dissociation threshold) and spatial variations of wave functions and energy level patterns. Our compositional search yields quantum circuits that produce VQE results with accuracy <1 cm−1 for ground and excited vibrational energy levels, illustrating the ability of VQEs to compute the rotational constants and vibrational anharmonicity, with between 2 and 9 entangling gates, for diatomic and triatomic molecules. For reference, previous VQE calculations of vibrational energy levels required extended quantum circuits with >200 (for CO, COH and O3 molecules17) or between 44 and 140292 (for CO2, H2CO and HCOOH molecules16) entangling gates.
The remainder of this article is organized as follows. After a brief introduction of VQEs, section 2.1 presents an algorithm to evaluate the DVR Hamiltonians with a polynomial number of measurements. Section 2.2 describes the algorithm for the compositional anzats optimization, which is followed by numerical results illustrating the efficiency and accuracy of the optimized Ansatze for Cr2 in seven different electronic states23 and van der Waals complexes Ar–HCl(1Σ) and Mg–NH(3Σ) in section 3. The work is summarized in section 4.
II. Theory
In VQEs, a quantum computer estimates the expectation value 〈ψ(φ)|Ĥ|ψ(j)〉, which is minimized by varying φ to yield the lowest eigenvalue Ei=0 and an approximate representation of the corresponding eigenvector of Ĥ. This method can be extended to compute excited states by optimizing2| | 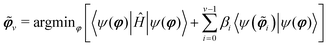 | (1) |
where βi ≥ Ei+1 − Ei and ![[small variant phi, Greek, tilde]](https://www.rsc.org/images/entities/b_i_char_e12c.gif) i denotes an optimal solution for the corresponding quantum state.
i denotes an optimal solution for the corresponding quantum state.
The quantum states |ψ(φ)〉 are obtained by quantum circuits acting on qubits, and the Hamiltonian is expanded in quantum operators. Most generally,
| | 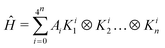 | (2) |
where
n is the number of qubits,
Kij ∈ {
σX,
σY,
σZ,
I} acting on qubit
j, {
σi} are the Pauli matrices, and
I is the identity matrix,
| | 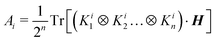 | (3) |
and
H is the Hamiltonian matrix in some basis. The computational complexity is determined by the number of non-zero terms in
eqn (2), which for a general matrix is 4
n; and the complexity of the quantum circuits yielding |
ψ(
φ)〉. In Section II.A, we show that the structure of DVR matrices allows an efficient quantum circuit representation of
Ĥ, scaling with
n as poly(
n).
A. Efficient measurement of DVR Hamiltonians
DVR is a finite basis representation, in which the coordinate operators (and consequently the potential energy) are diagonal. The DVR matrix of kinetic energy is not sparse. However, it has specific structure that is exploited here. We use the DVR introduced by Colbert and Miller21. Other DVR bases can be reduced to those in ref. 21 by coordinate transformations. As follows from ref. 21, the Hamiltonian matrix in the DVR representation has the following structure:| | 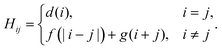 | (4) |
We use a number encoding to map the DVR basis states onto qubit states, where the indices of the flattened DVR basis states are mapped into their binary representation on the qubits. This allows VQE to compute the eigenvalues of H of size 2n × 2n using n qubits. As the number of DVR bases required for accurate results grows exponentially with the number of degrees of freedom of the system, the number of qubits necessary to represent the system grows polynomially with the size of the system.
As shown in Appendix A, the functions f and g in eqn (4) satisfy
| | 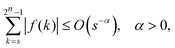 | (5) |
| | 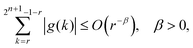 | (6) |
for 1 ≤
r ≪
N = 2
n and 1 ≤
s ≪
N = 2
n, where
N is the number of DVR states, assumed to be large.
Given a quantum state |ψ〉, our aim is to show that
| | 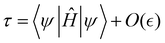 | (7) |
can be measured with the number of quantum circuits that scales polynomially with
n and
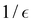
. We first use
eqn (5) and (6) to show that the expectation value
(7) can be approximated as
| | 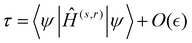 | (8) |
where
Ĥ(s,r) employs a truncated DVR matrix with the following elements:
| | 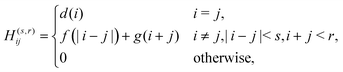 | (9) |
for
| | 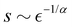 | (10) |
and
| | 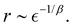 | (11) |
To prove eqn (8), we consider operators defined as a kth (anti)-diagonal matrix, with elements
where only + +, + − and − − combinations of signs are used. Since
Kk±± either permutes or shifts vector components, while either preserving the norm of vectors or truncating vectors in a finite Hilbert space, ‖
Kk±±ψ‖
2 ≤ ‖
ψ‖
2 (and the inequality is tight). This can be used in the Cauchy–Schwarz inequality |〈
ψ,
Kk±±ψ〉| ≤ ‖
ψ‖
2‖
Kk±±ψ‖
2 to bound the expectation value of
Kk±± as
| | | |〈ψ, Kk±±ψ〉| ≤ ‖ψ‖22. | (13) |
Using the triangle inequality and ‖
ψ‖
22 = 1, the approximation error can be bounded by
| | 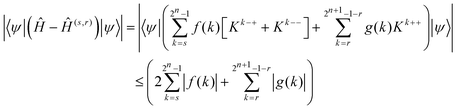 | (14) |
Due to
eqn (5), (6) and (8), the relationship holds for s and r given by
eqn (10) and (11).
We next seek to transform |ψ〉 by short-depth ![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) i, so that
i, so that
| | 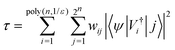 | (15) |
where
j is the index that enumerates the states of
n qubits in the computational
Z basis. We leverage
eqn (8) to decompose
τ into contributions from a diagonal matrix (
D),
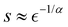
diagonal bands (
tk) and
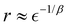
anti-diagonal components (
ak), as follows:
| | 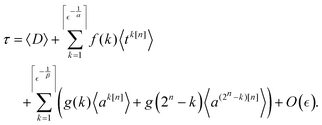 | (16) |
We note that the construction of
tk[n] and
ak[n] described below produces contributions to the main diagonal upon measurements. This is offset by a corresponding change in the diagonal matrix elements, as specified in Appendix B. The computation of the diagonal contribution 〈
D〉 is classically efficient. In this case, one can set {
Vi} = {1} and combine
w1i =
Dii from Appendix B with the weights given by the measurements in the computational basis.
