
Crystallization mechanism of organic compounds: the supramolecular cluster – a demarcation tool
Marcos A. P.
Martins
 *a,
Priscila S. V.
Lima
a,
Eudes F.
Silva
a,
Suzan K.
Kunz
a,
Tainára
Orlando
b,
Nilo
Zanatta
a,
Helio G.
Bonacorso
a and
Paulo R. S.
Salbego
c
*a,
Priscila S. V.
Lima
a,
Eudes F.
Silva
a,
Suzan K.
Kunz
a,
Tainára
Orlando
b,
Nilo
Zanatta
a,
Helio G.
Bonacorso
a and
Paulo R. S.
Salbego
c
aNúcleo de Química de Heterociclos (NUQUIMHE), Department of Chemistry, Federal University of Santa Maria (UFSM), 97105-900, Santa Maria, RS, Brazil. E-mail: martins.marcos@ufsm.br
bAcademic Department of Chemistry, Federal Technological University of Paraná (UTFPR), 85884-000 Medianeira Campus, PR, Brazil
cNúcleo de Química de Heterociclos (NUQUIMHE), Departamento de Engenharia e Tecnologia Ambiental (DETA), Federal University of Santa Maria (UFSM), 98400-000, Frederico Westphalen Campus, RS, Brazil
First published on 4th February 2026
Abstract
The formation of a crystal structure from molecules that self-assemble in solution until the final crystal formation constitutes the construction of a supramolecular structure. In this process, molecules interact to form a complex system. These self-organized systems, characterized as open, can organize spontaneously when exposed to a given gradient. Because this gradient is information-neutral, the organization emerges from within the system, resulting in “emergent” properties that are unpredictable and irreducible. This highlight review provides a concise guide to understanding the fundamentals of a holistic approach, where the emergent properties of the crystal system are considered in the study. It emphasizes that interactions between molecules result from the interaction of their surfaces with complementary electrostatic potentials; regions of high negative electrostatic potential on one molecule interact with regions of high positive electrostatic potential on another. According to the non-classical theory of nucleation, these complementary interactions between molecules lead to the formation of the “first building blocks” while still in solution. As the process progresses, interactions among the more robust and cooperative building blocks self-organize to form the three-dimensional supramolecular structure. Understanding the complexity inherent in the formation of supramolecular structures poses a major challenge, beginning with the difficulty of delimiting a portion of the supramolecular system that represents all the interactions existing in the crystal. This study considers a demarcation based on the “supramolecular cluster” formed by a central molecule (M1) and its neighbors (MN) in the first coordination sphere. Thus, the supramolecular cluster is conceptualized as the smallest portion of the crystal that contains all the intermolecular interactions present in the system. Through the concept of retrocrystallization, it is possible to identify the main building blocks of the crystal. Utilizing the energetic and topological data of the intermolecular interactions, we developed proposals for crystallization mechanisms. Some steps in these mechanisms are confirmed by 1H NMR and mass spectrometry, which identify initial nucleation blocks still in solution. In this highlight review, we present proposed crystallization mechanisms for more than 200 organic compounds with diverse molecular structures, identifying at least nine main crystallization mechanisms.
