Enhanced Cr(VI) adsorption on nitrogen and sulfur functionalized granular activated carbon
Received
5th July 2025
, Accepted 27th November 2025
First published on 2nd December 2025
Abstract
The effect of functionalizing a granular activated carbon (GAC) adsorbent with nitrogen and sulfur groups was investigated, with the aim of enhancing the adsorption capacity for hexavalent chromium (Cr(VI)) from aqueous solutions. The modification of GAC with nitrogen and sulfur groups was achieved in a one pot hydrothermal process using thiourea as the precursor. The amount of nitrogen and sulfur incorporated onto the GAC using this process was 1.6 at% and 1.0 at% based on XPS analysis, and 0.72 at% and 1.22 at% based on CHNS analysis, respectively. The modified GAC exhibited higher adsorption capacity compared with the unmodified GAC across all tested pH levels and ionic strengths with the highest value of 23.2 mg g−1 at pH 3, compared with 20.5 mg g−1 for the unmodified GAC. The N/S functionalized GAC favored the reduction of Cr(VI) to Cr(III) during the adsorption process, which is considered to have lower toxicity. XPS and FTIR analyses, together with desorption experiments, suggest that adsorption primarily occurs through hydrogen bonding and reduction–adsorption mechanisms for N/S functionalized GAC. Isotherm analysis revealed that Cr(VI) adsorbs predominantly as a monolayer of HCrO4− on the heterogeneous GAC surface (Sips model), whereas N/S-functionalized GAC follows the Freundlich model, indicating heterogeneous adsorption through the formation of Cr(III) hydroxides near functional groups. Kinetic studies indicated that the adsorption follows a revised pseudo-second order kinetic model on both surfaces, where chemisorption is the rate-controlling step. The rate of adsorption is similar for both types of GAC in the initial regime with fast kinetics. However, after 200 min, N/S functionalized GAC exhibits a higher uptake, likely associated with the additional Cr(VI) adsorption through a reduction process. Regeneration experiments revealed that NaOH and KOH solutions achieved significantly higher desorption efficiencies (>90% for unmodified GAC and >70% for N/S functionalized GAC) compared to acidic regeneration using HCl solution (<40% for GAC and <25% for N/S–GAC).
1. Introduction
Heavy metals are commonly used in numerous industries such as metal plating, battery manufacturing, and tanneries. The wastewater produced from these industries has high concentrations of metal ions which can pose severe health problems for humans and animals.1,2 Among the heavy metals, chromium is associated with particularly severe health hazards.3 Chromium from industrial effluents mainly exists in two oxidation states: trivalent chromium Cr(III) and hexavalent chromium Cr(VI). The latter is much more toxic and can cause DNA damage and cancer.4 Consequently, stringent regulations dictate a maximum allowable limit for chromium in industrial wastewater (for example, a maximum of 5 ppm for total Cr, set by the European Union Risk Assessment Report5) to safeguard the environment before any discharge takes place.
Adsorption is a well-developed method for treatment of groundwater and industrial wastewaters in general and for chromium removal in particular.2,3,6 Several natural and synthetic carbon-based materials are used for Cr adsorption studies,3,6,7 some of which include activated carbon,8 graphene, carbon aerogels and hydrogels and magnetic polymers.9,10 Activated carbon, with its high surface area, abundance of surface functional groups, low cost, and potential for regeneration, is one of the most widely used materials in industrial adsorption processes for a wide range of contaminants.11 Granular activated carbon (GAC), with particle sizes >100 µm, is ideal for fixed adsorption beds due to easy handling and minimal fine particle bleeding, while powdered activated carbon (PAC, d < 100 µm) offers faster kinetics and higher surface area, making it ideal for batch adsorption and flow beds.12–14
Understanding the adsorption mechanisms of Cr(VI) and the key parameters that influence adsorption behavior is essential for developing materials with enhanced adsorption capacities and designing efficient adsorption processes. In Cr(VI) adsorption, factors such as functional groups, solution pH and ionic strength are particularly significant, as they strongly impact the adsorption capacity of carbon-based materials.8,11,15,16 For example, in an acidic media, the surface functional groups of GAC are protonated,17,18 acquiring either a positive or neutral charge (e.g., –OH, –COOH, –NH3+), which attracts the dominant anionic Cr(VI) species through electrostatic interactions or hydrogen bonding. An ion exchange mechanism has been proposed where protonated functional groups such as C–OH2+ (protonated hydroxyl groups) interact with Cr(VI) ions.18 Additionally, reduction reactions can occur on the GAC surface, converting Cr(VI) to Cr(III), due to electron transfer facilitated by surface functional groups.16,19 Hydrogen bonding between oxygen or nitrogen functional groups and Cr(VI) species has also been proposed as one of the removal mechanisms on modified activated carbon surfaces.20–23
The adsorption capacity, as well as the conversion of Cr(VI) to Cr(III) of activated carbon, can be improved through chemical modifications that add functional groups, enhancing interactions with Cr(VI). Alkaline treatments introduce oxygen functional groups including hydroxyl and carboxyl groups, promoting ion exchange and electrostatic attraction.24,25 Nitric acid treatment for GAC has been found to enhance Cr(VI) removal efficiency by up to 70%, and increase the adsorption capacity by 2.5 times through the introduction of additional oxygen functional groups on the surface.26,27 The Langmuir model was reported to represent the adsorption isotherm data best, suggesting a monolayer adsorption process. Incorporating heteroatoms such as nitrogen and sulfur has also proven effective, as these atoms contribute additional adsorption or reactive sites, such as amine and sulfide groups, which aid Cr(VI) capture and conversion.8,15,16,19 Abushawish et al. showed that a 45% improvement in Cr adsorption capacity can be achieved with nitrogen-doped GAC, prepared by an impregnation method in an ethanol medium. The Sips and Redlich–Peterson models were found to fit the adsorption isotherm of the modified GAC, suggesting monolayer adsorption on a heterogeneous surface.8 Regeneration efficiency is a critical parameter in the development of new adsorbents, as the economic feasibility of the process depends on the reusability of the adsorbent material. Various desorbing solutions have been employed to remove Cr species from saturated adsorbents and restore adsorption capacity. For carbon-based materials, regeneration is commonly achieved using NaOH, KOH, HCl, or HNO3 solutions, typically in concentrations ranging from 0.1 to 1 M.3
While extensive research has explored the incorporation of nitrogen and sulfur heteroatoms into PAC, primarily during the synthesis stage using a carbon source and thiourea, studies on post-treatment modification of commercially available activated carbon remain scarce. Dual heteroatom doping (N/S) has been shown to improve Cr(VI) adsorption in various systems,16,28–30 but its application to GAC, particularly through post-synthetic modification, has not been systematically examined. This study investigates whether such post-treatment doping is effective for enhancing Cr(VI) adsorption on GAC under a range of environmental conditions. In addition to adsorption capacity, isotherms, and kinetics, we also explore the impact of ionic strength up to 0.5 M NaCl. Furthermore, desorbed species during the regeneration process are analyzed and discussed as complementary evidence to XPS results, offering new insights into the underlying adsorption mechanisms. To the best of our knowledge, this is the first comprehensive study to address these aspects in N/S functionalization of GAC systems for Cr(VI) removal.