To construct tk[n], we note that 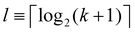 is the smallest number of qubits that allows the k-th band. We consider l-qubit entangled states produced from the Z basis by a sequence of CNOT gates shown in Fig. 1 (left panel). The resulting state is
is the smallest number of qubits that allows the k-th band. We consider l-qubit entangled states produced from the Z basis by a sequence of CNOT gates shown in Fig. 1 (left panel). The resulting state is
| | 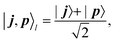 | (17) |
where
and
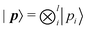
are the binary representations of the row (
p) and column (
j) indices of the DVR matrix, with
p =
j +
k, and
ji and
pi representing single-qubit states. The projection of an
n-qubit state onto the |
j,
p〉
l state has 2 non-zero off-diagonal matrix elements of magnitude 1/2. The binary representation of the row and column positions of these off-diagonal elements differ by
l bits. The position of the non-zero off-diagonal elements is controlled by the position of the CNOT gates in
Fig. 1. Thus, the expectation value of
tk[l] can be obtained by measurements with, at most,
k l-qubit entangled states.
 |
| | Fig. 1 Left panel: Preparation of state (17) from the computational basis. H denotes the Hadamard gate and the circles – the CNOT gates. Right: Measuring tk[n] that includes matrix elements on the main and kth diagonals. The open circles  are obtained from projections of (17), are obtained from projections of (17),  – via – via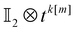 , and , and 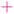 – by additional measurements in the entangled basis. – by additional measurements in the entangled basis. | |
We now observe that 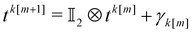 , where γk[m] represents k pairs of elements missing from the middle of the tensor product matrix (shown as pluses in Fig. 1, right panel). These elements can be directly targeted by additional measurements in an entangled basis, analogous to states in eqn (17) but with a different number q of qubits,
, where γk[m] represents k pairs of elements missing from the middle of the tensor product matrix (shown as pluses in Fig. 1, right panel). These elements can be directly targeted by additional measurements in an entangled basis, analogous to states in eqn (17) but with a different number q of qubits,
| | 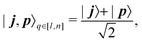 | (18) |
where
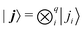
and
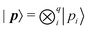
. These states can be constructed in the same way as states
(17). The missing elements can thus be obtained with, at most,
k measurements if performed element-wise.
The full algorithm to compute tk[n] thus includes: (i) at most 2l − k ≤ k expectation values via circuits of depth ≤l + 1 to obtain tk[l] (yielding elements represented by open circles in Fig. 1); (ii) filling the gap (pluses in Fig. 1) in 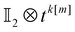 to produce tk[m+1], which requires measurements of at most k projections on entangled states via circuits of depth at most
to produce tk[m+1], which requires measurements of at most k projections on entangled states via circuits of depth at most 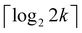 ; (iii) a total of n − l iterations to fill the entire band. The total number of measurement circuits for tk[n] is thus
; (iii) a total of n − l iterations to fill the entire band. The total number of measurement circuits for tk[n] is thus
| | 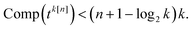 | (19) |
To construct the anti-diagonals ak[n], we first note that the expectation values of 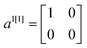 ,
, 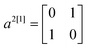 and
and 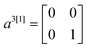 can be obtained by sampling one-qubit measurements of ψ in Z to get 〈a1[1]〉ψ and 〈a3[1]〉ψ and in X to get 〈a2[1]〉ψ. The elements ak[m+1] can be obtained by combining
can be obtained by sampling one-qubit measurements of ψ in Z to get 〈a1[1]〉ψ and 〈a3[1]〉ψ and in X to get 〈a2[1]〉ψ. The elements ak[m+1] can be obtained by combining 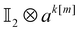 and
and 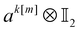 , which at most doubles the number of measurements.24
, which at most doubles the number of measurements.24
We limit the construction to 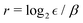 anti-diagonals. eqn (5) and (6) ensure that the remaining anti-diagonals will contribute less than ε to the expectation value (16). The circuits can be constructed from ak[1] by incrementally increasing the number of qubits to ak[r], which requires ≤2r bases in total. The contributions from the anti-diagonals are obtained by measuring n − r qubits in the computational basis and ak[r], thus constructed. For the n−s qubits, one needs to take into account only the all-↑ and all-↓ outputs, yielding the up-most and down-most 2r anti-diagonals. The total number of bases for this protocol is bound by
anti-diagonals. eqn (5) and (6) ensure that the remaining anti-diagonals will contribute less than ε to the expectation value (16). The circuits can be constructed from ak[1] by incrementally increasing the number of qubits to ak[r], which requires ≤2r bases in total. The contributions from the anti-diagonals are obtained by measuring n − r qubits in the computational basis and ak[r], thus constructed. For the n−s qubits, one needs to take into account only the all-↑ and all-↓ outputs, yielding the up-most and down-most 2r anti-diagonals. The total number of bases for this protocol is bound by
| | 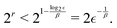 | (20) |
B. Algorithm for compositional ansatz optimization
In the second part of this work, we demonstrate that DVR Hamiltonians also lead to very efficient quantum circuits for representing vibrational states of molecules. In order to apply VQE, it is necessary to find an appropriate ansatz for |ψ〉. It is not always clear how to select the ansatz for |ψ〉. Previous work on the electronic structure proposed various types of Ansatzes for VQE with both fixed3,7,25–29 and adaptive structure.30–39 Unlike electronic structure problems, where interactions are pairwise additive, vibrational energy calculations are determined by a wide range of potential energy landscapes, which are highly molecule-specific. In order to obtain the most efficient quantum circuit representations of |ψ〉 for VQE with DVR matrices, we develop and illustrate an iterative algorithm for ansatz construction that minimizes the number of entangling gates for each specific molecule. This algorithm is inspired by work in ref. 40–42.
Our starting point is:7
| | 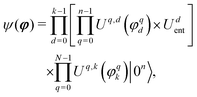 | (21) |
where
Uq,d(
φ) represent
RY = exp(−
iφσY/2) for qubit
q, and
k is the number of repetitions of the ansatz blocks. The operator
Udent introduces entanglement between qubits. The form of
Udent is determined by the ansatz construction algorithm that incrementally increases the complexity of the quantum circuits. More specifically, the ansatz construction starts with a non-entangled quantum state given by
eqn (21) with a predetermined number of blocks
k and
Udent set to identity. The method considers CNOT(
q,
p) ∀
q <
p as candidate gates for
Udent, with each
d segment treated independently. In each optimization step, the candidate gate that lowers the VQE energy is added without replacement until convergence. Here, we aim to converge the VQE calculation of the ground state either to 1 cm
−1 or 0.01 cm
−1, which yields quantum circuits of different complexity, denoted as
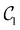
and

. This convergence error is with respect to the lowest eigenstate of the DVR matrix with the same number of DVR bases.
In order to benchmark the quantum circuits determined by this algorithm, we also use the following ansatz
| |  | (22) |
instead of
Udent. The ansatz
(22) is denoted hereafter as linear, as it linearly entangles adjacent qubits.
III. Numerical results
We calculate the vibrational energy levels of diatomic (Cr2) and triatomic (Ar–HCl and Mg–NH) systems. We consider these molecular systems because they exhibit vibrational states with widely different energies (from −55 to −15000 cm−1 from the dissociation threshold) and spatial variations of wave functions and energy level patterns. Our goal is to demonstrate that the same approach can be applied to these widely different molecular systems. For diatomic molecules, we use the DVR Hamiltonian from ref. 21. For triatomic complexes, we use the DVR approach by Choi and Light.22 We use two classical constrained optimization methods to optimize quantum circuit parameters: the bounded limited memory method of Broyden, Fletcher, Goldfarb, and Shanno,48,49 and sequential least squares programming.50 We benchmark the VQE results by the vibrational levels calculated using direct diagonalization with the converged DVR basis and previous literature results, where available.