1. Introduction – general overview
Imagine a solution from which the solvent is being removed by evaporation. As this occurs, the solute molecules begin to draw closer due to intermolecular interactions, eventually leading to the formation of aggregates. These aggregates maintain equilibrium with other aggregates because of the lability of intermolecular interactions, allowing the exchange of constituents and the formation of new supramolecular structures. This process, known as self-organization by design, aims to programmatically control the supramolecule. Thus, we have self-organization by selection, meaning that by acting on the dynamic constitutional diversity influenced by internal and external factors, this process aims to achieve an outcome comparable to Darwinian selection.1–12 As the solvent is gradually removed from the solution, the aggregates serve as nucleation centers for the future crystal.The supramolecular structure subsequently consolidates into a crystalline structure, with fixed intermolecular interactions and constituent positions. Interestingly, in self-assembled crystalline structures, the structural and functional properties of the supramolecular system are not solely depend on the properties of individual molecules.4,6–12 Certain properties, called “emergent”, can only be explained by the organizational framework of the molecules and are not predictable from a single component alone. These properties arise from the interactions during the self-assembly process, especially when nucleating aggregates remain in solution. Thus, a supramolecule is defined as a collective of interacting molecular entities. Although this phenomenon has long been known in the study of complex systems, it has recently gained attention in the natural sciences.1–12
The formation of a crystal structure from molecules that self-organize in solution, culminating in the final formation of the crystal, constitutes the development of a complex system. The emergent properties resulting from these interactions are characterized by their unpredictability and irreducibility.11,12 In other words, “emergent properties” cannot be predicted; that is, we cannot describe in advance their nature based solely on the characteristics of separate structures—the parts (molecules) or the whole (crystal)—or on the laws governing the interactions. Instead, attention must be directed towards the interactions themselves, specifically between the parts and between the parts and the whole. This approach involves considering both perspectives, parts and whole, with the aim of achieving a comprehensive understanding. It is an interaction-driven process that leads to emergence, wherein the whole manifests as more (or less) than the sum of all the parts. Thus, one of the main characteristics for adequately understanding an emergent property is its inherent unpredictability.12 It is well known that self-organized systems, characterized as open, can spontaneously organize when subjected to a given gradient. Since the imposed gradient is completely information-neutral, one can conclude that the organization emerges intrinsically within the system. Each molecule interacts with all its neighboring molecules, and each neighboring molecule reciprocates with its respective neighbors, and so on. Given that both the parts and the whole play a role in this process, the emergence of structural design occurs. Furthermore, it is worth noting that, currently, the findings of many researchers have suggested that understanding crystal design hinges on understanding the emergent properties involved during the formation process.1–12
At this point, we should contemplate a future question: what is the importance of investigating the nature and characteristics of emergent properties appearing during crystal formation? To assess the importance of this type of discovery, we must consider two aspects of the object: the molecule (the part) and the crystal (the whole).
There is no doubt that our understanding of molecules is highly advanced; chemists are capable of synthesizing almost any molecule with adequate stability and characterizing it using various physical and chemical methods. Likewise, on the other hand, when we consider a crystal possessing useful properties classifiable as material, researchers can characterize, modify, and use the different properties of this material.
By considering crystal as a material, we may already possess part of the answer to our question. Hence, is it possible to trace the origin of the properties of materials? These properties are likely inherent in the supramolecular system that makes up the material and thus are shaped by the essence of emergent properties that arise from intermolecular interactions.
Given this context, this article aimed to further advance the understanding of emergent properties in complex supramolecular systems, specifically, crystals of organic molecules. To this end, classical and non-classical nucleation, the characterization of intermolecular interactions, and the notion of intermolecular complementarity from the perspective of molecular electrostatic potential surfaces will be briefly discussed. These discussions will serve as a foundation for presenting a method utilizing the “supramolecular cluster” as a basic tool for demarcating a part of the crystal, enabling access to all interactions present within it. Subsequently, the data obtained will be used to infer the crystallization mechanisms involved in crystal formation.
2. Classical and non-classical nucleation at the supramolecular scale
Numerous studies in the literature, based on both theoretical and experimental data, have examined nucleation processes ranging from the supramolecular to the nanomaterial scale.13–26 These works discuss nucleation within the framework of two primary theories: classical nucleation theory (CNT) and non-classical nucleation theory (NCNT), as illustrated in Fig. 1(a). In CNT, the size of unstable nuclei is specific to each system and cannot be generalized. The critical nucleus size at realistic supersaturation levels is constituted by tens of molecules. CNT is grounded in the early 20th-century thermodynamic work of Gibbs.13 The central principles of CNT include the existence of a critical nucleus size: larger nuclei tend to persist, while smaller ones dissolve, where the process is described by a single-step barrier justified by thermodynamic arguments (Fig. 1). Nevertheless, CNT has been questioned by simulations demonstrating a multi-stage nucleation process, in which the first blocks form and then joined together to form larger and more ordered nuclei (i.e., NCNT). In NCNT, these first blocks are of a similar size and thermodynamically stable, and in many cases, detected in solution from experimental data.24,25 Stable nuclei are 1D or 2D aggregates with fixed molecules, which, when combined, form a 3D proto-crystal. Some researchers14 have suggested that the term “non-classical” should not be used, as this type of nucleation aligns with the framework of Gibbs' theory. | ||
| Fig. 1 (a) Schematic illustration of the nucleation mechanism according to classical nucleation theory (top) and non-classical nucleation theory (bottom); (b) thermodynamic allegory of different nucleation theories. Plots of the free energy change as a function of reaction coordinate in classical (red) and non-classical (green) growth models. | ||
NCNT merely proposes that the pathway from solution to solid does not rely on the formation of a critical nucleus as a fundamental and necessary transition state, which is emphasized in CNT. In NCNT processes, growth units larger than monomers (first blocks) or pre-nucleation clusters are thermodynamically stable, meaning they possess negative free energy of formation. These blocks then combine to form larger stable nuclei that define the design of the proto-crystal (Fig. 1).
