
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe crucial role of lattice water in directing supramolecular networks of deferiprone analogues: a combined X-ray and DFT study
Ángel
García-Raso
 *a,
Mariana
Rocha
b,
Ángel
Terrón
a,
Juan J.
Fiol
*a,
Adela
López-Zafra
a,
Carlos A.
Rodríguez
a,
Ezequiel M.
Vázquez-López
c,
Miquel
Barceló-Oliver
a and
Antonio
Frontera
*a
*a,
Mariana
Rocha
b,
Ángel
Terrón
a,
Juan J.
Fiol
*a,
Adela
López-Zafra
a,
Carlos A.
Rodríguez
a,
Ezequiel M.
Vázquez-López
c,
Miquel
Barceló-Oliver
a and
Antonio
Frontera
*a
aDepartament de Química, Universitat de les Illes Balears, Crta de Valldemossa km 7.5, 07122 Palma de Mallorca (Baleares), Spain. E-mail: toni.frontera@uib.es; jfa950@uib.es
bINBIOFAL (CONICET – UNT), Instituto de Química Orgánica. Facultad de Bioquímica, Química y Farmacia, Universidad Nacional de Tucumán, Ayacucho 471, T4000INI, San Miguel de Tucumán, Argentina
cDepartament de Química Inorgánica, Facultade de Química, Universidad de Vigo, Edificio Ciencias Experimentales, 36310 Vigo, Galicia, Spain
First published on 6th January 2026
Abstract
This work presents the synthesis, structural elucidation via X-ray diffraction, and density functional theory (DFT) investigation of three novel ligands based on deferiprone: maltol-TAU (3), maltol-histidine (4), and maltol-histamine (5), derived from taurine, histidine, and histamine, respectively. Additionally, the copper(II) complex of the histamine derivative, [Cu(maltol-HISTA)2(H2O)2]·2H2O·2HCl (6), was synthesized and characterized. In the solid state, the supramolecular architectures of ligands 3–5 and complex 6 are primarily stabilized by hydrogen bonding and $\pi$-stacking interactions. Theoretical modeling corroborated these observations, confirming that compounds 3, 4, and 5 possess protonated imidazole rings accompanied by sulfonate, carboxylate, and chloride counter-ions, respectively. A distinct conformational variation was observed regarding ring orientation: the hydroxypyridinone and imidazole rings are positioned nearly orthogonally in 4, whereas they adopt a parallel arrangement in 5. DFT calculations were further employed to analyze specific supramolecular assemblies, with a focus on the structural influence of co-crystallized water molecules. To rigorously characterize the H-bonding and non-covalent interactions, quantum theory of atoms in molecules (QTAIM) and non-covalent interaction (NCI) plot analyses were utilized, providing detailed insight into the electronic and structural features of these potential coordination chemistry candidates.
Introduction
The ability of hydroxypyridinones (HP) to form stable chelates with a wide range of metal ions has driven significant research into their medicinal applications.1–3 Among these, deferiprone (1,2-dimethyl-3-hydroxy-4-pyridinone) is prominent as an oral therapeutic for iron overload disorders such as hemochromatosis and thalassemia.4–6 To optimize the pharmacokinetic and pharmacodynamic behaviour of these chelators, various N-substituted pyridinones have been developed through specific functionalization strategies.7 Beyond the management of metal overload, the resulting metal complexes (typically ML2 and ML3 types) exhibit a broad spectrum of pharmacological potential, acting as antibacterial agents, imaging contrast agents, and potential treatments for Parkinson's disease. The crystallographic features of these bidentate (O,O) chelators coordinated with di- and trivalent metals are well-established in the literature.4–7 Furthermore, DFT calculations analyzing the metal ion selectivity of deferiprone complexes have demonstrated a preference for tricationic metal centers, such as Fe3+ and Al3+.8Recently we have studied the acid–base equilibrium in N-substituted 3-hydroxy-4-pyridinones, specifically the interconversion between the neutral form (1) and the “catechol” form (2) without an external proton source.9 To understand the influence of an acidic group on these equilibria, we prepared several ω-hydroxycarbonylalkyl-pyridones by reacting maltol with different ω-amino acids (glycine, β-alanine, γ-aminobutyric, and 6-aminocaproic acids). Our findings indicate that these two forms can be isolated by crystallization in different solvents. The “catechol” form was isolated in water (Scheme 1), while the neutral form was obtained in the aprotic polar solvent DMSO. For glycine, we were unable to isolate the neutral amino acid form, likely due to the higher acidity of its COOH group (pKa = 2.34) compared to the other amino acids: β-alanine (pKa = 3.55), γ-aminobutyric acid (pKa = 4.06), and 6-aminocaproic acid (pKa = 4.43) (see Scheme 1).
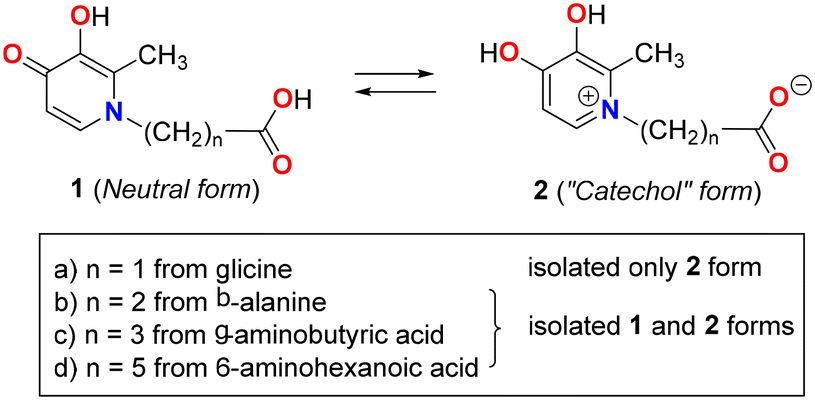 | ||
| Scheme 1 Molecular diagrams of ω-hydroxycarbonylalkyl-pyridinones previously described. | ||
With the aim of extending our previous study, we have prepared the corresponding products between maltol and taurine and two imidazole-containing derivatives: the amino acid L-histidine and its decarboxylated compound, the neurotransmitter histamine. Taurine presents a highly acidic SO3H group, suggesting that only the “catechol” form (2) should be obtained. By using histidine and histamine, we can evaluate how the acid–base equilibrium is modified in the presence of two proton acceptors: the carbonyl group of the HP and the imidazole ring of the lateral chain. In this paper, we report the synthesis and structural characterization of three new deferiprone derivatives: maltol-taurine [maltol-TAU·H2O, 2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)ethane-1-sulfonic acid monohydrate] [3·H2O], maltol-histidine [maltol-HISTI·2H2O, 2-(3-hydroxy-2-methyl-4-oxopyridin-1-(4H)-yl)-3-(1H-imidazole-4-yl)propanoic acid dihydrate] [4·2H2O], and maltol-histamine [maltol-HISTA·HCl·1.5H2O, 1-(2-(1H-imidazole-4-yl)ethyl)-3-hydroxy-2-methylpyridin-4(1H)-one hydrochloric acid sesquihydrate] [5·HCl·1.5H2O]. Additionally, we report the synthesis and structural characterization of the corresponding Cu(II) complex, 6, which is also a subject of the computational study (see Scheme 2).