2. Materials and methods
2.1. Materials
Acid-washed GAC (coconut shell, mesh size 8 × 30; Newterra Co.), denoted as GAC, was used as the starting material. Thiourea (Fisher Scientific) was used as the source of nitrogen and sulfur in the preparation of N/S-functionalized GAC (denoted as N/S–GAC). Potassium dichromate (Anachemia) was used to prepare Cr(VI) solutions. Sodium chloride (Sigma-Aldrich) was used to adjust the ionic strength of all solutions to values as indicated throughout this work. Hydrochloric acid (Sigma-Aldrich), sodium hydroxide (Sigma-Aldrich) and potassium hydroxide (Fisher Scientific) were used to adjust the pH of solutions and to prepare regeneration solutions.
2.2. Preparation of N/S functionalized GAC
N/S–GAC was prepared via a hydrothermal process in which 5 g of GAC was added to 100 mL of 100 g L−1 thiourea solution and in a Teflon container. The container was then put in a stainless steel autoclave and placed in a convection oven at 180 °C for 24 h to decompose the thiourea and functionalize the GAC surface with N- and S-containing groups. Then, after cooling at room temperature, the modified GAC was washed thoroughly with DI water to remove any excess materials (like unreacted thiourea or its products during decomposition in the hydrothermal process) and dried at 100 °C overnight.28
2.3. Characterization
Nitrogen adsorption analysis (Micromeritics TriStar II) was conducted to evaluate the pore-size distribution (the distribution of pore diameters calculated from nitrogen adsorption–desorption isotherms), pore volume and surface area of the sorbents. The Barrett–Joyner–Halenda (BJH) method31 was applied to calculate pore-size distribution (suitable for pores ∼1–50 nm), while the Brunauer–Emmett–Teller (BET) method was used to determine the specific surface area. SEM imaging (Quanta FEG 250 Field Emission SEM) was performed to obtain morphological information. FTIR analysis was conducted using the KBr pellet method with Nicolet 4700 at 32 scans and a resolution of 4 cm−1 for 500–4000 cm−1. Samples were crushed into fine powders and mixed with KBr powder in a 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volumetric ratio (KBr:GAC) and pressed into pellets with a 6-ton press load. Raman spectroscopy was performed using a confocal Raman microscope (WITec Alpha-300) equipped with a diode laser operating at a wavelength of 532 nm and a laser power of 5 mW. CHNS elemental analysis was performed using the ELEMENTAR UNICUBE analyzer. X-ray photoelectron spectroscopy (XPS, Thermo-Fisher ESCALAB QXi) was performed using a monochromatic Al Kα (1486.6 eV) radiation source and a passing energy of 150 eV for survey scans (step size of 1 eV) and 30 eV for high-resolution scans (step size of 0.1 eV). Adsorbents were placed on the sample holder in granular form. XPS data analysis was performed using CasaXPS software. All the spectra were calibrated with the C–C binding energy of 284.8 eV as the reference. Peak fitting was performed by constraining the FWHM and lineshape (Gaussian–Lorentzian and Lorentzian-asymmetric models), and by using the Shirley model for the background. Core-level peaks were added to each high-resolution spectrum through a thorough study of the literature.
:1 volumetric ratio (KBr:GAC) and pressed into pellets with a 6-ton press load. Raman spectroscopy was performed using a confocal Raman microscope (WITec Alpha-300) equipped with a diode laser operating at a wavelength of 532 nm and a laser power of 5 mW. CHNS elemental analysis was performed using the ELEMENTAR UNICUBE analyzer. X-ray photoelectron spectroscopy (XPS, Thermo-Fisher ESCALAB QXi) was performed using a monochromatic Al Kα (1486.6 eV) radiation source and a passing energy of 150 eV for survey scans (step size of 1 eV) and 30 eV for high-resolution scans (step size of 0.1 eV). Adsorbents were placed on the sample holder in granular form. XPS data analysis was performed using CasaXPS software. All the spectra were calibrated with the C–C binding energy of 284.8 eV as the reference. Peak fitting was performed by constraining the FWHM and lineshape (Gaussian–Lorentzian and Lorentzian-asymmetric models), and by using the Shirley model for the background. Core-level peaks were added to each high-resolution spectrum through a thorough study of the literature.
The surface charge of the materials (point of zero charge) as a function of pH was measured with a Malvern Zetasizer Nano-ZS using the dynamic light scattering (DLS) method. Prior to this measurement, samples were first crushed into fine powder and were then suspended in solutions with different pH values using an ultrasound bath. UV-vis absorption spectroscopy (Shimadzu UV-2600) at a wavelength of 350 nm was used to assess Cr(VI) concentrations and inductively coupled plasma optical emission spectroscopy (ICP-OES, iCAP 7000 Series) was carried out to measure total Cr concentrations in the solutions. For both UV-vis and ICP-OES analyses, samples were diluted with 2% nitric acid to ensure that Cr(VI) concentrations were below 50 ppm. The concentration of Cr(III) was then calculated by subtracting the Cr(VI) concentration from the total Cr concentration, assuming that no other chromium species were present.
2.4. Adsorption and regeneration experiments
To measure the equilibrium and time-dependent adsorption capacity of our materials, batch adsorption experiments with a magnetic stirrer (450 rpm) were performed. Samples of solutions were taken and analyzed for chromium concentration by ICP-OES. The equilibrium adsorption capacity (qe) of the adsorbents was calculated using eqn (1):| | 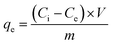 | (1) |
where Ci and Ce are the initial and equilibrium Cr concentration, V is the volume of the solution and m is the mass of the adsorbent.
Isotherm studies were conducted for Ci = 20–250 mg L−1 at pH = 5, in a 0.01 M NaCl solution at ambient temperature for an adsorption duration of 20 h. Kinetic experiments were carried out at Ci = 50 ppm, in a 0.01 M NaCl solution at pH = 5, with an adsorbent dosage of 2 g L−1 for a duration of 20 h. The effects of pH (from 3 to 8) and ionic strength (0 to 0.5 M NaCl) were studied.
Regeneration of adsorbents was performed using three desorption solutions, namely 1 M HCl, 0.1 M NaOH and 0.1 M KOH, for 16 h (two-step regeneration, each step 8 h). The regeneration (desorption) efficiency for each cycle was calculated as the ratio of the total amount of Cr species desorbed during regeneration to the total amount adsorbed.
3. Results and discussion
3.1. Incorporation of N-, S-groups in GAC
We incorporated N- and S-groups into GAC via the hydrothermal method described above. The physicochemical properties of GAC and N/S–GAC, including morphology, pore size distribution, surface area, and pore volume, play an important role in determining its effectiveness in the adsorption of Cr. The SEM micrographs of GAC and N/S–GAC (Fig. 1a and b) show the presence of large pores (a few microns) on the surface, however, no major difference can be observed between the two adsorbents. The N2 adsorption analyses show that N/S–GAC has lower surface area (by 15%) and pore volume (by 17%) than GAC (Table 1). Moreover, pore-size distribution plots in Fig. 1c suggest the presence of micropores for both adsorbents and that the proposed modification does not change the adsorbents significantly. This can be attributed to pore blockage by the products of thiourea decomposition, such as carbodiimide (HN![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CNH).32 XPS elemental analysis of the GAC and N/S–GAC, presented in Table 1, confirms the presence of nitrogen (1.64 at%), sulfur (1.04 at%), and oxygen (4.69 at%) on the N/S–GAC surface. In contrast, the GAC surface contained negligible sulfur (0.06 at%) and nitrogen (0.27 at%), with an oxygen content of 6.29 at%. CHNS analysis (Table 1) was conducted to compare the bulk composition with surface composition from XPS. Surprisingly, the bulk nitrogen content was significantly lower than that on the surface (0.72 at% vs. 1.64 at%), while the bulk sulfur content was slightly higher than that on the surface (1.23 at% vs. 1.04 at%). This implies that nitrogen species penetrated the pore structure less effectively than sulfur. To gain deeper insight into the nature of sulfur-, nitrogen-, and oxygen-containing functional groups on the GAC surface, high-resolution XPS analysis was conducted on both GAC and N/S–GAC (Fig. 2b–g). The XPS survey spectra of GAC and N/S–GAC, presented in the SI (Fig. S2), show the presence of O, N, and S atoms in addition to carbon. The C 1s spectra (Fig. 2b and c) confirm the presence of hydroxyl (286.3 eV), carbonyl (287.8 eV), and carboxylic (288.8 eV) groups.33,34 Deconvolution of the N 1s spectrum indicates that the majority of the nitrogen (69.5%) is present as amine groups, with a binding energy centered at 399.7 eV. Protonated amine groups made up the rest of the incorporated nitrogen at 402.5 eV (30.5%). The S 2p spectra show the doublet peaks at 164.3 eV and 165.5 eV, attributed to the C–S–C bonding environment (S 2p3/2 and S 2p1/2), as well as the doublet at 168.4 eV and 169.6 eV attributable to sulfonic groups (–SOx, 35%).