Table 1 demonstrates the performance of VQE for vibrational states v = 0–5 of seven electronic states of Cr2 with zero rotational angular momentum. We use the interaction potentials from ref. 23, illustrated in Fig. 2. The VQE calculations use 16 DVR points placed to span the range, including the minimum of the potential energy. Apart from the DVR parameters, the circuit optimization algorithm is applied identically to all seven electronic states. The Hamiltonian is represented by expansion (2) including ≈130 Pauli terms. Table 1 displays VQE results obtained with three types of quantum circuits: the linear ansatz (22) with 3 repetitions, and optimized circuits 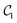 (shown in Fig. 2) and
(shown in Fig. 2) and 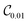 (shown in Fig. 3).
(shown in Fig. 3).
Table 1 Vibrational energy (in cm−1) of Cr2 (v = 0–5) in different electronic states. The benchmark (BM) results are obtained with a converged DVR basis. Exact energies obtained from the truncated DVR basis are displayed as Eν (DVR). VQEs use quantum circuits as shown in Fig. 2 (for 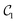 ) and Fig. 3 (for
) and Fig. 3 (for 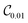 )
)
| Electronic state |
v
|
BM |
E
v
(DVR) |
E
v
|
|

|
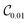
|
Linear |
|
1Σ+g |
0 |
−15358.94 |
−15358.99 |
−15358.87 |
−15358.99 |
−15358.99 |
| 1 |
−14846.67 |
−14846.96 |
−14838.70 |
−14846.75 |
−14846.96 |
| 2 |
−14333.21 |
−14332.81 |
−14310.29 |
−14332.80 |
−14332.82 |
| 3 |
−13826.93 |
−13827.29 |
−13797.65 |
−13826.87 |
−13827.29 |
| 4 |
−13334.02 |
−13335.45 |
−13275.12 |
−13318.77 |
−13335.45 |
| 5 |
−12861.37 |
−12868.75 |
−12897.70 |
−12871.32 |
−12868.75 |
|
|
|
3Σ+u |
0 |
−9862.07 |
−9862.14 |
−9861.37 |
−9862.14 |
−9862.14 |
| 1 |
−9559.46 |
−9559.41 |
−9538.97 |
−9556.67 |
−9559.41 |
| 2 |
−9300.92 |
−9300.88 |
−9240.74 |
−9266.82 |
−9300.88 |
| 3 |
−9080.60 |
−9080.42 |
9068.09 |
−9079.26 |
−9085.40 |
| 4 |
−8897.58 |
−8896.82 |
−8866.09 |
−8870.57 |
−8896.82 |
| 5 |
−8747.53 |
−8742.68 |
−8729.41 |
−8750.18 |
— |
|
|
|
5Σ+g |
0 |
−7566.53 |
−7566.50 |
−7565.88 |
−7566.50 |
−7566.50 |
| 1 |
−7416.28 |
−7416.29 |
−7397.28 |
−7416.28 |
−7416.29 |
| 2 |
−7264.92 |
−7264.69 |
−7181.98 |
−7264.60 |
−7264.69 |
| 3 |
−7114.40 |
−7118.43 |
−7075.88 |
−7117.83 |
−7118.43 |
| 4 |
−6965.91 |
−6958.08 |
−7001.98 |
−6953.74 |
−6958.08 |
| 5 |
−6820.04 |
−6840.02 |
−6873.37 |
−6837.02 |
−6840.02 |
|
|
|
7Σ+u |
0 |
−6519.01 |
−6519.04 |
−6519.04 |
−6519.04 |
| 1 |
−6350.36 |
−6350.11 |
−6350.11 |
−6530.11 |
| 2 |
−6183.38 |
−6185.17 |
−6185.17 |
−6185.17 |
| 3 |
−6018.09 |
−6018.28 |
−6018.12 |
−6018.28 |
| 4 |
−5854.50 |
−5849.10 |
−5848.69 |
−5849.10 |
| 5 |
−5692.63 |
−5731.33 |
−5728.00 |
−5731.33 |
|
|
|
9Σ+g |
0 |
−5348.79 |
−5348.82 |
−5348.82 |
−5348.82 |
| 1 |
−5175.81 |
−5175.51 |
−5175.51 |
−5175.51 |
| 2 |
−5005.17 |
−5008.56 |
−5008.55 |
−5008.56 |
| 3 |
−4836.85 |
−4829.92 |
−4829.80 |
−4829.92 |
| 4 |
−4670.91 |
−4683.68 |
−4683.42 |
−4683.68 |
| 5 |
−4507.31 |
−4530.16 |
−4526.72 |
−4612.83 |
|
|
|
11Σ+u |
0 |
−3677.68 |
−3677.68 |
−3677.00 |
−3677.68 |
−3677.68 |
| 1 |
−3507.89 |
−3507.82 |
−3489.51 |
−3507.82 |
−3507.82 |
| 2 |
−3341.77 |
−3341.40 |
−3253.24 |
−3341.26 |
−3341.40 |
| 3 |
−3180.16 |
−3186.93 |
−3133.59 |
−3185.51 |
−3186.93 |
| 4 |
−3023.07 |
−3012.82 |
−3061.61 |
−3001.04 |
−3012.82 |
| 5 |
−2870.22 |
−2874.49 |
−2904.46 |
−2866.05 |
−2874.54 |
|
|
|
13Σ+g |
0 |
−548.68 |
−548.68 |
−548.65 |
−548.67 |
−548.68 |
| 1 |
−497.16 |
−497.15 |
−496.47 |
−496.84 |
−497.15 |
| 2 |
−449.18 |
−449.26 |
−443.48 |
−448.36 |
−449.26 |
| 3 |
−404.71 |
−404.67 |
−382.88 |
−390.96 |
−404.67 |
| 4 |
−363.58 |
−362.99 |
−369.09 |
−360.62 |
−362.99 |
| 5 |
−325.67 |
−325.72 |
−315.46 |
−310.29 |
−325.72 |
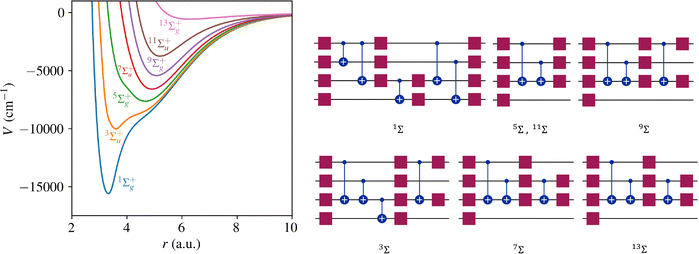 |
| | Fig. 2 Left panel: Potential energy for Cr2 from ref. 23. Right: Quantum circuits for VQE yielding the ground state energy with error ≤1 cm−1. The squares represent the RY gates and the circles show the entangling CNOT gates. | |
 |
| | Fig. 3 Quantum circuits for VQE yielding the ground state energy with error ≤0.01 cm−1. The squares represent the RY gates and the circles show the entangling CNOT gates. | |
The results in Table 1 and Fig. 2, 3 show that VQE can be used to compute the vibrational levels of diatomic molecules with high precision using shallow quantum circuits. It is particularly instructive to analyze the difference between the quantum circuits displayed in Fig. 2 and 3. The quantum circuits in Fig. 3 yield a much better convergence (within 0.01 cm−1) and a significantly higher accuracy than the quantum circuits in Fig. 2 at the expense of a small number of additional quantum gates. For example, for the ground vibrational state of Cr2 in the 3Σ+g electronic state, adding two entanglement gates reduces the error of the computation from 0.69 cm−1 to 0.07 cm−1, with respect to the benchmark calculation result (labeled BM).