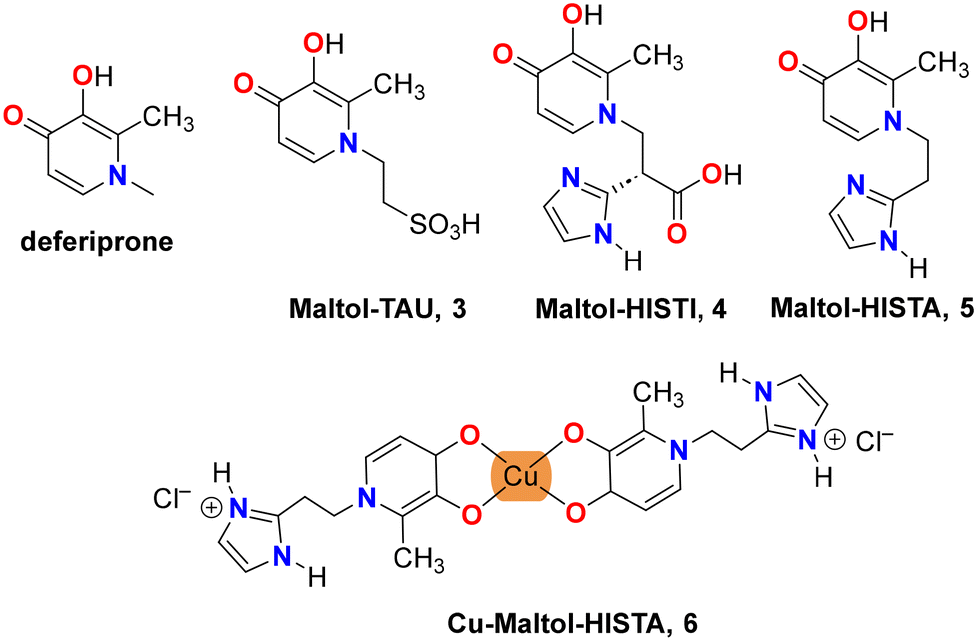 | ||
| Scheme 2 Structures of deferiprone and deferiprone derivatives: maltol-taurine 3, maltol-histidine 4, maltol-histamine 5 and its Cu-complex 6. | ||
Density functional theory (DFT) calculations were used to evaluate the formation energy of several supramolecular assemblies, which were further characterized using the Bader's theory of “atoms-in-molecules” (QTAIM) and the noncovalent interaction (NCI) plot analysis. We specifically investigated the H-bonding assemblies where co-crystallized water molecules extend the supramolecular networks. These were analyzed energetically and by using the QTAIM analysis and various methods to estimate H-bonding strengths.
Methods
Materials and measurements
All reagents were purchased from Sigma-Aldrich and used as received without further purification. Elemental analyses (C, H, N) were performed using a Carlo-Erba 1108 and a Thermo Finnigan Flash 1112 microanalyzer. Thermogravimetric analysis (TGA) was conducted on a DSC-TGA/TA Instruments 2690 SDT under a nitrogen atmosphere at a heating rate of 2 °C min−1. High-resolution mass spectra (ESI-HRMS) were acquired in methanol using an AUTOSPECT 3000. Solid-state FT-IR spectra were recorded in the 4000–400 cm−1 range using the attenuated total reflectance (ATR) accessory of a Bruker Tensor 27 spectrometer. 1H and 13C NMR spectra were obtained at room temperature on a Bruker AMX 300 spectrometer operating at 300 MHz and 75 MHz, respectively. Chemical shifts are reported relative to the residual solvent signals of DMSO-d6 (1H δ = 2.50 ppm; 13C δ = 40 ppm).Synthesis
Compounds 1b, 1c and 1d and 2b, 2c and 2d were previously synthesized and characterized as mentioned in ref. 9 (see Scheme 1).Synthesis of [maltol-TAU·H2O, 2-(3-hydroxy-2-methyl-4-oxopyridin-1(4H)-yl)ethane-1-sulfonic acid monohydrate] [3·H2O]. A mixture of 1.26 g (10 mmol) of maltol and 2.50 g (20 mmol) of taurine in 20 mL of distilled water was heated at 90 °C in a water bath with stirring until total dissolution and basified to pH = 12 with NaOH 6N. The solution, with a yellowish color, was transferred to a Parr pump and kept in an oven at 130 °C for 24 hours. After this period, it was allowed to cool down to room temperature, and the content of the pump was transferred to a beaker (dark brown). It was acidified with stirring to pH = 2 with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of HCl (37%) and H2O. Under these conditions the light brown reaction crude precipitated. The mixture was stirred for 10–15 minutes, filtered under vacuum, airdried and collected (0.93 g, 40% yield). The product was recrystallized in hot water (90 °C). Brownish monocrystals suitable for X-ray diffraction can be obtained from liquid mother or by crystallization in hot water (“zwitterion” form, 2b·1.5H2O) Mp: 181–184 °C. Anal. calc. for [3·H2O], C8H11NO5S·1H2O (251): C, 38.24; H, 5.22; N, 5.57. Found: C, 38.42; H, 5.14; N, 5.45. TGA: a weight loss is observed between 80–120 °C corresponding to one water molecule per formula unit (theoretical 7.17%, experimental 7.13%). ESI-HRMS (negative ions): [C8H10NO5S]− (experimental mass: 232.0281; theoretical mass: 232.02852). IR (cm−1): 3051br,w [ν(O–H)], 1635m [ν(C
:1 mixture of HCl (37%) and H2O. Under these conditions the light brown reaction crude precipitated. The mixture was stirred for 10–15 minutes, filtered under vacuum, airdried and collected (0.93 g, 40% yield). The product was recrystallized in hot water (90 °C). Brownish monocrystals suitable for X-ray diffraction can be obtained from liquid mother or by crystallization in hot water (“zwitterion” form, 2b·1.5H2O) Mp: 181–184 °C. Anal. calc. for [3·H2O], C8H11NO5S·1H2O (251): C, 38.24; H, 5.22; N, 5.57. Found: C, 38.42; H, 5.14; N, 5.45. TGA: a weight loss is observed between 80–120 °C corresponding to one water molecule per formula unit (theoretical 7.17%, experimental 7.13%). ESI-HRMS (negative ions): [C8H10NO5S]− (experimental mass: 232.0281; theoretical mass: 232.02852). IR (cm−1): 3051br,w [ν(O–H)], 1635m [ν(C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O)], 1540w, 1508m, 1338m, 1254m, 1190m, 1128s [ν(SO2)sym], 1029vs [ν(SO2)sym], 817m, 620m, 523s, 486m. 1H-NMR (DMSO-d6) δ (300 MHz): 8.25d [1H, C(3)-H], 7.07d [1H, J = 7.5 Hz, C(4)–H], 4.56t [2H, J = 7.2 Hz, C(2)–H], 2.96t [2H, J = 7.2 Hz, C(1)–H], 2.57s [3H, C(8)–H]. 13C-NMR δ (ppm) (75 MHz; DMSO-d6): tentative assignation, 158.1 [C(5)], 142.4 [C(7)], 142.2 [C(6)], 138.9 [C(3)], 110.2 [C(4)], 52.9 [C(1)], 50.1 [C(2)], 12.5 [C(8)].