CNH).32 XPS elemental analysis of the GAC and N/S–GAC, presented in Table 1, confirms the presence of nitrogen (1.64 at%), sulfur (1.04 at%), and oxygen (4.69 at%) on the N/S–GAC surface. In contrast, the GAC surface contained negligible sulfur (0.06 at%) and nitrogen (0.27 at%), with an oxygen content of 6.29 at%. CHNS analysis (Table 1) was conducted to compare the bulk composition with surface composition from XPS. Surprisingly, the bulk nitrogen content was significantly lower than that on the surface (0.72 at% vs. 1.64 at%), while the bulk sulfur content was slightly higher than that on the surface (1.23 at% vs. 1.04 at%). This implies that nitrogen species penetrated the pore structure less effectively than sulfur. To gain deeper insight into the nature of sulfur-, nitrogen-, and oxygen-containing functional groups on the GAC surface, high-resolution XPS analysis was conducted on both GAC and N/S–GAC (Fig. 2b–g). The XPS survey spectra of GAC and N/S–GAC, presented in the SI (Fig. S2), show the presence of O, N, and S atoms in addition to carbon. The C 1s spectra (Fig. 2b and c) confirm the presence of hydroxyl (286.3 eV), carbonyl (287.8 eV), and carboxylic (288.8 eV) groups.33,34 Deconvolution of the N 1s spectrum indicates that the majority of the nitrogen (69.5%) is present as amine groups, with a binding energy centered at 399.7 eV. Protonated amine groups made up the rest of the incorporated nitrogen at 402.5 eV (30.5%). The S 2p spectra show the doublet peaks at 164.3 eV and 165.5 eV, attributed to the C–S–C bonding environment (S 2p3/2 and S 2p1/2), as well as the doublet at 168.4 eV and 169.6 eV attributable to sulfonic groups (–SOx, 35%).
 |
| | Fig. 1 SEM micrographs of (a) GAC and (b) N/S–GAC. Large pores (micron-sized) can be seen on both materials, suggesting the presence of macropores which could not be detected with N2 adsorption analysis; (c) pore-size distribution of GAC and N/S–GAC obtained using fitting of N2 adsorption data by the BJH model. The figure shows that for both materials, micropores (pore-size less than 2 nm) are dominant and almost no mesopores (pore-size between 2 and 50 nm) are seen. | |
Table 1 Structural properties and elemental composition of adsorbents
|
|
N2 adsorption |
Elemental analysis (at%) – CHNS |
Elemental analysis (at%) – XPS |
|
S
total (m2 g−1) |
V
total (cm3 g−1) |
C |
N |
S |
O |
C |
N |
S |
O |
| GAC |
987.93 |
0.42 |
94.81 |
0.23 |
0 |
4.73 |
93.38 |
0.27 |
0.06 |
6.29 |
| N/S–GAC |
835.54 |
0.35 |
95.52 |
0.72 |
1.23 |
2.53 |
92.63 |
1.64 |
1.04 |
4.69 |
 |
| | Fig. 2 (a) FTIR spectra of GAC and N/S–GAC, before and after adsorption. Enhancement in peaks at 1060–1160 cm−1 and 3300–3500 cm−1 for N/S–GAC, compared with GAC, are attributed to the presence of SO and C–N and N–H bonding, respectively. (b)–(g) High-resolution XPS spectra of (b) and (c) C 1s for GAC and N/S–GAC, (d) and (e) O 1s for GAC and N/S–GAC, and (f) and (g) N 1s and S 2p for N/S–GAC. | |
We also employed FTIR and Raman spectroscopy to identify different heteroatoms and their groups and structural disorders in GAC and N/S–GAC (Fig. 2a and Fig. S1). For the unmodified GAC, three major FTIR peaks can be seen: ∼1060–1160 cm−1 attributed to C–O stretching,35 ∼1575 cm−1 attributed to CC stretching, and ∼3440 cm−1 related to O–H stretching (hydroxyl groups).36 After incorporating N and S, a more pronounced peak at 3400–3500 cm−1 is associated with N–H stretching (amine group). At 1060–1160 cm−1, SO stretching (sulfonic group) and C–N stretching (amine group) are responsible for the peak broadening and increased intensity.35 Raman spectra of GAC and N/S–GAC are shown in Fig. S1. The two main Raman peaks of carbon-based materials, the D-band at ∼1340 cm−1 and the G-band at ∼1600 cm−1 are present as expected. The D-band represents the degree of disorder in sp2-hybridized carbon caused by edge sites, functional groups and pores, while the G-band represents in-plane stretching vibrations of the sp2-hybridized graphitic structure. The ID/IG ratio for GAC and N/S–GAC was 1.15 ± 0.02 and 1.19 ± 0.03, respectively. The slight increase in the ID/IG ratio in the N/S–GAC is likely due to the defects added through functionalization with N and S.