These results also illustrate the utility of the ansatz optimization algorithm developed here. By construction, the algorithm aims to produce quantum circuits of the lowest complexity for a particular accuracy target or particular convergence threshold. This can be exploited to examine the role of specific gates or gate combinations in determining the expressivity of quantum circuits for representing the vibrational states of molecules. Given that the optimized quantum circuits illustrated in Fig. 2 and 3 are very shallow, the results of Table 1 suggest that current quantum computing devices can already be used for high-precision computation of vibrational energy levels.
For tri-atomic complexes, we use 32 DVR basis states and accurate atom–molecule potential energy surfaces by Hutson for Ar–HCl43 and by Soldán et al. for Mg–NH.51 We keep both HCl and NH in the ground vibrational state and compute the vibrational states supported by the atom–molecule interaction potential. We obtain the DVR points for the triatomic systems by diagonalizing the coordinate representations. The parameters to generate the DVR points are selected to cover the low-energy regions of the potential energy surface. The DVR Hamiltonians are represented by 165 (Ar–HCl) and 170 (Mg–NH) Pauli terms in eqn (2) acting on 5 qubits. As above, we construct three types of quantum circuits: 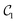 ,
, 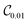 and the linear ansatz (22) with k repetitions.
and the linear ansatz (22) with k repetitions.
We note that the triatomic molecular systems considered here represent van der Waals complexes. Accurate calculations of ro-vibrational energies for van der Waals complexes are generally challenging due to the weak binding of the underlying potential energy surface and a large spatial extent of the corresponding vibrational states. Typically, ro-vibrational energy levels of van der Waals complexes are computed by solving a system of coupled differential equations, using, for example, a log-derivative propagation method. Fig. 4 and Table 2 show that accurate results for these weakly bound molecular complexes can be obtained by VQE with very shallow circuits. Fig. 5 shows the fidelity of the ground and excited states obtained from 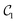 and
and 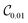 , with respect to the corresponding DVR eigenstates and demonstrates that the quantum circuits not only provide accurate energies, but also a good representation of the underlying eigenstates. We observe that a small number of entangling gates is sufficient to ensure accurate calculations for both the ground and excited state energy. We also observe that some of the optimized circuits in Fig. 2 and 4 are only partially entangled, which indicates that accurate VQE results can be obtained with ensembles of unentangled quantum circuits. This further decreases the complexity of the quantum computations.
, with respect to the corresponding DVR eigenstates and demonstrates that the quantum circuits not only provide accurate energies, but also a good representation of the underlying eigenstates. We observe that a small number of entangling gates is sufficient to ensure accurate calculations for both the ground and excited state energy. We also observe that some of the optimized circuits in Fig. 2 and 4 are only partially entangled, which indicates that accurate VQE results can be obtained with ensembles of unentangled quantum circuits. This further decreases the complexity of the quantum computations.
 |
| | Fig. 4 Left panel: VQE error for the lowest energy of Ar–HCl (open symbols) and Mg–NH (full symbols) computed with optimized quantum circuits in the right panel. Circles – with 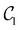 circuits; squares – with circuits; squares – with 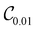 circuits; triangles – with ansatz (21) using (left to right) k = 1, 2, 3 and 4 and eqn (22). The circuits; triangles – with ansatz (21) using (left to right) k = 1, 2, 3 and 4 and eqn (22). The 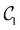 Ansatz for Mg–NH excludes the gate shown in green. Ansatz for Mg–NH excludes the gate shown in green. | |
Table 2 Vibrational energy (in cm−1) of Ar–HCl and Mg–NH by VQEs with 32 DVR points and 5-qubit circuits
| Molecule |
v
|
E
v
(experiment) |
E
v
(computed in ref. 43) |
E
v
(classical, present) |
E
v
(DVR) |
E
v
(VQE) |
|
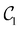
|
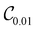
|
Linear |
| ArHCl |
0 |
−114.744 |
−115.151 |
−115.265 |
−115.178 |
−114.645 |
−115.169 |
−115.171 |
| 1 |
−91.0445,46 |
−91.485 |
−91.642 |
−90.959 |
−80.824 |
−90.485 |
−90.929 |
| 2 |
−82.2647 |
−82.717 |
−82.825 |
−82.727 |
−75.900 |
−82.986 |
−82.650 |
|
|
| MgNH |
0 |
— |
— |
−88.227 |
−88.196 |
−87.650 |
−88.191 |
−88.190 |
| 1 |
— |
— |
−63.603 |
−62.928 |
−56.050 |
−62.730 |
−62.664 |
| 2 |
— |
— |
−55.461 |
−54.961 |
−55.145 |
−54.850 |
−54.866 |
 |
| | Fig. 5 Fidelity of the vibrational ground and excited states calculated with respect to the corresponding DVR wave functions for Ar–HCl (left panel) and Mg–NH (right panel). | |
In order to assess whether the present method is practical for NISQ-era devices, we also ran the circuit optimization algorithm for the Ar–HCl complex with a realistic noise model. Specifically, we repeat the full compositional ansatz search using the Sherbrooke noise model in Qiskit, which captures gate and readout errors of a superconducting quantum processor. Fig. 6 shows the error of the ground state VQE calculation during the circuit optimization algorithm for circuits with k = 1 in eqn (21), and two circuits obtained after 4 steps of the greedy search algorithm. We find that the greedy optimization continues to identify shallow circuits yielding spectroscopic accuracy for multiple excited vibrational states. While the circuits obtained under noise may differ from those found in noiseless simulations, their accuracy is comparable, indicating the existence of multiple noise-resilient shallow representations of the same vibrational eigenstates. The persistence of successful optimization under realistic noise and the existence of multiple shallow optima suggest that the DVR-based ansatz space avoids the effects typically associated with barren plateaus in deep, unstructured circuits. Further analysis of the robustness of the method to noise is left for future work.
 |
| | Fig. 6 (Top panel) VQE error during the greedy circuit optimization for Ar–HCl using k = 1 in eqn (21). (Bottom panel) The circuits obtained after adding 3 entangling gates with and without the noise model for Ar–HCl. | |
IV. Conclusions
We have shown that the structure of DVR matrices can be leveraged to represent molecular Hamiltonians by the number of quantum circuits that grows polynomially with the number n of qubits. Given the binary basis encoding yielding N = 2n, this offers an exponential reduction of the measurement complexity for matrices with N DVR basis states. This has significant implications for the potential quantum advantage of VQEs for ro-vibrational computations. Exponential scaling of VQE is a significant and common challenge, particularly relevant for unstructured Hamiltonians such as the vibrational eigenvalue problem in an unstructured basis. For quantum chemistry calculations as well as for ro-vibrational problems, this is usually dealt with by second quantization. However, these approaches often lead to extended quantum circuits that require a large number of qubits and gates.