O)], 1540w, 1508m, 1338m, 1254m, 1190m, 1128s [ν(SO2)sym], 1029vs [ν(SO2)sym], 817m, 620m, 523s, 486m. 1H-NMR (DMSO-d6) δ (300 MHz): 8.25d [1H, C(3)-H], 7.07d [1H, J = 7.5 Hz, C(4)–H], 4.56t [2H, J = 7.2 Hz, C(2)–H], 2.96t [2H, J = 7.2 Hz, C(1)–H], 2.57s [3H, C(8)–H]. 13C-NMR δ (ppm) (75 MHz; DMSO-d6): tentative assignation, 158.1 [C(5)], 142.4 [C(7)], 142.2 [C(6)], 138.9 [C(3)], 110.2 [C(4)], 52.9 [C(1)], 50.1 [C(2)], 12.5 [C(8)].
Synthesis of [maltol-HISTI·2H2O, 2-(3-hydroxy-2-methyl-4-oxopyridin-1-(4H)-yl)-3-(1H-imidazole-4-yl)propanoic acid dihydrate] [4·2H2O], and [maltol-HISTA·HCl·1.5H2O, 1-(2-(1H-imidazole-4-yl)ethyl)-3-hydroxy-2-methylpyridin-4(1H)-one hydrochloric acid sesquihydrate] [5·HCl·1.5H2O]. Maltol (0.64 g, 5 mmol) and 10 mmol of L-histidine (1.55 g) or histamine·2HCl (1.84 g) were added to 15 ml of distilled water and heated at 80 °C in a water bath with stirring until total dissolution. The solution was subsequently basified up to pH = 11 with NaOH 6N. The mixture was transferred to a Parr pump, with addition of 2 ml of distilled water, which was maintained in an oven at 130 °C for 48 hours. The resulting dark solution was acidified with stirring to a pH of 3 and a beige solid product was observed. The resulting suspension is left with stirring for 24 hours, filtered under vacuum, washed with 3 ml of cold distilled water and 1 ml of cold acetone and allowed to air dry. Grayish beige solids [(550 mg) of [4·2H2O], (36%) and (850 mg) of [5·HCl·1.5H2O], (52%)] were obtained. [Mp for [4·2H2O]: 218–221 °C; Mp for [5·HCl·1.5H2O]: 243–246 °C]. TGA: weight loss is observed for 4 between 30 and 150 °C corresponding to 1.5 water molecules per formula unit (theoretical 9.3%, experimental 8.4%) and for [5·HCl·1.5H2O] between 30 and 260 °C corresponding to 1.5 water molecules and 1 HCl per formula unit (theoretical 22.5%, experimental 23.3%). Recrystallization with boiling water and active C yields brown twins and monocrystals useful for X-ray diffraction. IR for [4·2H2O]: (cm−1): 3406br,m [ν(N–H)], 3080–2900br,w [ν(O–H)], 1632m [ν(CO) + ν(COO−)asym], 1605s, 1578s, 1495vs, 1449w, 1429m, 1414s [ν(COO−)sym], 1334vs, 1310m, 1286m, 1167w, 1142w, 1123m, 1063m, 959m, 867s, 820m, 805s, 628s, 527vs. 1H-NMR δ (ppm) (300 MHz; DMSO-d6): 7.70s [1H, C(12)–H], 7.61d [1H, J = 7.5 Hz, C(6)–H], 6.73s [1H, C(10)–H], 6.10d [1H, J = 7.5 Hz, C(5)–H], 5.24dd [1H, J = 9.6 and 5.4 Hz, C(7)–H], 3.40dd [1H, J = 15.3 and 5.4 Hz, C(8)–H], 3.32dd [1H, J = 15.3 and 9.6 Hz, C(8′)–H], 2.14s [3H, CH3]; 13C-NMR δ (ppm) (75 MHz; DMSO-d6): tentative assignation, 171.4 [COOH], 169.5 [C(4)], 145.3 [C(6)], 135.7 [C(3)], 134.9 [C(12)], 133.0 [C(9)], 131.9 [C(2)], 117.4 [C(10)], 111.7 [C(5)], 38.4 [C(7)], 29.6 [C(8)], 12.6 [CH3].
IR for [5·HCl·1.5H2O]: (cm−1): 3200–2800br,m [ν(N–H) + ν(O–H)], 1688w, 1620m [ν(CO)], 1556m, 1507m, 1438w, 1394w, 1359w, 1250–1200vs, 1072w, 1049m, 1017w, 903w, 821s, 777w, 736w, 664w, 630s, 593m, 571s, 534m, 507vs, 432w. 1H-NMR δ (ppm) (300 MHz; DMSO-d6): 8.14s [1H, C(12)–H], 7.46d [1H, J = 7.5 Hz, C(6)–H], 7.00s [1H, C(10)–H], 6.08d [1H, J = 7.5 Hz, C(5)–H], 4.21t [2H, J = 7.2 Hz, C(7)–H], 2.95t [2H, C(8)–H], 2.32s [3H, CH3]; 13C-NMR δ (ppm) (75 MHz; DMSO-d6): tentative assignation, 168.9 [C(4)], 145.8 [C(6)], 138.1 [C(3)], 134.7 [C(12)], 130.7 [C(9)], 130.5 [C(2)], 117.5 [C(10)], 111.3 [C(5)], 52.6 [C(7)], 26.7 [C(8)], 12.0 [CH3]. ESI-HRMS (positive ions): [C11H14N3O2]+ (experimental mass: 220.1078; theoretical mass: 220.1081).
Synthesis of [Cu(maltol-HISTA)2(H2O)2]·2H2O·2HCl 6. To a solution of 0.25 mmol (60 mg) of [5·HCl·1.5H2O] in 8 mL of distilled water was added 0.5 mmol (90 mg) of CuCl2·H2O in 2 ml of H2O. The resulting solution was allowed to react at room temperature with stirring for about 30 min. It was left to stand, and after 33 days emerald-colored twins were obtained (10% yield). Few crystals useful for X-ray diffraction analysis were separated. IR: (cm−1): 3450–3400br,m [ν(O–H),], 3141 m, 2859 m, 1629w, 1600s, 1543s, 1503s, 1485vs, 1469s, 1357s, 1306w, 1279vs, 1248s, 1171m, 1089w, 1062m, 1011w, 976w, 913w, 831vs, 803m, 763w, 738w, 705w, 653w, 630s, 584s, 560s, 515br,m, 493m.