3.2. Performance
3.2.1. The effect of pH and ionic strength.
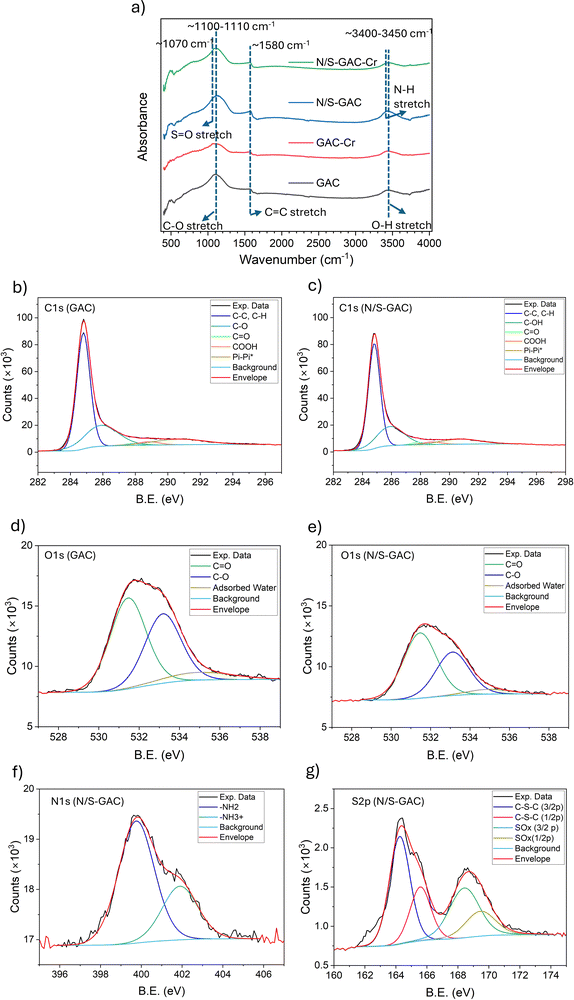
The pH of a solution determines the Cr speciation and charge state of the adsorbents’ surface species, both of which can impact the adsorption process and performance. We investigated the effect of the initial pH of the solution on Cr(VI) removal for unmodified and modified GAC adsorbents over a range from pH 3 to 8, using an initial Cr(VI) concentration of 50 ppm and a 0.01 M NaCl background solution. The results, shown in Fig. 3a, suggest that lower initial pH (below 4) is favorable for the removal of Cr(VI) for both GAC and N/S–GAC. The adsorption capacity significantly decreased as the pH increased from 3 to 8 (from 20.0 to 1.0 mg g−1 for GAC and 23.3 mg g−1 to 4.6 mg g−1 for N/S–GAC). Similar trends were observed in previous studies of carbon-based adsorbents, where low initial pH in the range of 2–4 resulted in higher adsorption capacity, with a significant (3- to 5-fold) decrease in capacity as the pH increased.8,15,19,21 Possible reasons for higher capacity at lower pH generally include: (1) positive surface charge (positive zeta potential) which can result in electrostatic attraction; (2) specific interactions between functional groups and Cr(VI) species (such as hydrogen bonding or ion exchange); and (3) reduction of Cr(VI) due to the abundance of H+. Zeta potential measurements (Fig. S3) show that point of zero charge for unmodified GAC and N/S–GAC was around pH 2.0 and 3.0, respectively. Above this pH, the surface is negatively charged and tends repel anions, including all Cr(VI) ions (Fig. S4). The acid dissociation constant (pKa) for protonation of oxygen functional groups such as hydroxyl groups (–OH2+/–OH) is typically very low (pKa < 0),37 hence, electrostatic attraction is not a dominant mechanism in this system. Hydrogen bonding between HCrO4− and oxygen functional groups such as –COOH and –OH is the most probable interaction mechanism, since hydrogen bonding energy can be larger than that of the electrostatic repulsion (see calculations in the SI, Section S1). As the pH increases, CrO42− becomes the dominant Cr(VI) species, and the capacity decreases due to weaker hydrogen bonding between –OH and –OCr, the abundance of OH− as competing anions, and larger electrostatic repulsion as the surface charge and the anion charge both increase. At acidic pH, adsorption through reduction reaction pathways may also occur, since the H+ and surface functional groups that act as electron donors are both abundant. This will lead to formation of Cr(III) species,16,38 which are positively charged at pH < 6 (provided electrostatic attraction to the adsorbent surface), and precipitate as Cr(OH)3 at higher pH values.39Reactions (1)–(3) summarize the mechanism of reduction and Cr(OH)3 formation.| | | 3R–H + 2HCrO4− + 8H+ → 3R–OH + 2Cr3+ + 5H2O | (1) |
 |
| | Fig. 3 (a) Cr loading as a function of pH (conditions: Ci = 50 ppm, t = 20 h, adsorbent dosage = 2 g L−1, and ionic strength = 0.01 M NaCl); (b) Cr loading as a function of ionic concentration (conditions: pH = 5, Ci = 50 ppm, t = 20 h, and adsorbent dosage = 2 g L−1); high-resolution Cr 2p spectra of (c) GAC and (d) N/S–GAC after adsorption and (e) N 1s and (f) S 2p spectra of N/S–GAC after adsorption (pHi = 5, Ci = 50 ppm, t = 20 h, and adsorbent dosage = 2 g L−1). | |
(Redox reactions:
| 3R−H + 3H2O → 3R−OH + 6H+ + 6e− |
| 2HCrO4− + 14H+ + 6e− → 2Cr3+ + 8H2O) |
| | | 6R–OH + 2HCrO4− + 8H+ → 6RO + 2Cr3+ + 2H2O | (2) |
(Redox reactions:
| 3R−OH → 3RO + 3H+ + 3e− |
| HCrO4− + 7H+ + 3e− → Cr3+ + 4H2O) |
The adsorption results indicate that N and S functionalization improves the capacity of the GAC at all pH studied, with the highest capacity of 23.3 mg g−1 obtained at pH 3. N-Functional groups can be protonated at low pH16 (e.g. amine groups with pKa(–NH3+/–NH2) ≈ 937,40), leading to a more positive surface charge compared to the unmodified GAC, and thus enhanced electrostatic attraction of Cr(VI) anions, resulting in a higher adsorption capacity.17 N and S functional groups also take part in hydrogen bonding with Cr(VI) species, which is not readily affected by pH change.21,22 N and S functional groups can also act as electron donors for the reduction of Cr(VI) to Cr(III)16,19,38 in the presence of H+, further increasing the adsorption capacity.
The Cr 2p XPS spectra of adsorbents after Cr adsorption are shown in Fig. 3c and d. The presence of Cr(III) was confirmed for both GAC and N/S–GAC, accounting for 66.6% and 83.2% of the total Cr, respectively. Previous studies have reported similar XPS spectra for Cr adsorbed on carbon materials, indicating reduction reaction as one of the main adsorption mechanisms.19,41 FTIR analysis (Fig. 2a) shows that several peaks relating to oxygen, nitrogen and sulfur groups were reduced in size (peaks at 1060–1160 cm−1 and 3300–3500 cm−1 exhibit lower intensities) for the adsorbents after Cr adsorption, suggesting that functional groups play an important role in the adsorption process.8,42,43 The N 1s and S 2p spectra of N/S–GAC after adsorption are presented in Fig. 3e and f, respectively. A notable decrease in the proportion of protonated amine groups (–NH3+) was observed after Cr adsorption. Protonated amine groups accounted for 14.6% of the total nitrogen after Cr adsorption on N/S–GAC, compared to 30.5% prior to adsorption, indicating their involvement in the adsorption process. Protonated amine groups can act as proton reservoirs, providing H+ for the reduction reaction, while oxygen and sulfur functional groups such as –OH, –SH and C–S–C act as electron donors. The content of oxidized sulfur species (–SOx) increased from 35.0% to 60.6% after Cr adsorption, suggesting the formation of additional sulfur–oxygen functionalities. Fig. 3f shows that some S–H/C–S–C groups remain unutilized after adsorption. This may be attributed to diffusion limitations, either due to blockage of underneath S groups by Cr(III) hydroxide formed by reduction reactions, or restricted accessibility due to the microporous structure of the N/S–GAC (Fig. 1c) for HCrO4− anions. Possible reduction reactions that may occur in the system based on these observations include:
| | 2HCrO4−![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) + 8RNH3+ + 3RCH2OH → 2Cr3+ + 8H2O + 3RCHO + 8RNH2 + 8RNH3+ + 3RCH2OH → 2Cr3+ + 8H2O + 3RCHO + 8RNH2 | (4) |
| (Oxidation: 3RCH2OH → 3RCHO + 6H+ + 6e− |
| Reduction: 2HCrO4− + 14H+ + 3e− → Cr3+ + 4H2O |
| Proton supply: RNH3+ ⇌ RNH2 + H+) |
| | | 2HCrO4− + 7RNH3+ + RSH → 2Cr3+ + 5H2O + RSO3− + 7RNH2 | (5) |
| (Oxidation: RSH + 3H2O → RSO3− + 7H+ + 6e− |
| Reduction: HCrO4− + 7H+ + 3e− → Cr3+ + 4H2O |
| Proton supply: RNH3+ ⇌ RNH2 + H+) |
| | | 2HCrO4− + 8RNH3+ + 3R2S → 2Cr3+ + 5H2O + 3R2SO + 8RNH2 | (6) |
| (Oxidation: 3R2S + 6H2O → 3R2SO2 + 12H+ + 12e− |
| Reduction: 2HCrO4− + 14H+ + 6e− → 2Cr3+ + 8H2O |
| Proton supply: RNH3+ ⇌ RNH2 + H+) |
| | | R–SH + 2HCrO4− + 7H+ → R–SO3− + 2Cr3+ + 5H2O | (7) |
| (Oxidation: R–SH + 3H2O → R–SO3− + 7H+ + 6e− |
| Reduction: 2HCrO4− + 14H+ + 6e− → 2Cr3+ + 8H2O) |
The O 1s spectra in Fig. S6 for both GAC and N/S–GAC show an increase in the CO and Cr–OH components around ∼531.7 eV, indicating the formation of these species during adsorption. The –NH2 peak was observed to shift slightly, from 399.7 to 400.3 eV, during the Cr adsorption on N/S–GAC. This may be due to hydrogen bonding between N–H and HCrO4−, which withdraws electron density from the nitrogen group and increases the binding energy. Section S2 of the SI provides additional details on the influence of nitrogen functional groups and solution pH on the reduction mechanism.