We have also demonstrated that DVR leads to efficient quantum circuits for VQE computations of vibrational energy levels. To show this, we have introduced a general approach for constructing the quantum ansatz by combining DVR with VQE and a greedy search in the space of gate permutations. The results yield compact representations of vibrational states by quantum circuits of a gate-based quantum computer. We have shown that both the ground and excited vibrational energies can be computed with a relative accuracy of <1% using very simple, in some cases, partially entangled circuits. The accuracy of 1 cm−1 can be achieved with <20 (<5 entangling) gates and 4 qubits for diatomic molecules and <30 (<9 entangling) gates with 5 qubits for triatomic van der Waals complexes. This should be compared with previous VQE calculations of vibrational energy levels that required extended quantum circuits with >200 (for CO, COH and O3 molecules17) or between 44 and 140292 (for CO2, H2CO and HCOOH molecules16) entangling gates.
Reducing the complexity of quantum circuits is particularly important for VQE approaches that are known to be affected by barren plateaus, which limit the quantum advantage of variational quantum computations. Although barren plateaus are formally an asymptotic effect, they can become relevant already for systems of moderate size. While a comprehensive treatment of barren plateaus is beyond the scope of the present work, our results provide several encouraging indications that this issue may be mitigated for vibrational energy calculations using the present framework. In particular, we observe no signs of barren plateaus for the systems considered here, including in simulations that explicitly incorporate realistic noise. Moreover, the anzatz construction algorithm introduced in this work consistently yields accurate vibrational energies and wave functions using extremely shallow circuits, some of which are only partially entangled. These observations suggest that VQE computations of vibrational energies may be scalable to larger systems than can be anticipated based on the previous calculations.16,17 This must be confirmed by a more systematic numerical investigation, which we leave for future work.
Finally, we note that DVR does not require global fits of potential energy surfaces or integrals over the potential energy for the construction of the Hamiltonian matrix. The implementation of VQEs with DVR in the first quantization does not require normal-mode analysis for the construction of the quantum ansatz. For these reasons, the present approach can be readily extended beyond molecular dynamics. For example, this method can be directly applied to designing efficient quantum circuits for variational computations of the eigenspectra of semiconductor quantum dots.52 Finally, our calculations suggest that quantum circuit representations of vibrational states can be used as efficient (small number of free parameters) Ansatze for classical variational calculations.
Author contributions
All authors have designed the concept of this work and co-wrote the manuscript. The algorithm presented in Section IIA was developed by DB. The algorithm for Ansatz optimization was developed by KA and RVK and the numerical calculations were performed by KA.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data that support the findings of this study are available within the article.
Appendices
A Scaling of DVR matrix bands
The goal of this Appendix is to prove eqn (5) and (6). Specifically, we aim to show that| | 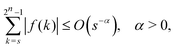 | (A1) |
| | 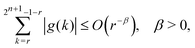 | (A2) |
where r ≪ N and s ≪ N, and N = 2n is assumed to be large.
We consider a particle with mass m on a grid of x = [a, b] with the uniform spacing Δx, yielding
| | | xj = b + jΔx, j = 1,…,N − 1. | (A3) |
Given an orthonormal basis {
ϕn}, the matrix elements of the kinetic energy can be written as
| | 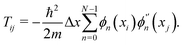 | (A4) |
Fourier grid DVR uses the basis
21| | 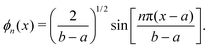 | (A5) |
For this basis, the sum can be evaluated analytically, yielding
21| | 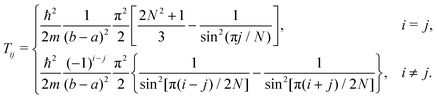 | (A6) |
Following Colbert and Miller,21 we now consider the cases of infinite lattices, relevant for radial molecular coordinates, and a case of finite a and b.
1. Infinite lattice [a = −∞, b = ∞].
The finite grid spacing 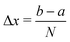 requires N → ∞. In this limit, eqn (A6) becomes
requires N → ∞. In this limit, eqn (A6) becomes| | 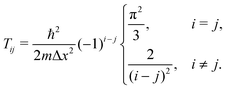 | (A7) |
Note that in this case g(i + j) vanishes. It is evident that| | 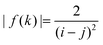 | (A8) |
satisfies eqn (A1). To prove this, we note that for functions that are (strictly) monotonically decaying in absolute value| | 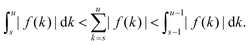 | (A9) |
For our choice of f(·) this yields the bound| | 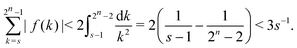 | (A10) |
2. Infinite lattice [a = 0, b = ∞].
For the lattice with [a = 0, b = ∞], the matrix elements| | 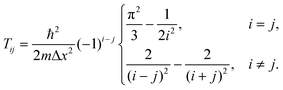 | (A11) |
include g(k). Since eqn (A10) applies to g(k), the arguments in subsection (8.1) apply to eqn (12).
3. Finite lattice [a, b].
We note that the prefactor in eqn (A6) satisfies 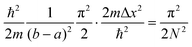 . For the following functions
. For the following functions| | 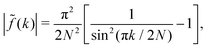 | (A12) |
| | 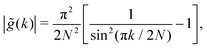 | (A13) |
Eqn (A9) yields| | 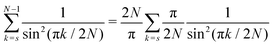 | (A14) |
| | 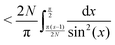 | (A15) |
| | 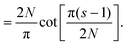 | (A16) |
Using the Laurent series for cot(y) around y = 0, we can write| | 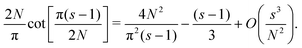 | (A17) |
This yields| | 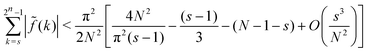 | (A18) |
| | 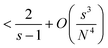 | (A19) |
For g(k), we exploit the symmetry of sin(·) to write| | 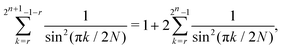 | (A21) |
and apply eqn (A19)–(A21).
B Representation of H(s,r) in terms of tk[n] and ak[n]
We leverage the structure of the truncated DRV matrix by representing it as a sum of a diagonal matrix D and kth diagonal and anti-diagonal components,| | 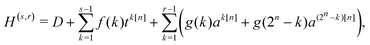 | (B1) |
where| | 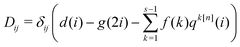 | (B2) |
| | | tk[n]ij = δ|i−j|,k + δijqk[n](i), | (B3) |
and qk[n](i) can be computed by an efficient Algorithm 1. Our strategy is to measure the expectation values of each of the {D, tk[n], ak[n]} separately using the induction over n and sum them with the corresponding weights.
In addition to kth diagonals, the construction of tk[n] adopted in this work introduces additional contributions qk[n](i) to the matrix elements on the main diagonal. These contributions can be deduced from the measurement protocol for tk[n] as described in Algorithm 1.