Crystallographic analyses
Single crystals of 3 and 4 were selected, covered with Parabar 10320 (formally known as Paratone N) and mounted on a cryoloop on a D8 Venture diffractometer, with a Photon III 14 detector, using an Incoatec high brilliance IμS DIAMOND Cu tube (CuKα, λ = 1.54178 nm) equipped with an Incoatec Helios MX multilayer optics. The crystals were kept at 100 K during data collection. Data reduction and cell refinements were performed using the Bruker APEX4 program.10 Scaling and absorption corrections were carried out using the SADABS program in all cases.10 Using Olex2,11 the structure was solved with the ShelXT structure solution program12 using intrinsic phasing and refined with the ShelXL refinement package12 using least squares minimization. All non-hydrogen atoms were refined with anisotropic thermal parameters by full-matrix least-squares calculations on F2.The crystallographic data for 5 and 6 were collected at 100 K using a Bruker D8 Venture diffractometer with a Photon 100 CMOS detector and Mo Kα radiation (λ = 0.71073 Å) generated by an Incoatec high-brillance microfocus source equipped with Incoatec Helios multilayer optics. The software APEX3 (ref. 10) was used to collect frames of data, index reflections, and determine the lattice parameters. SAINT was used for the integration of the intensity of reflections and SADABS for scaling and empirical absorption correction.10 The structures were solved by direct methods by using the program ShelXT.12 All non-hydrogen atoms were refined on F2 with anisotropic thermal parameters by using ShelXL.12
Hydrogen atoms were generally inserted at calculated positions and refined as riders, except for those bound to oxygen atoms (–OH groups and water molecules), which were located using a Fourier difference map and refined isotropically. The structures were checked for higher symmetry with help of the program PLATON.13 The graphical material has been prepared with the help of Mercury software.14
A summary of the key crystallographic information is given in Table S1. CCDC 2505171–2505174 contain the SI crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre viahttps://ww.ccdc.cam.ac.uk/structures.
Theoretical methods
The computational methodology was designed to evaluate non-covalent interactions within the context of the experimentally determined solid-state structures. All calculations were performed using Gaussian 16 program package15 at the PBE0-D3/def2-TZVP level of theory.16–18 This theoretical approach was selected due to its successful application in the literature for investigating similar molecular systems.19–22 Interaction energies for π-stacked dimers were determined by calculating the energy difference between the isolated monomers and the assembled dimer. These values were corrected for basis set superposition error (BSSE) using the standard Boys–Bernardi counterpoise technique.23 The crystallographic coordinates served as the initial geometry; the positions of the H-atoms were optimized, while non-hydrogen atoms were constrained to their experimental positions.To provide a deeper characterization of the interactions, a topological analysis of the electron density was carried out. We utilized the AIMall software package23 to implement the quantum theory of atoms-in-molecules (QTAIM).24 Additionally, to visually identify the nature of these non-covalent interactions in real space, we employed the NCIPLOT index.25 This visualization method, based on the reduced density gradient (RDG) and its relationship to the electron density,26 distinguishes between attractive and repulsive interactions. In the 3D plots, the sign of the second Hessian eigenvalue, multiplied by the electron density [sign(λ2)ρ], was used to colour-code the isosurfaces: blue-green for attractive/stabilizing interactions and yellow-red for repulsive ones. For the plots displayed in Fig. 8, the following parameters were applied: RDG = 0.5, a density cutoff (ρ cut off) of 0.04 a.u., and a colour scale ranging from −0.035 a.u. to 0.035 a.u. for sign(λ2)ρ. The energies of the H-bonding assemblies were also computed using a specific methodology proposed by Espinosa et al.,27 which is based on the potential kinetic energy density. This approach has been successfully applied to estimate H-bond strengths in the X-ray structures of various salts where Coulombic effects are dominant.28
Results and discussion
Syntheses
The deferiprone derivatives 3, 4, 5 have been obtained by direct reaction of maltol [3-hydroxi-2-methyl-(4H)-pyranone] and an excess of amino acid (1:2) in one-pot at pH = 11–12 (NaOH 6N addition), 130 °C in Parr high pressure digestion vessel during one to three days (see Experimental details and ref. 8, for further information). The synthetic method reported herein eliminates additional steps described previously in the literature,5–7 involving protection and subsequent deprotection of the maltol hydroxyl group, achieving reasonable yields between 36–52%.
Description of the structures
The asymmetric units of the crystal structures of compounds 3, 4 and 5 are shown in Fig. 1. The taurine derivative 3, show a positive catecholic form with deprotonated sulfonate group while compounds 4 and 5 exhibit protonated imidazole ring with the negative charge localized into carboxylate 4 or chloride anion 5. | ||
| Fig. 1 X-ray geometries of compounds 3 (a), 4 (b) and 5 (c) with the atomic numbering scheme. | ||
In case of hydroxypyridinone and imidazole compounds the X-ray structure reveals that both rings are nearly orthogonal in 4, while almost parallel in 5.
The structures of the three new deferiprone analogs are primarily dominated by hydrogen bonding (H-bonding) interactions and π stacking (4 and 5), which have been further analysed using theoretical calculations (vide infra). Selected hydrogen bonds are given in Table 1.
| D–H⋯A | d(D⋯A) (Å) | D–H–A (°) |
|---|---|---|
| 11 − x, + y, 1/2 − z; 21 − x, 1 − y, 1 − z; 3+x, −1 + y, + z; 4+x, 2 − y, 1/2 + z; 5+ x, 2 − y, −1/2 + z; 63/2 − x, 5/2 − y, 1 − z; 73/2 − x, −1/2 + y, 1/2 − z; 83/2 − x, 3/2 − y, 1 − z; 91 − x, 1 + y, 1/2 − z; 101 − x, 2 − y, 1 − z; 11+x, 1 + y, +z; 121 − x, −1 + y, 1/2 − z. | ||
| Compound 3 | ||
| C5C–H5C⋯O111 | 3.198(3) | 158.4 |
| O31C–H31C⋯O132 | 2.629(2) | 144.6 |
| O41C–H41C⋯O1W | 2.573(2) | 165.6 |
| O1W–H1WA⋯O123 | 2.748(2) | 172.0 |
| O1W–H1WB⋯O121 | 2.759(2) | 174.2 |
| 11 − x, 1/2 + y, 1/2 − z; 2−1/2 + x, 1/2 − y, 1 − Z; 31/2 − x, 1 − y, 1/2 + z | ||
| Compound 4 | ||
| N13–H13⋯O721 | 2.6764(19) | 168.6 |
| N11–H11⋯O2W2 | 2.753(2) | 136.8 |
| O31–H31⋯O723 | 2.8366(17) | 159.4 |
| O1W–H1WA⋯O2W4 | 2.7930(19) | 166.9 |
| O1W–H1WB⋯O73 | 2.7548(18) | 172.3 |
| O2W–H2WA⋯O1W | 2.821(2) | 169.2 |
| 11 + x, + y, + z; 22 − x, 2 − y, 2 − z; 31 − x, 1 − y, 1 − z; 42 − x, 1 − y, 2 − z; 52 − x, 1 − y, 1 − z | ||
| Compound 5 | ||
| O1W–H1WA⋯Cl2 | 3.1384(13) | 172(2) |
| O1W–H1WB⋯Cl1 | 3.1205(13) | 171(2) |
| O2W–H2WA⋯O3W1 | 2.7900(18) | 173(3) |
| O2W–H2WB⋯O41B2 | 2.9565(18) | 176(3) |
| O3W–H3WA⋯O41A | 2.7117(16) | 172(2) |
| O3W–H3WB⋯O31A3 | 3.2000(16) | 128(2) |
| O3W–H3WB⋯O41B3 | 3.0613(18) | 164(3) |
| O31A–H31A⋯O41B | 2.6207(15) | 157(2) |
| C21A–H21B⋯Cl14 | 3.7185(16) | 141.9 |
| C21A–H21C⋯Cl1 | 3.8545(16) | 148.4 |
| C7A–H7AA⋯Cl23 | 3.5845(15) | 162.4 |
| C8A–H8AB⋯Cl25 | 3.4596(15) | 111.8 |
| C10A–H10A⋯O1W3 | 3.324(2) | 146.7 |
| C12A–H12A⋯Cl26 | 3.7147(16) | 157.7 |
| N13A–H13A⋯O1W | 2.6928(18) | 176(2) |
| N11A–H11A⋯Cl17 | 3.1928(13) | 133(2) |
| N11A–H11A⋯Cl28 | 3.2504(14) | 129.4(19) |
| O31B–H31B⋯O41A | 2.6396(16) | 159(2) |
| C21B–H21E⋯O31B2 | 3.296(2) | 131.1 |
| C21B–H21F⋯O3W1 | 3.359(2) | 129.0 |
| C21B–H21F⋯O31B1 | 3.503(2) | 146.0 |
| C6B–H6B⋯O3W9 | 3.3109(19) | 138.6 |
| C7B–H7BA⋯O41A1 | 3.3606(18) | 150.7 |
| C7B–H7BB⋯O3W9 | 3.469(2) | 155.3 |
| C8B–H8BB⋯O41B10 | 3.3793(18) | 171.1 |
| N13B–H13B⋯O2W | 2.7607(18) | 173.5(19) |
| N11B–H11B⋯Cl11 | 3.4953(15) | 125(2) |
| N11B–H11B⋯Cl210 | 3.2199(13) | 145(2) |
| C10B–H10B⋯O2W11 | 3.440(2) | 141.2 |
| C12B–H12B⋯Cl112 | 3.4840(16) | 163.5 |
Single-crystal X-ray analysis revealed that compounds 3, 4, and 5 crystallize in the orthorhombic, triclinic, and monoclinic systems, respectively. Their corresponding space groups are P212121 for 3, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) for 4, and P2/c for 5.