Proponents of CNT have suggested that the concentrations of first blocks and stable nuclei are very low within the solution, reducing the likelihood of collisions between these blocks or stable nuclei and similar species.14 Consequently, given the low concentration, classical theory assumes that nucleation progresses through the successive assembly of single molecules (Fig. 1, top). This assumption, commonly referred to in the nucleation literature as the Szilard postulate, is violated in non-classical nucleation pathways involving intermediate aggregated phases, as highlighted in Faraday Discussions on crystal nucleation.14 Recent evidence has indicated that crystals can nucleate within liquid droplets, amorphous aggregates, mesoscopic clusters, and other assemblies of solutes.14 One potential reason supporting multistep nucleation thermodynamically is that stable blocks or nuclei in these droplets exist at high concentration in this microsystem, making them preferable to direct nucleation in dilute solution.27,28 It is important to note that Gibbs proposed a thermodynamic framework for nucleation in liquid droplets within supersaturated vapors based on the second law of thermodynamics, which he reformulated in terms of thermodynamic potentials, providing guidance for systems under isothermal and isobaric conditions commonly found in laboratories, nature, and industry. Since all processes adhere to the second law, it is no mystery that all nucleation processes align with Gibbs' general theory.
Our studies have demonstrated that first blocks (dimers and tetramers, among others) or stable nuclei (1D) exist in solution and can be detected using experimental methods such as concentration dependent 1H nuclear magnetic resonance (CD-NMR)29–37 and mass spectrometry.35
3. Intermolecular interactions
The nature of intermolecular interactions has been extensively studied, leading researchers to classify these interactions based on the interacting atoms or atomic groups. Thus, several named interactions exist, including hydrogen bonds, π–π, CH–π, lp–σ-hole, and H–H interactions, among others (Fig. 2).39–51 | ||
| Fig. 2 A selection of intermolecular interactions. | ||
Despite major technological advancements in acquiring structural data through single-crystal X-ray diffraction in recent decades, self-assembled supramolecular systems continue to be studied as simple systems under a Cartesian reductionist conception. There is a plethora of papers presenting the study of crystal structures based on the principle that the “driving forces of defining crystal design” lies in a few principal intermolecular interactions.
Generally, studies have systematically presented data on these interactions across various molecular structures. It is also important to note that some interactions, such as hydrogen and halogen bonds, are presented as determinants in directing the geometry observed in crystal structures. These interactions are characterized by a range of angles and distances, and in some cases, the energetic data of these “main” interactions are included.39–51
The reporting of many interactions identified in the literature has led to the publication of numerous articles, sometimes with repetitive approaches, emphasizing one or another of these interactions as “directors” of the crystallization of organic molecules. Many of these works, which initially only considered distances and angles of interactions, were used to define the most important interactions in the crystalline system. Based on this prioritization, methods were developed to approach data obtained by X-ray diffraction. Among such methods, the one that defines supramolecular synthons should be mentioned.52–59
The supramolecular synthon is defined as a molecular fragment linked by intermolecular interactions that reasonably approximate the organization of the entire crystal. The identification of supramolecular synthons arises from distances predetermined by investigators based on the van der Waals radii. Thus, it has been established that intermolecular interactions, termed “non-covalent”, including hydrogen bonds, tend to form robust supramolecular synthons, which in turn direct the crystal packing.52–59 This approach, however, has proven to be insufficient because it excludes interactions considered “weaker” than hydrogen bonds. Generally, these so-called weak interactions, often referred to as dispersive forces, were considered of minor importance. Furthermore, topological factors were neglected in the synthon approach, causing researchers to disregard molecules that, although present in the crystal, were misaligned with the “appropriate direction” indicated by their respective molecular fragments. In essence, this approach focused only on interactions that effectively participate in the so-called “robust synthons” to form dimers, chains, layers, or macrocycles.