Algorithm 1 Computing qk[n](i) from binary representation of i
define
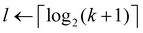 , , |
| last_l_bits ← i mod 2l, |
| anti_last_l_bits ← 2l − last_l_bits; |
|
initialize
output ← 1, |
| j ← l + 1; |
|
while
j ≤ ndo |
| ifjth bit of i is 1 then |
| iflast_l_bits < k or anti_last_l_bits < lthen |
| output → out + 1; |
| j → j + 1; |
|
return
output. |
Acknowledgements
This work was supported by NSERC of Canada and the Stewart Blusson Quantum Matter Institute.
References
-
J. Tilly, H. Chen, S. Cao, D. Picozzi, K. Setia, Y. Li, E. Grant, L. Wossnig, I. Rungger, G. H. Booth and J. Tennyson, The variational quantum eigensolver: a review of methods and best practices, arXiv, 2021, preprint DOI:10.48550/arXiv.2111.05176.
- O. Higgott, D. Wang and S. Brierley, Variational Quantum Computation of Excited States, Quantum, 2019, 3, 156 CrossRef.
- A. Peruzzo, J. McClean, P. Shadbolt, M.-H. Yung, X.-Q. Zhou, P. J. Love, A. Aspuru-Guzik and J. L. O'Brien, A variational eigenvalue solver on a photonic quantum processor, Nat. Commun., 2014, 5, 4213 CrossRef CAS.
- J. R. McClean, J. Romero, R. Babbush and A. Aspuru-Guzik, The theory of variational hybrid quantum-classical algorithms, New J. Phys., 2016, 18, 023023 CrossRef.
- P. J. J. O'Malley, R. Babbush, I. D. Kivlichan, J. Romero, J. R. McClean, R. Barends, J. Kelly, P. Roushan, A. Tranter, N. Ding, B. Campbell, Y. Chen, Z. Chen, B. Chiaro, A. Dunsworth, A. G. Fowler, E. Jeffrey, E. Lucero, A. Megrant, J. Y. Mutus, M. Neeley, C. Neill, C. Quintana, D. Sank, A. Vainsencher, J. Wenner, T. C. White, P. V. Coveney, P. J. Love, H. Neven, A. Aspuru-Guzik and J. M. Martinis, Scalable quantum simulation of molecular energies, Phys. Rev. X, 2016, 6, 031007 Search PubMed.
- Y. Shen, X. Zhang, S. Zhang, J.-N. Zhang, M.-H. Yung and K. Kim, Quantum implementation of the unitary coupled cluster for simulating molecular electronic structure, Phys. Rev. A, 2017, 95, 020501 CrossRef.
- A. Kandala, A. Mezzacapo, K. Temme, M. Takita, M. Brink, J. M. Chow and J. M. Gambetta, Hardware-efficient variational quantum eigensolver for small molecules and quantum magnets, Nature, 2017, 549, 242–246 CrossRef CAS PubMed.
- J. I. Colless, V. V. Ramasesh, D. Dahlen, M. S. Blok, M. E. Kimchi-Schwartz, J. R. McClean, J. Carter, W. A. de Jong and I. Siddiqi, Computation of molecular spectra on a quantum processor with an error-resilient algorithm, Phys. Rev. X, 2018, 8, 011021 CAS.
- C. Hempel, C. Maier, J. Romero, J. McClean, T. Monz, H. Shen, P. Jurcevic, B. P. Lanyon, P. Love, R. Babbush, A. Aspuru-Guzik, R. Blatt and C. F. Roos, Quantum chemistry calculations on a trapped-ion quantum simulator, Phys. Rev. X, 2018, 8, 031022 CAS.
-
Y. Nam, J.-S. Chen, N. C. Pisenti, K. Wright, C. Delaney, D. Maslov, K. R. Brown, S. Allen, J. M. Amini, J. Apisdorf, K. M. Beck, A. Blinov, V. Chaplin, M. Chmielewski, C. Collins, S. Debnath, A. M. Ducore, K. M. Hudek, M. Keesan, S. M. Kreikemeier, J. Mizrahi, P. Solomon, M. Williams, J. D. Wong-Campos, C. Monroe and J. Kim, Ground-state energy estimation of the water molecule on a trapped ion quantum computer, arXiv, 2019, preprint DOI:10.48550/arXiv.1902.10171.
- S. McArdle, S. Endo, A. Aspuru-Guzik, S. C. Benjamin and X. Yuan, Quantum computational chemistry, Rev. Mod. Phys., 2020, 92, 015003 CrossRef CAS.
- C. Kokail, C. Maier, R. van Bijnen, T. Brydges, M. K. Joshi, P. Jurcevic, C. A. Muschik, P. Silvi, R. Blatt, C. F. Roos and P. Zoller, Self-verifying variational quantum simulation of lattice models, Nature, 2019, 569, 355–360 CrossRef CAS.
- C.-K. Lee, C.-Y. Hsieh, S. Zhang and L. Shi, Variational quantum simulation of chemical dynamics with quantum computers, J. Chem. Theory Comput., 2022, 18, 2105–2113 CrossRef CAS.
-
J. Apanavicius, Y. Feng, Y. Flores, M. Hassan and M. McGuigan, Morse potential on a quantum computer for molecules and supersymmetric quantum mechanics, arXiv, 2021, preprint DOI:10.48550/arXiv.2102.05102.
- S. McArdle, A. Mayorov, X. Shan, S. Benjamin and X. Yuan, Digital quantum simulation of molecular vibrations, Chem. Sci., 2019, 10, 5725–5735 RSC.
- P. J. Ollitrault, A. Baiardi, M. Reiher and I. Tavernelli, Hardware efficient quantum algorithms for vibrational structure calculations, Chem. Sci., 2020, 11, 6842–6855 RSC.
- N. P. D. Sawaya, F. Paesani and D. P. Tabor, Near- and long-term quantum algorithmic approaches for vibrational spectroscopy, Phys. Rev. A, 2021, 104, 062419 CrossRef CAS.
- E. Lötstedt, K. Yamanouchi and Y. Tachikawa, Evaluation of vibrational energies and wave functions of CO2 on a quantum computer, AVS Quantum Sci., 2022, 4, 036801 CrossRef.
- E. Lötstedt, K. Yamanouchi, T. Tsuchiya and Y. Tachikawa, Calculation of vibrational eigenenergies on a quantum computer: application to the Fermi resonance in CO2, Phys. Rev. A, 2021, 103, 062609 CrossRef.
- M. T. Nguyen, Y.-L. Lee, D. Alfonso, Q. Shao and Y. Duan, Description of reaction and vibrational energetics of CO2–NH3 interaction using quantum computing algorithms, AVS Quantum Sci., 2023, 5, 013801 CrossRef CAS.
- D. T. Colbert and W. H. Miller, A novel discrete variable representation for quantum mechanical reactive scattering via the s-matrix kohn method, J. Chem. Phys., 1992, 96, 1982–1991 CrossRef CAS.
- S. E. Choi and J. C. Light, Determination of the bound and quasibound states of Ar–HCl van der Waals complex: discrete variable representation method, J. Chem. Phys., 1990, 92, 2129–2145 CrossRef CAS.