for 4, and P2/c for 5.
In the solid state of compound 3, the crystal packing is dominated by hydrogen-bonding interactions, as depicted in Fig. 2. Each molecular unit forms a strong hydrogen bond with a neighbouring molecule through the interaction of the O(3)–H group and a sulfonate oxygen atom, with an O–H⋯Osulfonate distance of 1.90 Å. Furthermore, each unit is stabilized by interactions with three water molecules: one through the O(4)−H group (1.75 Å), and two via the sulfonate group (1.88 Å and 1.89 Å). Beyond the primary hydrogen bonds, two longer C–H⋯O hydrogen bond-like contacts are observed between the sulfonate group (O3S) of a neighbouring molecule and a C–H group (C1) of another. These contacts, with distances of 2.64 Å and 2.75 Å, further contribute to the overall stabilization of the crystal lattice. Detailed geometric features of all these interactions can be found in Table 1.
 | ||
| Fig. 2 H-bonds contacts in 3 (distances in Å). | ||
In compound 4, the crystal structure is characterized by a protonated imidazole ring and a corresponding carboxylate anion. The planes of the hydroxypyridinone (HP) and imidazole rings are nearly orthogonal, with an angle of 78.6°. This structural arrangement, including the specific bond lengths and angles, is consistent with other previously described structures.6,9 The packing is stabilized by several key interactions (Fig. 3 and 4). Dimeric units form through bifurcated hydrogen bonds involving the carboxylate group, with [(Im)N–H⋯OCO⋯H–O(3)–(HP)] interactions (1.81 Å, 105.9° and 2.03 Å, 124.7°). Additionally, there are significant stacking interactions between HP rings, both within the same dimer (3.44 Å) and between different dimers (3.32 Å), as shown in Fig. 3a. In a separate part of the structure, four water molecules generate tetrameric units (columns) via a network of hydrogen bonds: [[(Im)N–H⋯OH–H⋯OH–H⋯OC(O)]] (2.04 Å, 136.8°; 1.94 Å, 106.7° and 1.89 Å, 121.0°). Finally, stacking is also observed between two imidazole rings with a distance of 3.39 Å, as depicted in Fig. 3b.
 | ||
| Fig. 3 (a) Partial view of the X-ray structure of 4 showing the π⋯π stacking interactions between dimers of maltol-histidine 4; (b) tetrameric water cluster bridging four molecules of maltol-histidine. | ||
 | ||
| Fig. 4 3D packing of compound 4 along b axis. | ||
In compound 5, the structure is a hydrochloride salt, with a protonated imidazole ring and a chloride anion. The crystal packing is defined by a network of hydrogen bonds. This includes a notable double hydrogen bond between the hydroxyl and carbonyl groups of two different molecules, specifically the O(31)–H⋯O(41) atoms, with geometric parameters of 1.82 Å (156.7°) and 1.91 Å (159.1°). Each carbonyl group also extends its network by interacting with two water molecules. For example, the O(4) carbonyl group interacts with two other units via a chain of two water molecules [O(4)⋯H–OH(w1)⋯H–OH(w2)⋯O(4)], with bond lengths and angles of 1.97 Å (171.5°), 1.96 Å (172.7°), and 2.17 Å (176.0°). Additionally, the water molecule w2 forms a bifurcated hydrogen bond with the imidazole NH group (1.88 Å, 173.3°). Other important interactions include alternating N–H⋯Cl⋯N–H contacts (2.51 Å, 144.7° and 2.63 Å, 129.2°), as well as Cl⋯H–O–Hwater⋯Cl interactions (2.34 Å, 172.1° and 2.36 Å, 171.0°) (Fig. 5).
 | ||
| Fig. 5 Partial view of the solid state of compound 5 with indication of the H-bonds. Distances in Å. | ||
In compound [Cu(maltol-HISTA)2(H2O)2]·2H2O·2HCl (6), the copper(II) ion, Cu(1), presents a highly distorted octahedral geometry (O6 coordination sphere), which is characteristic of d9 systems due to the Jahn–Teller effect.
The equatorial plane is defined by four oxygen atoms (O4) originating from the two maltol-HISTA ligands, forming a chelate ring with the copper centre via the hydroxyl O(4) and carbonyl O(3) oxygen atoms. The equatorial bond distances, Cu(1)–O(3) and Cu(1)–O(4), are 1.9396(8) and 1.9573(8) Å, respectively (see Fig. 6). The coordination sphere is completed by two H2O molecules (O(1W) and O(2W)) situated in the axial positions. These axial Cu(1)–O(W) distances are significantly elongated to approximately 2.65 Å, a clear manifestation of the Jahn–Teller distortion (4 + 2 geometry).
 | ||
| Fig. 6 X-ray geometry of compound 6 with the atomic numbering scheme. | ||
The coordination of the maltol moiety to the Cu2+ ion causes a measurable change in the intramolecular bond lengths, consistent with electron density transfer upon chelation. The C(4)O bond distance is slightly lengthened to 1.2965(13) Å in the complex compared to 1.2803(17) Å in the free ligand, while the C(3)–O bond distance is shortened to 1.3320(13) Å from 1.3602(17) Å in the ligand. This structural perturbation is corroborated by the IR spectroscopic data:29 the characteristic carbonyl stretching vibration (νCO) is shifted to lower wavenumbers upon coordination (1629 cm−1 w, 1600 cm−1 s in the complex vs. 1689 cm−1 w, 1620 cm−1 m in the ligand), confirming the involvement of the CO group in the metal coordination.