Wastewater effluents usually contain dissolved ions such as chloride (200–4000 ppm) and sulfate (150–1000 ppm) in addition to the heavy metals.44,45 The presence of co-ions in the solution typically reduces the adsorption capacity of the adsorbent. For Cr(VI), since it exists in an anionic form, other anions may influence the adsorption capacity due to competition for active sites. In this study, the effect of NaCl concentration on Cr(VI) adsorption was investigated at pH 5 and an initial Cr(VI) concentration of 50 ppm, as shown in Fig. 3b. As expected, the highest Cr adsorption capacity (17.9 mg g−1 for N/S–GAC and 13.7 mg g−1 for GAC) was observed when no NaCl was present. As the salt concentration was increased, the Cr adsorption capacity decreased, reaching 8.5 and 5.3 mg g−1 at 0.5 M NaCl for N/S–GAC and GAC, respectively. The rate of decrease in Cr(VI) adsorption capacity at NaCl concentrations higher than 0.1 M drops significantly. Similar trends have previously been reported,46,47 with the adsorption capacity of activated carbon reaching a plateau at NaCl concentrations greater than 0.3 M NaCl.
3.2.2. Regeneration.
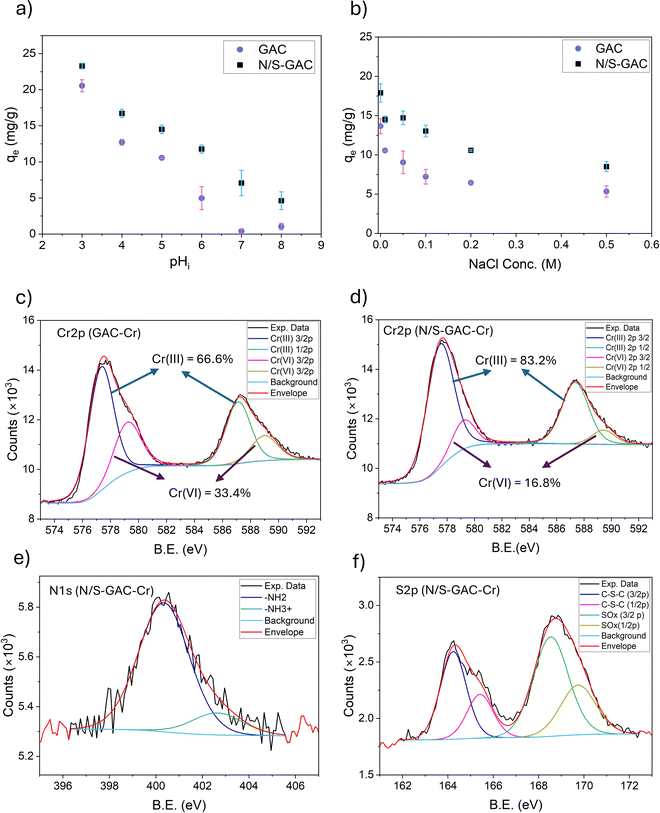
The regeneration of adsorbents serves as one of the benchmarks for designing a practical adsorption system. Often, adsorption capacity declines with each cycle, resulting in progressively worse performance. Both alkaline and acid solutions were evaluated for adsorbent regeneration, including 0.1 M NaOH, 0.1 M KOH, and 1 M HCl solutions. Fig. 4a shows the result of desorption of chromium from GAC and N/S–GAC for the first cycle. On GAC, the 0.1 M NaOH and 0.1 M KOH solutions performed similarly with Cr desorption efficiencies of >90%. This can be explained by electrostatic repulsion as follows: at high pH, surface functional groups (OH, COOH, and NH3+) become deprotonated, making the surface highly negative. This results in increased electrostatic repulsion between chromate anions and negatively charged surface which in turn either eliminates or reduces the extent of the attractive hydrogen bond network. On the other hand, 1 M HCl solution showed significantly lower regeneration performance (<40% desorption for 1st cycle). This is most likely due to the stability of hydrogen bonds at low pH. In the case of NaOH and KOH, both Cr(VI) and Cr(III) were desorbed. However, when a 1 M HCl solution was used, most of the desorbed Cr species was in the form of Cr(III). For GAC, the total amount of Cr(III) in the regeneration solution was substantially higher when using an HCl solution than that obtained with alkaline solutions (33% vs. 3.5% of total Cr adsorbed). The increased presence of Cr(III) with the HCl regenerant can be attributed to a reduction reaction occurring during desorption, as the abundance of H+ ions create a favorable environment for Cr(VI) reduction (providing H+ for reactions (1) and (2)).
 |
| | Fig. 4 (a) Desorption efficiencies and amount of Cr species desorbed for regeneration solutions, 0.1 M KOH, 0.1 M NaOH and 1 M HCl; conditions: two-step desorption, each step Vsol = 50 mL and t = 8 h, (b) cyclic regeneration performance of GAC and N/S–GAC with KOH 0.1 M as the regeneration solution; conditions: two-step desorption, each step Vsol = 50 mL and t = 8 h. | |
Table S1 provides more detailed analysis of desorbed species by ICP–OES and UV-vis, and comparison with XPS data. For GAC, UV-vis analysis shows that ∼9.3 mg g−1 of Cr is desorbed as Cr(VI) during regeneration, corresponding to ∼90% of the total adsorbed Cr. The remaining fraction is mainly desorbed as Cr(III), accounting for ∼10% of the total. However, speciation results for Cr on GAC from XPS (Fig. 3c and Fig. S6) indicate that Cr(III) species dominate (∼67% after adsorption and ∼81% after regeneration), which contradicts the ICP-OES and UV-vis results for the desorbed species. This discrepancy can be attributed to the preferential formation of Cr(III) hydroxides on the external surface of GAC granules. Since XPS is highly surface-sensitive, probing only the top few atomic layers, it detects these surface species more readily than those confined within the micro-, meso- and macropores of GAC.