- Z. Pavlović, R. V. Krems, R. Côté and H. R. Sadeghpour, Magnetic feshbach resonances and Zeeman relaxation in bosonic chromium gas with anisotropic interaction, Phys. Rev. A: At., Mol., Opt. Phys., 2005, 71, 061402 CrossRef.
- SI'.
- J. Romero, R. Babbush, J. R. McClean, C. Hempel, P. J. Love and A. Aspuru-Guzik, Strategies for quantum computing molecular energies using the unitary coupled cluster ansatz, Quantum Sci. Technol., 2018, 4, 014008 CrossRef.
- D. Wecker, M. B. Hastings and M. Troyer, Progress towards practical quantum variational algorithms, Phys. Rev. A: At., Mol., Opt. Phys., 2015, 92, 042303 CrossRef.
- J. Lee, W. J. Huggins, M. Head-Gordon and K. B. Whaley, Generalized unitary coupled cluster wave functions for quantum computation, J. Chem. Theory Comput., 2019, 15, 311–324 CrossRef CAS PubMed.
- P. K. Barkoutsos, J. F. Gonthier, I. Sokolov, N. Moll, G. Salis, A. Fuhrer, M. Ganzhorn, D. J. Egger, M. Troyer, A. Mezzacapo, S. Filipp and I. Tavernelli, Quantum algorithms for electronic structure calculations: particle–hole Hamiltonian and optimized wave-function expansions, Phys. Rev. A, 2018, 98, 022322 CrossRef CAS.
- R. Wiersema, C. Zhou, Y. de Sereville, J. F. Carrasquilla, Y. B. Kim and H. Yuen, Exploring entanglement and optimization within the Hamiltonian variational ansatz, PRX Quantum, 2020, 1, 020319 CrossRef.
- H. R. Grimsley, S. E. Economou, E. Barnes and N. J. Mayhall, An adaptive variational algorithm for exact molecular simulations on a quantum computer, Nat. Commun., 2019, 10, 3007 CrossRef PubMed.
- A. M. Romero, J. Engel, H. L. Tang and S. E. Economou, Solving nuclear structure problems with the adaptive variational quantum algorithm, Phys. Rev. C, 2022, 105, 064317 CrossRef CAS.
- H. L. Tang, V. Shkolnikov, G. S. Barron, H. R. Grimsley, N. J. Mayhall, E. Barnes and S. E. Economou, Qubit-ADAPT-VQE: an adaptive algorithm for constructing hardware-efficient ansätze on a quantum processor, PRX Quantum, 2021, 2, 020310 CrossRef.
- I. G. Ryabinkin, R. A. Lang, S. N. Genin and A. F. Izmaylov, Iterative qubit coupled cluster approach with efficient screening of generators, J. Chem. Theory Comput., 2020, 16, 1055–1063 CrossRef CAS PubMed.
- Y. Zhang, L. Cincio, C. F. A. Negre, P. Czarnik, P. J. Coles, P. M. Anisimov, S. M. Mniszewski, S. Tretiak and P. A. Dub, Variational quantum eigensolver with reduced circuit complexity, npj Quantum Inf., 2022, 8, 96 CrossRef.
- M. Ostaszewski, E. Grant and M. Benedetti, Structure optimization for parameterized quantum circuits, Quantum, 2021, 5, 391 CrossRef.
- M. Bilkis, M. Cerezo, G. Verdon, P. J. Coles and L. Cincio, A semi-agnostic ansatz with variable structure for variational quantum algorithms, Quantum Mach. Intell., 2023, 5, 43 CrossRef.
-
A. G. Rattew, S. Hu, M. Pistoia, R. Chen and S. Wood, A domain-agnostic, noise-resistant, hardware-efficient evolutionary variational quantum eigensolver, arXiv, 2020, preprint DOI:10.48550/arXiv.1910.09694.
-
D. Chivilikhin, A. Samarin, V. Ulyantsev, I. Iorsh, A. R. Oganov and O. Kyriienko, MoG-VQE: Multiobjective genetic variational quantum eigensolver, arXiv, 2020, preprint DOI:10.48550/arXiv.2007.04424.
- Y. Du, T. Huang, S. You, M.-H. Hsieh and D. Tao, Quantum circuit architecture search for variational quantum algorithms, npj Quantum Inf., 2022, 8, 62 CrossRef.
- H. R. Grimsley, S. E. Economou, E. Barnes and N. J. Mayhall, An adaptive variational algorithm for exact molecular simulations on a quantum computer, Nat. Commun., 2019, 10, 3007 CrossRef PubMed.
- E. Torabian and R. V. Krems, Compositional optimization of quantum circuits for quantum kernels of support vector machines, Phys. Rev. Res., 2023, 5, 013211 CrossRef CAS.
- X. Guo, J. Dai and R. V. Krems, Benchmarking of quantum fidelity kernels for Gaussian process regression, Mach. Learn.: Sci. Technol., 2024, 5, 035081 Search PubMed.
- J. M. Hutson, Vibrational dependence of the anisotropic intermolecular potential of argon-hydrogen chloride, J. Phys. Chem., 1992, 96, 4237–4247 CrossRef CAS.
- A. S. Pine and B. J. Howard, Hydrogen bond energies of the HF and HCl dimers from absolute infrared intensities, J. Chem. Phys., 1986, 84, 590–596 CrossRef CAS.
- K. L. Busarow, G. A. Blake, K. Laughlin, R. Cohen, Y. Lee and R. Saykally, Tunable far-infrared laser spectroscopy in a planar supersonic jet: the σ bending vibration of Ar35 HCl, Chem. Phys. Lett., 1987, 141, 289–291 CrossRef CAS.
- R. L. Robinson, D.-H. Gwo and R. J. Saykally, Far infrared laser stark spectroscopy of the σ bending vibration of ArHCl, Mol. Phys., 1988, 63, 1021–1029 CrossRef CAS.
- R. L. Robinson, D. Gwo and R. J. Saykally, The high-resolution far infrared spectrum of a van der Waals stretching vibration: the ν3 band of Ar–HCL, J. Chem. Phys., 1987, 87, 5156–5160 CrossRef CAS.
- R. H. Byrd, P. Lu, J. Nocedal and C. Zhu, A limited memory algorithm for bound constrained optimization, SIAM J. Sci. Comput., 1995, 16, 1190–1208 CrossRef.
- C. Zhu, R. H. Byrd, P. Lu and J. Nocedal, Algorithm 778: L-BFGS-B: Fortran subroutines for large-scale bound-constrained optimization, ACM Trans. Math. Softw., 1997, 23, 550–560 CrossRef.
-
D. Kraft, A Software Package for Sequential Quadratic Programming, Deutsche Forschungs- und Versuchsanstalt für Luft- und Raumfahrt Köln: Forschungsbericht (Wiss. Berichtswesen d. DFVLR), 1988 Search PubMed.
- P. Soldán, P. S. Żuchowski and J. M. Hutson, Prospects for sympathetic cooling of polar molecules: NH with alkali-metal and alkaline-earth atoms – a new hope, Faraday Discuss, 2009, 142, 191–201 RSC.