A network of hydrogen bonds that implies the ligands, water molecules and chloride counterions are present in compound 6, as shown in the DFT study. The relevant geometric features of the hydrogen bonds are given in Table 2.
| D–H⋯A | d(D⋯A) (Å) | D–H–A (°) |
|---|---|---|
| 11 − x, 1 − y, 1 − z; 2−1 + x, −1 + y, −1 + z; 3−1 + x, +y, +z; 4+x, 1 + y, 1 + z; 51 + x, 1 + y, 1 + z. | ||
| O11–H1WA⋯Cl11 | 3.2202(10) | 177.9(19) |
| O1W–H1WB⋯Cl12 | 3.2767(10) | 176(2) |
| O2W–H2WA⋯Cl11 | 3.1894(10) | 168.5(18) |
| O2W–H2WB⋯O31 | 2.6937(12) | 177(2) |
| C21–H21C⋯O2W1 | 3.5740(15) | 159.3 |
| C5–H5⋯O2W3 | 3.4698(14) | 153.9 |
| C10–H10⋯O1W4 | 3.3939(15) | 143.2 |
| C12–H12⋯O415 | 3.0689(14) | 124.5 |
| C12–H12⋯O1W5 | 3.1081(15) | 142.4 |
| N13–H13⋯Cl1 | 3.1650(10) | 172.6(16) |
| N11–H11⋯O2W4 | 2.7189(13) | 165.6(18) |
DFT study
Density functional theory (DFT) calculations were conducted to evaluate the non-covalent interactions and supramolecular assemblies observed in the experimentally determined solid-state structures, with a particular focus on the prominent role of co-crystallized water molecules. The computational methodology utilized the PBE0-D3/def2-TZVP level of theory, a well-established approach for similar systems. The primary goal was to investigate how these water molecules influence the crystal packing and the arrangement of the deferiprone derivatives through extensive hydrogen-bonding networks. To provide a detailed characterization of these interactions, we performed a topological analysis of the electron density using the quantum theory of atoms in molecules (QTAIM) and employed the non-covalent interaction (NCI) plot index to visualize the nature of these contacts in real space. The strength of the hydrogen bonds was also estimated using QTAIM parameters. These computational tools collectively offer new insights into the structural and electronic properties of these deferiprone derivatives and highlight their potential in crystal engineering.A molecular electrostatic potential (MEP) surface analysis was performed to investigate the most nucleophilic (electron-rich) and electrophilic (electron-deficient) regions of compounds 3, 4, and 5. The results, shown in Fig. 7, rationalize the supramolecular behaviour observed in their solid-state structures. Fig. 7a depicts the MEP surface for compound 3, a deferiprone analogue of taurine. As a zwitterion, it exhibits large MEP values, indicating the potential for strong hydrogen bonds. The MEP maxima are located at the phenolic protons (84.1 kcal mol−1 and 69.0 kcal mol−1), while the minimum is at the sulfonate group (−74.0 kcal mol−1). The MEP surface for compound 4, a histidine analogue, is shown in Fig. 7b. Here, the molecule's zwitterionic nature is also evident. The MEP maxima are situated at the NH groups of the imidazolium ring (94.1 and 93.5 kcal mol−1), while the MEP minimum is found at the oxygen atoms of the carboxylate group. For this compound, the hydroxypyridinone is neutral, resulting in a small MEP value at the hydroxyl hydrogen atom (11.9 kcal mol−1) and a large negative value at the carbonyl oxygen atom (−61.5 kcal mol−1). Finally, for compound 5, the hydrochloride salt of the deferiprone analogue derived from histamine, the MEP surface is shown in Fig. 7c. The MEP minimum is located at the chloride anion (−72.2 kcal mol−1), while the maximum is at the imidazolium ring (94.8 kcal mol−1). The MEP at the carbonyl oxygen atom of the hydroxypyridinone is −43.3 kcal mol−1, and at the hydrogen atom of the hydroxyl group is 40.2 kcal mol−1. This analysis highlights the molecules' significant capacity for hydrogen-bonding interactions, suggesting that these assemblies are expected to be energetically favorable.
 | ||
| Fig. 7 MEP surfaces of compounds 3 (a), 4 (b), and 5 (c). Selected MEP values are indicated in kcal mol−1. The isovalue is set to 0.001 a.u., with red representing negative (nucleophilic) and blue for positive (electrophilic) MEP values. | ||
The MEP surface of compound 6, a Cu(II) complex of the histamine deferiprone derivative in its hydrochloride form, is shown in Fig. 8. This surface reveals a MEP maximum at the NH imidazolium group (87.9 kcal mol−1), and minima at the anionic oxygen atoms coordinated to Cu(II) (−72.8 kcal mol−1) and the chloride ion (−70.3 kcal mol−1). The H-atoms of the coordinated water molecules show positive MEP values (28.9 kcal mol−1), while their O-atoms have negative values (−48.7 kcal mol−1), confirming that these water molecules are both strong hydrogen bond donors and acceptors.
 | ||
| Fig. 8 MEP surface of the Cu(II) complex 6. Selected MEP values are indicated in kcal mol−1. The isovalue is set to 0.001 a.u., with red representing negative MEP values (nucleophilic regions), and blue representing positive values (electrophilic regions). | ||
The intermolecular interactions in the solid-state structure of compound 3 were further analysed using a combined QTAIM/NCIplot approach. Fig. 9a illustrates a homodimer stabilized by a strong O–H⋯OSO2R hydrogen bond and π-based interactions, which are a combination of π⋯π and lone pair (LP⋯π) contacts. The presence and distribution of these non-covalent interactions are confirmed by the bond critical points (BCPs) shown as red spheres and their corresponding bond paths (orange lines). The π-interactions are visualized as a large, green reduced density gradient (RDG) isosurface embracing the π-system of one pyridinium ring. In contrast, the O–H⋯O hydrogen bond is represented by a disk-shaped, blue RDG isosurface, which indicates a strong interaction. An ancillary C–H⋯O contact is also observed, characterized by a BCP, a bond path, and a corresponding green RDG isosurface. The total interaction energy for this dimer is −13.45 kcal mol−1. To isolate the contribution of the hydrogen bonds from the π-interactions, a theoretical model was computed where the C–C torsion angle of the ethanesulfonate group was rotated to eliminate the π-interactions, as shown in Fig. 9b. This rotation reduced the interaction energy to −12.40 kcal mol−1, confirming the strong and dominant role of the hydrogen bonds.