For N/S–GAC, ∼3.8 mg g−1 of Cr(VI) was desorbed during regeneration based on analysis of desorbed species, corresponding to 23% of the total adsorbed Cr. In addition, ∼7.6 mg g−1 of Cr was desorbed as Cr(III), accounting for 45% of the total. The remaining 32% of Cr on N/S–GAC was identified by XPS analysis, as shown in Fig. S6, which indicated that the majority was present as Cr(III) (85% of Cr remained after regeneration). These species do not dissolve during regeneration and remain irreversibly bound to the surface. Comparison of desorption data from ICP-OES and UV-vis analyses with XPS results after adsorption (Fig. 3d) shows a slight discrepancy with the Cr species, which again arises from the surface sensitivity of XPS and its strong response to Cr(III) species concentrated on the outer surface. Overall, these findings suggest that for GAC, hydrogen bonding of HCrO4− anions could be considered as the dominant mechanism with minimal reduction happening on the outer surface, whereas for N/S–GAC, reduction is the main mechanism of Cr(VI) removal during the first adsorption cycle, with Cr(III) hydroxides heterogeneously distributed on the outer surface and bulk of the adsorbent. Some reduced species remaining irreversibly bound and others extractable by KOH and HCl regeneration. This is attributed to the functional groups on N/S–GAC that promote the reduction of Cr(VI) during the adsorption process.16,19,41 This difference is also reflected in isotherm analysis in Section 3.2.3. It should be noted that the formation of surface Cr(III) hydroxides may lead to biased XPS results, and thus complementary techniques such as ICP-OES and UV-vis provide more reliable information by accounting for both surface and bulk contributions. Scheme 1 summarizes the adsorption mechanisms for the GAC and N/S–GAC discussed above. A summary of XPS analyses for GAC and N/S–GAC has been provided in Fig. S8 and S9, respectively.
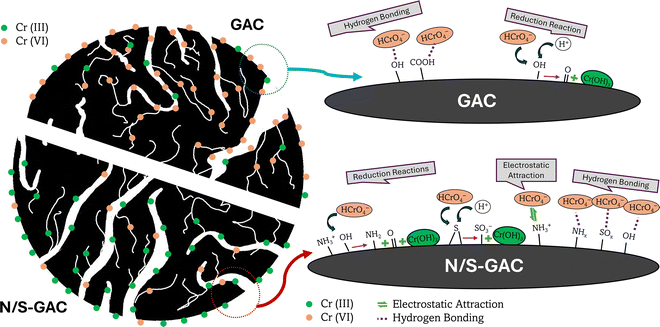 |
| | Scheme 1 Schematic of the adsorption process on GAC and N/S–GAC. For GAC, Cr(VI) is primarily bound to the surface through hydrogen bonding, with only a small fraction reduced to Cr(III). In contrast, for N/S–GAC, reduction is the dominant pathway during the first adsorption cycle, while from the second cycle onwards, hydrogen bonding and electrostatic attraction become the main mechanisms. | |
The regeneration performance of GAC and N/S–GAC was investigated over multiple adsorption–desorption cycles using 0.1 M KOH as the regeneration solution (Fig. 4b). The regeneration efficiency for GAC had a gradual decrease with each cycle, with the initial capacity of 10.6 mg g−1 decreasing to 8.1 mg g−1 on the fourth cycle. For the GAC, as expected, Cr(VI) is the predominant species in the regeneration solutions throughout the multiple cycle regeneration. For N/S–GAC, while new N and S functional groups showed significant improvement in the first cycle, their participation in adsorption reduced in the subsequent cycles, which led to a decrease in the regeneration efficiency, and the performance of N/S–GAC and GAC became similar after the second cycle. This decrease in performance may be attributed to a decrease in the reducing ability of N/S–GAC after the first cycle, as the regeneration process does not reactivate these reduction sites (Fig. S6 and S10). This reduction in adsorption was reported for N-doped GAC previously, where 15% to 20% of the initial capacity is lost after the first cycle.8,15
Addressing irreversible oxidation is critical for practical deployment, as it directly influences cost-effectiveness, lifespan of the adsorbent, and process sustainability. Some strategies for preserving the N,S-functionality include (i) more robust regeneration strategies that can partially restore or protect the active sites, such as electro-regeneration where the oxidized sites can be reduced or (ii) periodic re-functionalization of the adsorbent, if possible.
3.2.3. Adsorption isotherm and kinetics.
Isotherm and kinetic studies were carried out for Cr(VI) adsorption on both GAC and N/S–GAC to validate the adsorption mechanisms proposed in the preceding sections. As shown in Fig. 2, the initial solution pH has a significant influence on Cr(VI) uptake by both adsorbents. In the absence of pH adjustment, solutions with initial Cr(VI) concentrations ranging from 10 to 100 mg L−1 exhibited pH values between 4.5 and 5. Accordingly, a pH of 5 was selected for the isotherm and kinetic experiments. As shown in Fig. 5a, the adsorption capacity increased with initial Cr(VI) concentration without reaching a plateau, indicating unsaturated active sites across the tested range. N/S–GAC consistently outperformed GAC, which was attributed to the presence of N and S functional groups.
 |
| | Fig. 5 (a) Adsorption data and best-fitted isotherms for GAC and N/S–GAC. (Conditions: Ci = 25–250 ppm, pH = 5, adsorbent dosage = 2 g L−1, and T = 22 °C); (b) adsorption kinetics and best fitted model (conditions: Ci = 50 ppm, pH = 5, adsorbent dosage = 2 g L−1, and T = 22 °C). Shaded areas are representative of 95% confidence intervals for the fits. | |
Isotherm data were fitted using non-linear regression to common models: Freundlich, Langmuir, Temkin, Sips and Redlich–Peterson (Table S2 and Fig. S11). Table S3 summarizes the model parameters obtained from each fitting, along with the 95% confidence intervals, correlation coefficients (R2), and sum of square root of errors (SSE). The Sips model best described the adsorption behavior of GAC, showing high R2, low SSE, and narrow confidence intervals, suggesting a monolayer adsorption process on a surface that may be heterogeneous or homogeneous, with heterogeneity arising from the distribution of adsorption energies among surface active sites. This is consistent with the regeneration results, which indicate relatively uniform adsorption of Cr(VI) as HCrO4− on the GAC surface. In contrast, for N/S–GAC, although both the Sips and Redlich–Peterson models produced good fits (low SSE and high R2), the parameters exhibited very broad 95% confidence intervals (Table S3), indicating that the additional parameter does not provide meaningful interpretive value. This likely reflects strong correlation among the model parameters.48 The Freundlich model, in turn, offered a better balance of fit quality and parameter reliability, with higher R2, lower SSE, and comparatively narrow confidence intervals, making it the most suitable model for describing adsorption on N/S–GAC. This finding is consistent with previous observations of heterogeneous Cr(III) hydroxide formation near N and S functional groups, as confirmed by ICP-OES and UV-vis analyses.