- Y. Wang, J. P. Dehollain, F. Liu, U. Mukhopadhyay, M. S. Rudner, L. M. K. Vandersypen and E. Demler, Ab initio exact diagonalization simulation of the nagaoka transition in quantum dots, Phys. Rev. B, 2019, 100, 155133 CrossRef CAS.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab,
D.
Bondarenko
*ab,
D.
Bondarenko







 . We first use eqn (5) and (6) to show that the expectation value (7) can be approximated as
. We first use eqn (5) and (6) to show that the expectation value (7) can be approximated as





 diagonal bands (tk) and
diagonal bands (tk) and  anti-diagonal components (ak), as follows:
anti-diagonal components (ak), as follows:
 is the smallest number of qubits that allows the k-th band. We consider l-qubit entangled states produced from the Z basis by a sequence of CNOT gates shown in Fig. 1 (left panel). The resulting state is
is the smallest number of qubits that allows the k-th band. We consider l-qubit entangled states produced from the Z basis by a sequence of CNOT gates shown in Fig. 1 (left panel). The resulting state is
 and
and  are the binary representations of the row (p) and column (j) indices of the DVR matrix, with p = j + k, and ji and pi representing single-qubit states. The projection of an n-qubit state onto the |j, p〉l state has 2 non-zero off-diagonal matrix elements of magnitude 1/2. The binary representation of the row and column positions of these off-diagonal elements differ by l bits. The position of the non-zero off-diagonal elements is controlled by the position of the CNOT gates in Fig. 1. Thus, the expectation value of tk[l] can be obtained by measurements with, at most, k l-qubit entangled states.
are the binary representations of the row (p) and column (j) indices of the DVR matrix, with p = j + k, and ji and pi representing single-qubit states. The projection of an n-qubit state onto the |j, p〉l state has 2 non-zero off-diagonal matrix elements of magnitude 1/2. The binary representation of the row and column positions of these off-diagonal elements differ by l bits. The position of the non-zero off-diagonal elements is controlled by the position of the CNOT gates in Fig. 1. Thus, the expectation value of tk[l] can be obtained by measurements with, at most, k l-qubit entangled states.

 are obtained from projections of (17),
are obtained from projections of (17),  – via
– via , and
, and  – by additional measurements in the entangled basis.
– by additional measurements in the entangled basis. , where γk[m] represents k pairs of elements missing from the middle of the tensor product matrix (shown as pluses in Fig. 1, right panel). These elements can be directly targeted by additional measurements in an entangled basis, analogous to states in eqn (17) but with a different number q of qubits,
, where γk[m] represents k pairs of elements missing from the middle of the tensor product matrix (shown as pluses in Fig. 1, right panel). These elements can be directly targeted by additional measurements in an entangled basis, analogous to states in eqn (17) but with a different number q of qubits,
 and
and  . These states can be constructed in the same way as states (17). The missing elements can thus be obtained with, at most, k measurements if performed element-wise.
. These states can be constructed in the same way as states (17). The missing elements can thus be obtained with, at most, k measurements if performed element-wise.
 to produce tk[m+1], which requires measurements of at most k projections on entangled states via circuits of depth at most
to produce tk[m+1], which requires measurements of at most k projections on entangled states via circuits of depth at most  ; (iii) a total of n − l iterations to fill the entire band. The total number of measurement circuits for tk[n] is thus
; (iii) a total of n − l iterations to fill the entire band. The total number of measurement circuits for tk[n] is thus
 ,
,  and
and  can be obtained by sampling one-qubit measurements of ψ in Z to get 〈a1[1]〉ψ and 〈a3[1]〉ψ and in X to get 〈a2[1]〉ψ. The elements ak[m+1] can be obtained by combining
can be obtained by sampling one-qubit measurements of ψ in Z to get 〈a1[1]〉ψ and 〈a3[1]〉ψ and in X to get 〈a2[1]〉ψ. The elements ak[m+1] can be obtained by combining  and
and  , which at most doubles the number of measurements.24
, which at most doubles the number of measurements.24 anti-diagonals. eqn (5) and (6) ensure that the remaining anti-diagonals will contribute less than ε to the expectation value (16). The circuits can be constructed from ak[1] by incrementally increasing the number of qubits to ak[r], which requires ≤2r bases in total. The contributions from the anti-diagonals are obtained by measuring n − r qubits in the computational basis and ak[r], thus constructed. For the n−s qubits, one needs to take into account only the all-↑ and all-↓ outputs, yielding the up-most and down-most 2r anti-diagonals. The total number of bases for this protocol is bound by
anti-diagonals. eqn (5) and (6) ensure that the remaining anti-diagonals will contribute less than ε to the expectation value (16). The circuits can be constructed from ak[1] by incrementally increasing the number of qubits to ak[r], which requires ≤2r bases in total. The contributions from the anti-diagonals are obtained by measuring n − r qubits in the computational basis and ak[r], thus constructed. For the n−s qubits, one needs to take into account only the all-↑ and all-↓ outputs, yielding the up-most and down-most 2r anti-diagonals. The total number of bases for this protocol is bound by

 and
and  . This convergence error is with respect to the lowest eigenstate of the DVR matrix with the same number of DVR bases.
. This convergence error is with respect to the lowest eigenstate of the DVR matrix with the same number of DVR bases.

 (shown in Fig. 2) and
(shown in Fig. 2) and  (shown in Fig. 3).
(shown in Fig. 3). ) and Fig. 3 (for
) and Fig. 3 (for  )
)




 ,
, 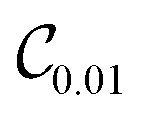 and the linear ansatz (22) with k repetitions.
and the linear ansatz (22) with k repetitions. and
and  , with respect to the corresponding DVR eigenstates and demonstrates that the quantum circuits not only provide accurate energies, but also a good representation of the underlying eigenstates. We observe that a small number of entangling gates is sufficient to ensure accurate calculations for both the ground and excited state energy. We also observe that some of the optimized circuits in Fig. 2 and 4 are only partially entangled, which indicates that accurate VQE results can be obtained with ensembles of unentangled quantum circuits. This further decreases the complexity of the quantum computations.
, with respect to the corresponding DVR eigenstates and demonstrates that the quantum circuits not only provide accurate energies, but also a good representation of the underlying eigenstates. We observe that a small number of entangling gates is sufficient to ensure accurate calculations for both the ground and excited state energy. We also observe that some of the optimized circuits in Fig. 2 and 4 are only partially entangled, which indicates that accurate VQE results can be obtained with ensembles of unentangled quantum circuits. This further decreases the complexity of the quantum computations.
 circuits; squares – with
circuits; squares – with  circuits; triangles – with ansatz (21) using (left to right) k = 1, 2, 3 and 4 and eqn (22). The
circuits; triangles – with ansatz (21) using (left to right) k = 1, 2, 3 and 4 and eqn (22). The  Ansatz for Mg–NH excludes the gate shown in green.
Ansatz for Mg–NH excludes the gate shown in green.








 requires N → ∞. In this limit, eqn (A6) becomes
requires N → ∞. In this limit, eqn (A6) becomes




 . For the following functions
. For the following functions










 ,
,