 | ||
| Fig. 9 (a) Combined QTAIM/NCIplot analysis of a homodimer of 3 stabilized by a strong O–H⋯OSO2R hydrogen bond (H-bond) and π-interactions. Bond critical points (BCPs) are shown as red spheres with corresponding bond paths (orange lines). (b) A theoretical model of the homodimer from (a) with a rotated C–C torsion angle to eliminate π-stacking. (c) QTAIM/NCIplot analysis of a supramolecular assembly where three organic molecules are linked by a central water molecule. | ||
Fig. 9c presents the QTAIM/NCIplot analysis of a supramolecular assembly where the organic molecules are connected via a central water molecule. This water molecule forms three strong hydrogen bonds: two as a donor with the sulfonate groups and one as an acceptor with the phenolic group. An additional C–H⋯O contact is observed between an aromatic C–H bond and the oxygen atom of the water molecule. The NCIplot and QTAIM analyses also reveal other interactions, such as C–H⋯OSO2R contacts. The total interaction energy for this assembly is −68.2 kcal mol−1, highlighting the crucial role of the lattice water molecule in stabilizing the solid-state structure of compound 3. Interestingly, the estimated hydrogen-bonding contribution using QTAIM parameters and Espinosa's equation is only −33.93 kcal mol−1, which is considerably smaller than the total interaction energy. This difference suggests that the large formation energy of this assembly is primarily due to the zwitterionic nature of compound 3 and the formation of electrostatically enhanced hydrogen bonds, a factor not fully captured by Espinosa's equation,27 which does not account for pure coulombic effects.
Fig. 10 displays three different trimeric assemblies of compound 4, each featuring a tetrameric water cluster (W4) as a central entity. The four oxygen atoms of the W4 cluster are essentially coplanar and are interconnected by four O–H⋯O hydrogen bonds, each characterized by a bond critical point (BCP) and a bond path, which together form a R44(8) synthon. The formation energy of this W4 cluster is −25.26 kcal mol−1, as calculated using Espinosa's formula and −23.98 using the supramolecular approach, giving reliability to the energy predictor in neutral assemblies. This W4 cluster plays a prominent role by bridging the maltol-histidine units in three distinct binding modes. In “Trimer-A” (Fig. 10a), the cluster connects two molecules via two strong O–H⋯O hydrogen bonds with the keto groups of the deferiprone ring. In “Trimer-B” (Fig. 10b), the cluster bridges the maltol-histidine units through strong hydrogen bonds with the carboxylate groups. Finally, in “Trimer-C” (Fig. 8c), the cluster acts as a hydrogen bond acceptor, forming bonds with the imidazolium rings of the maltol-histidine units. All the OH⋯O and NH⋯O contacts are characterized by the corresponding BCPs, bond paths, and blue RDG isosurfaces. The most energetically favoured assembly is “Trimer-B”, with a total interaction energy of −59.8 kcal mol−1 (including the energy of the water cluster). The other two assemblies, “Trimer-A” and “Trimer-C”, have similar formation energies of −42.88 kcal mol−1 and −41.93 kcal mol−1, respectively. These large interaction energies and the participation of the W4 cluster in forming three different assemblies that bridge the maltol-histidine units further emphasize the crucial role of water in the solid-state structure of compound 4.
 | ||
| Fig. 10 QTAIM/NCIplot analysis of trimeric assemblies of compound 4 involving a tetrameric water cluster. (a) Trimer-A: the W4 cluster connects two molecules via hydrogen bonds with the keto groups. (b) Trimer-B: the W4 cluster bridges molecules through strong hydrogen bonds with the carboxylate groups. (c) Trimer-C: the W4 cluster connects to the imidazolium rings via hydrogen bonds. | ||
Fig. 11 shows a supramolecular assembly of compound 5 where two homodimers, each characterized by a R22(10) motif, are linked by a chain of two water molecules. This representation highlights the significant role of co-crystallized water molecules in interconnecting the maltol-histamine units. The chloride atoms have been omitted for clarity. The R22(10) motifs are energetically favoured, with each having an energy of approximately −15 kcal mol−1, as indicated by the blue RDG isosurfaces that characterize the strong O–H⋯O hydrogen bonds. The hydrogen bonds involving the lattice water molecules contribute an additional −9.23 kcal mol−1 to the network. The total interaction energy for this entire assembly is −39.65 kcal mol−1, which underscores the robust nature of this hydrogen-bonding network.
 | ||
| Fig. 11 QTAIM/NCIplot analysis of a supramolecular assembly of compound 5. | ||
Fig. 12 presents the QTAIM and NCIplot analyses of the Cu(II) complex 6, which shows an intricate assembly evidencing a complex network of hydrogen bonds and secondary interactions. Here, coordinated water molecules and chloride anions form an interesting R44(8) synthon that connects two Cu(II) complexes. Lattice water molecules interact with the protonated imidazolium rings via strong N–H⋯O bonds. Beyond these, additional water molecules bridge the Cu(II) complexes through O–H⋯O and O–H⋯Cl hydrogen bonds, in addition to C–H⋯Cl contacts. This extensive hydrogen-bonding network is further stabilized by anion–π interactions (Cl⋯π),30 as revealed by the QTAIM analysis showing a BCP and bond path connecting the chloride atom to a carbon atom of the deferiprone ring. The total interaction energy of this assembly is very large (−67.32 kcal mol−1) due to the numerous and strong hydrogen bonds between the Cu(II) complexes, counterions, and lattice water molecules.
 | ||
| Fig. 12 QTAIM and NCIplot analyses of the Cu(II) complex 6. The coordinated water molecules and chloride anions form an R44(8) synthon. This network is stabilized by numerous hydrogen bonds (including N–H⋯O, O–H⋯O, O–H⋯Cl, and C–H⋯Cl) and an anion–π interaction (Cl⋯π). | ||
Concluding remarks
This study successfully reports the synthesis, X-ray characterization, and DFT analysis of three novel deferiprone analogues derived from taurine, histidine, and histamine. The solid-state structures of these compounds (3, 4, and 5) are primarily governed by hydrogen-bonding and π-stacking interactions. Theoretical calculations confirmed these interactions, with compounds 3 and 4 exhibiting zwitterionic forms, while compound 5 is a hydrochloride salt. A key structural difference was found in the orientation of the rings: they are nearly orthogonal in compound 4 but almost parallel in compound 5. Additionally, we synthesized and structurally characterized the Cu(II) complex 6, which showed a distorted square-planar geometry with varying degrees of hydration.The DFT calculations provided crucial insights into the supramolecular behaviour of these compounds, as well as the Cu(II) complex 6. Molecular electrostatic potential (MEP) surfaces revealed the most nucleophilic and electrophilic regions, highlighting the strong capacity for hydrogen-bonding interactions. The combined quantum theory of atoms in molecules (QTAIM) and non-covalent interaction (NCI) plot analyses provided a detailed energetic and topological characterization of these assemblies. A central finding of this study is the prominent and crucial role of co-crystallized water molecules in influencing the crystal packing and arrangement of the deferiprone derivatives through extensive hydrogen-bonding networks. This is evident in compound 3, where a central water molecule forms a highly stable assembly. For compound 4, a tetrameric water cluster (W4) acts as a versatile bridge, linking the organic units in three different binding modes.
For the Cu(II) complex, the QTAIM/NCIplot analysis further corroborated the importance of these networks by revealing an intricate network of strong hydrogen bonds and secondary anion–π interactions involving coordinated water molecules, chloride anions, and the complexes themselves. Collectively, these findings provide new insights into the structural and electronic properties of deferiprone derivatives, highlighting their potential in coordination chemistry and crystal engineering.