The kinetics of Cr adsorption exhibit two regimes (Fig. 5b), a rapid initial regime during the first 200 min, followed by a slower adsorption towards equilibrium. In the fast regime, adsorption rates on GAC and N/S–GAC are similar (∼0.108 mg (g min)−1vs. 0.113 mg (g min)−1, respectively). On the other hand, Cr adsorption is faster on N/S–GAC than on GAC in the slow regime (∼0.001 mg (g min)−1vs. 0.003 mg (g min)−1, respectively), with the higher adsorption capacities on N/S–GAC becoming more apparent. Given that the enhanced adsorption capacity on N/S–GAC arises from its adsorption–reduction ability rather than differences in surface area or porosity, the faster adsorption rate at longer adsorption times in N/S–GAC is attributed to the adsorption–reduction process. Similar kinetic trends have been reported for activated carbon systems. The kinetic data were analyzed using pseudo-first order, pseudo-second order (PSO), revised pseudo-second order (r-PSO, removes the necessity of constant adsorbate concentration in PSO49), Elovich, Boyd–Reichberg, and Weber–Morris models (described in Table S4). Among these, r-PSO provided the best fit for both GAC and N/S–GAC, based on R2 and SSE values, indicating chemisorption as the rate-limiting step (please refer to Table S5 and Fig. S12). The slow regime (200 min < t) has been fitted with the models separately and the rate constants have been included in Table S6 in the SI. The Weber–Morris model (Fig. S13) also shows that there are clearly two stages involved in the adsorption process; the first stage can be attributed to the film diffusion with larger constant rates, and the second stage with smaller constant rates assigned to the pore-diffusion of adsorbate species. Prior studies on N and S-doped activated carbon have also identified PSO as the most appropriate model, supporting chemisorption as the dominant mechanism.16,19,21,28–30 Notably, given the small pore sizes (<2 nm) of both GAC and N/S–GAC (Fig. S1), intraparticle diffusion likely governs the initial adsorption stage. However, the assumption of constant adsorbate concentration during the adsorption process in conventional diffusion models (and also PSO model) may not fully apply under the experimental conditions.50
4. Conclusions
In this study, we investigated the effect of simultaneous nitrogen and sulfur incorporation into commercial GAC using a simple one-pot hydrothermal post-treatment method for Cr(VI) adsorption. Characterization by FTIR and XPS confirmed the successful formation of functional groups such as amines, sulfides, and sulfonic group, which enhanced adsorption performance. Although the modification resulted in a 15% reduction in surface area which is usually unfavorable, the N/S–GAC showed improved Cr(VI) adsorption across a broad pH range (3–8) and in the presence of high ionic strength (up to 0.5 M NaCl), with the highest uptake at pH 3. Isotherm modeling suggested a shift in the adsorption mechanism: the Sips model best described unmodified GAC, while the Freundlich model better fit the N/S–GAC data, indicating a transition from predominantly monolayer physisorption of HCrO4− to heterogeneous adsorption involving chemical reduction. Kinetic analysis using the revised pseudo-second-order model pointed to chemisorption as the rate-limiting step.
To gain further insight into the adsorption mechanism, we compared surface species identified by XPS with desorbed species analyzed by ICP-OES and UV-vis after regeneration. XPS results showed the presence of Cr(III) on the adsorbents’ surface, suggesting reduction of Cr(VI) for both GAC and N/S–GAC which happens along with hydrogen bonding of Cr(VI) species to O–H/N–H groups and electrostatic attraction of Cr(VI) species to the protonated amine group (–NH3+). The analysis of the desorption solutions revealed that Cr(VI) was the dominant species released from GAC, indicating that reduction was limited or did not occur extensively. This discrepancy underscores the limitations of relying solely on XPS for speciation and highlights the importance of complementary solution-phase analysis. Our approach, combining surface and desorbed species analysis, provides a more accurate understanding of the reduction behavior and adsorption mechanism, and to the best of our knowledge, has not previously been applied to N/S-functionalized activated carbon systems. Regeneration studies further showed that alkaline desorption (using NaOH or KOH) was more effective than acidic conditions, although some Cr remained irreversibly bound, leading to performance decline over successive cycles and apparent loss of reduction capability. This points to the need for improved regeneration strategies; ongoing work is exploring electrochemical regeneration to address this limitation.
Overall, our findings demonstrate that post-synthetic N/S-functionalization of GAC is a promising route to enhance Cr(VI) adsorption. Moreover, the integration of desorbed species analysis with surface characterization provides deeper insight into adsorption mechanisms and reveals that Cr(VI) reduction on activated carbon may be less extensive than previously assumed. These results contribute to the development of more effective adsorbents for water treatment and set a foundation for more robust mechanistic investigations.
Conflicts of interest
There are no conflicts to declare.
Data availability
The data supporting this article have been included in the main text and the supplementary information (SI). The supplementary information includes additional calculations, materials characterization data, and isotherm and kinetic modeling results. See DOI: https://doi.org/10.1039/d5cp02560e.
Acknowledgements
This work was funded in part by a National Science and Engineering Research Council Innovation Links grant, Alberta Innovates Clean Resources (Project number: 202102514) and Imperial Oil Ltd. J. B. was supported by an NSERC Undergraduate Student Research Award (2024). A. B. acknowledges support from the Alberta Innovates – Technology Scholarship and NOVA Chemicals Graduate Scholarship. A. M. B. acknowledges support from an NSERC Discovery Grant (RGPIN-2024-04246). S. P. acknowledges support from an NSERC Alliance Grant (ALLRP 586637).
References
- W. S. Chai, J. Y. Cheun, P. S. Kumar, M. Mubashir, Z. Majeed, F. Banat, S.-H. Ho and P. L. Show, J. Cleaner Prod., 2021, 296, 126589 CrossRef.
- I. Ihsanullah, A. Abbas, A. M. Al-Amer, T. Laoui, M. J. Al-Marri, M. S. Nasser, M. Khraisheh and M. A. Atieh, Sep. Purif. Technol., 2016, 157, 141–161 CrossRef.
- A. U. Rajapaksha, R. Selvasembian, A. Ashiq, V. Gunarathne, A. Ekanayake, V. O. Perera, H. Wijesekera, S. Mia, M. Ahmad, M. Vithanage and Y. S. Ok, Sci. Total Environ, 2022, 809, 152055 CrossRef PubMed.
- M. Iyer, U. Anand, S. Thiruvenkataswamy, H. W. S. Babu, A. Narayanasamy, V. K. Prajapati, C. K. Tiwari, A. V. Gopalakrishnan, E. Bontempi, C. Sonne, D. Barceló and B. Vellingiri, Sci. Total Environ, 2023, 882, 163483 CrossRef PubMed.
- E. Vaiopoulou and P. Gikas, Chemosphere, 2020, 254, 126876 CrossRef PubMed.
- B. Liu, Y.-N. Xin, J. Zou, F. Khoso, Y.-P. Liu, X.-Y. Jiang, S. Peng and J.-G. Yu, Molecules, 2023, 28, 639 CrossRef PubMed.
- K. Z. Elwakeel, A. M. Elgarahy, Z. A. Khan, M. S. Almughamisi and A. S. Al-Bogami, Mater. Adv., 2020, 1, 1546–1574 RSC.
- A. Abushawish, I. W. Almanassra, S. N. Backer, L. Jaber, A. K. A. Khalil, M. A. Abdelkareem, E. T. Sayed, H. Alawadhi, A. Shanableh and M. A. Atieh, Mater. Chem. Phys., 2022, 291, 126758 CrossRef.
- H. M. Alghamdi, R. M. Mohammad and K. Z. Elwakeel, J. Polym. Environ., 2024, 32, 6522–6543 CrossRef.
- K. Z. Elwakeel, Desalination, 2010, 250, 105–112 CrossRef.
- D. Mohan and C. U. Pittman Jr., J. Hazard. Mater., 2006, 137, 762–811 CrossRef PubMed.
-
A. S. Mestre, M. Campinas, R. M. C. Viegas, E. Mesquita, A. P. Carvalho and M. J. Rosa, in Advanced Materials for Sustainable Environmental Remediation, Elsevier, 2022, pp. 433–475 Search PubMed.
- E. I. Ugwu and J. C. Agunwamba, Environ. Monit. Assess., 2020, 192, 240 CrossRef PubMed.
- S. H. Kim, H. K. Shon and H. H. Ngo, J. Ind. Eng. Chem., 2010, 16, 344–349 CrossRef.
- H. Liang, R. Sun, B. Song, Q. Sun, P. Peng and D. She, J. Hazard. Mater., 2020, 387, 121987 CrossRef PubMed.
- H. Li, N. Li, P. Zuo, S. Qu and W. Shen, Colloids Surf., A, 2021, 618, 126502 Search PubMed.
- C. Jung, J. Heo, J. Han, N. Her, S.-J. Lee, J. Oh, J. Ryu and Y. Yoon, Sep. Purif. Technol., 2013, 106, 63–71 CrossRef.
- S.-J. Park and Y.-S. Jang, J. Colloid Interface Sci., 2002, 249, 458–463 CrossRef PubMed.
- Q. Zhu, H. Gao, Y. Sun, Y. Xiang, X. Liang, A. Ivanets, X. Li, X. Su and Z. Lin, Sci. Total Environ, 2022, 834, 155312 CrossRef PubMed.
- Y.-J. Ko, K. Choi, S. Lee, K.-W. Jung, S. Hong, H. Mizuseki, J.-W. Choi and W.-S. Lee, Water Res., 2018, 145, 287–296 Search PubMed.
- J. Chen, M. Yuan, W. Cai, J. Wei, J. Zhou, P. Liu, Z. Yang, J. Luo, Q. Xia and Z. Cai, Chem. Eng. J., 2021, 422, 130153 CrossRef.
- H. Dong, L. Zhang, P. Shao, Z. Hu, Z. Yao, Q. Xiao, D. Li, M. Li, L. Yang, S. Luo and X. Luo, J. Hazard. Mater., 2023, 441, 129945 Search PubMed.
- M. Yang, R. Lu, Y. Wei, Y. Guo, L. Lan, J. Lu, Y. Liao, H. Li, P. Lan and Z. Kou, Process Saf. Environ. Prot., 2025, 195, 106736 CrossRef.
- M. Mariana, H. P. S. Abdul Khalil, E. M. Mistar, E. B. Yahya, T. Alfatah, M. Danish and M. Amayreh, J. Water Process Eng., 2021, 43, 102221 CrossRef.
- X. Yang, Y. Wan, Y. Zheng, F. He, Z. Yu, J. Huang, H. Wang, Y. S. Ok, Y. Jiang and B. Gao, Chem. Eng. J., 2019, 366, 608–621 CrossRef PubMed.
- S. Babel and T. A. Kurniawan, Chemosphere, 2004, 54, 951–967 CrossRef PubMed.
- G. Huang, J. X. Shi and T. A. G. Langrish, Chem. Eng. J., 2009, 152, 434–439 CrossRef.
- F. Chen, M. Zhang, L. Ma, J. Ren, P. Ma, B. Li, N. Wu, Z. Song and L. Huang, Sci. Total Environ, 2020, 730, 138930 CrossRef PubMed.
- Y. Liu, T. Wang, N. Song, Q. Wang, Y. Wu, Y. Zhang and H. Yu, Sci. Total Environ, 2023, 860, 160360 Search PubMed.
- X. Yang, B. Wang, P. Zhang, X. Song and F. Cheng, Sci. Total Environ, 2024, 912, 169265 Search PubMed.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, J. Am. Chem. Soc., 1951, 73, 373–380 Search PubMed.
- Z. D. Wang, M. Yoshida and B. George, Comput. Theor. Chem., 2013, 1017, 91–98 Search PubMed.
- M. Ayiania, M. Smith, A. J. R. Hensley, L. Scudiero, J.-S. McEwen and M. Garcia-Perez, Carbon, 2020, 162, 528–544 Search PubMed.
- M. C. Biesinger, Appl. Surf. Sci., 2022, 597, 153681 CrossRef.
- A. P. Terzyk, Colloids Surf., A, 2001, 177, 23–45 CrossRef.
- S. He, G. Chen, H. Xiao, G. Shi, C. Ruan, Y. Ma, H. Dai, B. Yuan, X. Chen and X. Yang, J. Colloid Interface Sci., 2021, 582, 90–101 Search PubMed.
-
J. Clayden, N. Greeves and S. Warren, Organic chemistry, Oxford university press, Oxford, 2nd edn, 2012 Search PubMed.
- J. He, Y. Long, Y. Wang, C. Wei and J. Zhan, ACS Appl. Mater. Interfaces, 2016, 8, 16699–16707 CrossRef PubMed.
- G.-R. R. Bernardo, R.-M. J. Rene and A.-D. L. T. Ma Catalina, J. Hazard. Mater., 2009, 170, 845–854 Search PubMed.
- D. Kurniawan, B. A. Anjali, O. Setiawan, K. K. Ostrikov, Y. G. Chung and W.-H. Chiang, ACS Appl. Mater. Interfaces, 2022, 14, 1670–1683 CrossRef PubMed.
- F. Chen, S. Guo, Y. Wang, L. Ma, B. Li, Z. Song, L. Huang and W. Zhang, Front. Environ. Sci. Eng., 2022, 16, 57 CrossRef.
- L. Jaber, I. Ihsanullah, I. W. Almanassra, S. N. Backer, A. Abushawish, A. K. A. Khalil, H. Alawadhi, A. Shanableh and M. A. Atieh, Sustainability, 2022, 14, 10877 Search PubMed.
- S. Singha, U. Sarkar and P. Luharuka, Sci. Total Environ, 2013, 447, 472–487 Search PubMed.
- G. Dönmez and Z. Aksu, Process Biochem., 2002, 38, 751–762 CrossRef.
- C. Fabiani, F. Ruscio, M. Spadoni and M. Pizzichini, Desalination, 1997, 108, 183–191 CrossRef.
- F. Dinatale, A. Lancia, A. Molino and D. Musmarra, J. Hazard. Mater., 2007, 145, 381–390 CrossRef PubMed.
- V. Lopez-Ramon, C. Moreno-Castilla, J. Rivera-Urtilla and L. R. Radovic, Carbon, 2003, 41, 2020–2022 CrossRef.
- N. Tolazzi, E. Steffani, E. Barbosa-Coutinho, J. B. Severo Júnior, J. C. Pinto and M. Schwaab, Chem. Eng. Res. Des., 2018, 138, 144–157 Search PubMed.
- J. C. Bullen, S. Saleesongsom, K. Gallagher and D. J. Weiss, Langmuir, 2021 DOI:10.1021/acs.langmuir.1c00142.
- Y. Wang, C. Wang, X. Huang, Q. Zhang, T. Wang and X. Guo, Chemosphere, 2024, 349, 140736 CrossRef PubMed.
|
| This journal is © the Owner Societies 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Linda J.
Eastcott
b,
Krista
Stevenson
b,
Edward P. L.
Roberts
a,
Linda J.
Eastcott
b,
Krista
Stevenson
b,
Edward P. L.
Roberts