Author contributions
Á. Terrón and A. López-Zafra: experimental chemistry, investigation, supervision. M. Rocha: theoretical calculations. C. A. Rodríguez: experimental chemistry. M. Barceló-Oliver: X-ray acquisition and analysis. A. Garcia-Raso: experimental chemistry, investigation, supervision, writing – original draft. J. J. Fiol: Experimental chemistry, investigation, supervision, writing – original draft. A. Frontera: computational chemistry, investigation, visualization, writing – original draft.Conflicts of interest
There are no conflicts to declare.Data availability
All software packages utilized for the theoretical calculations are thoroughly described and appropriately cited in the Theoretical calculations section. X-ray coordinates were employed for the calculations and can be accessed from CIF files. Additionally, CIF format crystallographic data have been included as supplementary information (SI).Supplementary information: Table S1. See DOI: https://doi.org/10.1039/d5ce01136a.
CCDC 2505171–2505174 (3–6) contain the supplementary crystallographic data for this paper.31a–d
Acknowledgements
We acknowledge the grant PID2023-148453NB-I00 funded by MICIU/AEI/10.13039/501100011033 and by “ERDF/EU”. We thank the METALBIO Spanish Thematic Network RED2022-134091-T for financial support. We thank the CTI (UIB) for computational facilities. M. Rocha thanks CONICET for financial support.Notes and references
- S. Chaves, L. Piemontese, A. Hiremathad and M. A. Santos, Curr. Med. Chem., 2018, 25, 97–112 CrossRef CAS PubMed.
- M. A. Santos and S. Chaves, Future Med. Chem., 2015, 7, 381–408 Search PubMed.
- (a) M. A. Santos, K. Chand and S. Chaves, Coord. Chem. Rev., 2016, 327–328, 287–303 CrossRef CAS; (b) M. A. Santos, S. M. Marques and S. Chaves, Coord. Chem. Rev., 2012, 256, 240–259 CrossRef CAS.
- S. Enterzari, S. M. Haghi, N. Norouzkhani, B. Sahebnazar, F. Vosoughian, D. Akbarzadeh, M. Islampanah, N. Naghsh, M. Abbasalizadeh and N. Deravi, J. Toxicol., 2022, 2022, 4911205 Search PubMed.
- A. Irto, P. Cardiano, K. Chand, R. M. Cigala, F. Crea, C. De Stefano, L. Gano, S. Sammartano and M. A. Santos, J. Inorg. Biochem., 2018, 183, 116–129 CrossRef PubMed.
- R. C. Hider, P. D. Taylor, M. Walkinshaw, J. L. Wang and D. van der Helm, J. Chem. Res., 1990, 316–317 CAS.
- (a) R. C. Chisté, D. Ribeiro, M. Freitas, A. Leite, T. Moniz, M. Rangel and E. Fernandes, J. Inorg. Biochem., 2016, 155, 9–16 CrossRef PubMed; (b) M. A. Santos, A. Irto, P. Buglyó and S. Chaves, Molecules, 2022, 27, 1966 CrossRef CAS PubMed.
- (a) S. Kaviani, M. Izadyar and M. R. Housaindokht, Comput. Biol. Chem., 2020, 86, 107267 CrossRef CAS PubMed; (b) S. Kaviani, M. Izadyar and M. R. Housaindokht, J. Mol. Graphics Modell., 2018, 80, 182–189 CrossRef CAS PubMed; (c) S. Kaviani, M. Izadyar and M. R. Housaindokht, Polyhedron, 2016, 117, 623–627 CrossRef CAS.
- A. García-Raso, A. Terrón, J. J. Fiol, A. López-Zafra, M. Herreros, I. Capilla, M. A. Domínguez, M. Barceló-Oliver, B. Spingler and A. Frontera, J. Mol. Struct., 2025, 1326, 141123 CrossRef.
- Bruker, APEX4, SAINT and SADABS, Bruker AXS Inc., Madison, Wisconsin, USA, 2021 Search PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–11 CrossRef CAS.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226–235 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16, Gaussian, Inc., Wallingford CT, 2016 Search PubMed.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6169 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- F. Weigend, Phys. Chem. Chem. Phys., 2006, 8, 1057–1065 RSC.
- P. Chakraborty, S. Purkait, S. Mondal, A. Bauzá, A. Frontera, C. Massera and D. Das, CrystEngComm, 2015, 17, 4680–4690 RSC.
- A. Das, S. R. Choudhury, B. Dey, S. K. Yalamanchili, M. Helliwell, P. Gamez, S. Mukhopadhyay, C. Estarellas and A. Frontera, J. Phys. Chem. B, 2010, 114, 4998–5009 CrossRef CAS PubMed.
- M. Mirzaei, H. Eshtiagh-Hosseini, A. Bauzá, S. Zarghami, P. Ballester, J. T. Mague and A. Frontera, CrystEngComm, 2014, 16, 6149–6158 RSC.
- D. Sadhukhan, M. Maiti, G. Pilet, A. Bauzá, A. Frontera and S. Mitra, Eur. J. Inorg. Chem., 2015, 2015, 1958–1972 CrossRef CAS.
- R. F. W. Bader, J. Phys. Chem. A, 1998, 102, 7314–7323 CrossRef CAS.
- T. A. Keith, AIMAll (Version 13.05.06), TK Gristmill Software, Overland Park, KS, 2013 Search PubMed.
- J. Contreras-García, E. R. Johnson, S. Keinan, R. Chaudret, J.-P. Piquemal, D. N. Beratan and W. Yang, J. Chem. Theory Comput., 2011, 7, 625–632 CrossRef PubMed.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, J. Am. Chem. Soc., 2010, 132, 6498–6506 CrossRef CAS PubMed.
- E. Espinosa, E. Molins and C. Lecomte, Chem. Phys. Lett., 1998, 258, 170–173 CrossRef.
- (a) E. Muñoz-Hernández, C. Alarcón-Payer, A. Frontera, R. Prohens, R. Barbas, F. J. Acebedo-Martínez, A. Domínguez-Martín and D. Choquesillo-Lazarte, CrystEngComm, 2026, 28, 88–100 RSC; (b) F. J. Acebedo-Martínez, C. Alarcón-Payer, A. Domínguez-Martín, A. Frontera, R. Prohens, R. Barbas and D. Choquesillo-Lazarte, Cryst. Growth Des., 2025, 25, 9137–9149 CrossRef.
- J. M. Mir, R. C. Maurya and P. K. Vishwakarma, Karbala Int. J. Mod. Sci., 2017, 3, 212–223 CrossRef.
- C. Estarellas, A. Bauzá, A. Frontera, D. Quiñonero and P. M. Deyà, Phys. Chem. Chem. Phys., 2011, 13, 5696–5702 RSC.
- (a) CCDC 2505171: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2q2tzm; (b) CCDC 2505172: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2q2v0p; (c) CCDC 2505173: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2q2v1q; (d) CCDC 2505174: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2q2v2r.
| This journal is © The Royal Society of Chemistry 2026 |