In the early 21st century, a more complex approach began to take shape, as some paradigms of the synthon approach based only on distances and interaction angles underwent revision.60,61 For instance, atom–atom distances smaller than the sum of the van der Waals radii did not imply large stabilization energies. Likewise, distances larger than the sum of the atomic radii could contain significant stabilization energy.
Describing crystal interactions only in terms of short interatomic contacts as the main component of structural bonding was recognized as simplistic and insufficient.60,61 Unfortunately, most studies continued with a linear-Cartesian perception of the phenomenon, innovating only by including the stabilization energies of the so-called “robust” intermolecular interactions. Consequently, these studies continued to report the geometric data—now complemented by energetic information—of the interactions present in supramolecular synthons, reaffirming the idea that such interactions would be obligatory and sufficient for defining the crystal design.
In parallel with this apparent evolution, our research group has been developing a complex approach that considers all interactions present in the crystal. In 2014, our first results were published, which convincingly demonstrated that crystal design depends on interactions across the entire molecular surface.62 We demonstrated that there was no scientific justification for “isolating” a certain face of the molecule and claiming it is responsible for the crystal design. Based on this principle, advances in our work, which uses data on intermolecular interaction energies and molecular topology, have demonstrated that the distances and angles of interactions are, in fact, just consequences of crystal design. This design results from the emergent properties of the supramolecular system that derive mainly from the energies of intermolecular interactions and molecular topologies.29–38,62–76
4. Molecular electrostatic potential complementarity
The concept of electrostatic potential complementarity60–62,77–79 is based on the principle that crystal formation results from the exchange of intermolecular interactions between solute and solvent (which is displaced from the medium) by intermolecular interactions of self-associating solute molecules. This initially produces the “first blocks” in solution (Fig. 1), where regions of high negative electrostatic potential on the surface of one molecule interact with regions of high positive electrostatic potential on the surface of another. As the process continues, some “first blocks”, those most robust and cooperative (reminiscent of Darwinian selection), interact with neighboring blocks and arrange themselves to form the 3D supramolecular structure (the proto-crystal, Fig. 1). This perception aligns with a more systemic and satisfactory approach to understanding crystal structure formation, as a linear-Cartesian approach does not consider only specific atoms of the molecule that supposedly constitute the synthons and the named interactions (Fig. 2).Over the past decade, our research group has been dedicated to developing a systemic approach toward complexity in crystal studies.29–38,62–76 From a linear-Cartesian perspective, the crystal is considered from its constituent parts (i.e., the molecules), which present regions with varying magnitudes of negative and positive electrostatic potentials. In this view, attention tends to focus solely on the so-called “robust” synthons, apparently responsible for the strongest interactions.
Conversely, from a systemic perspective, the crystal must be understood as an integrated whole, where regions of different electrostatic potentials of a given molecule participate in a harmonious network of complementary interactions. This network, along with neighboring molecules, results in a stabilizing equilibrium. Furthermore, from the formation of the first stable blocks and nuclei to the proto-crystal (Fig. 1), the supramolecular system becomes increasingly complex as emergent properties begin to arise. In this process, molecules continuously interact, leading to the redistribution of molecular electrostatic potentials up to the proto-crystal stage. For example, when a molecule aggregates with another to form a dimer, all regions of the two molecules have their potential redistributed, and this process continues (Fig. 3). As an example of this behavior, 3-amino-4-iodo-5-methylisoxazole was used to represent the redistribution of electrostatic potential during the formation of molecular aggregates.
 | ||
| Fig. 3 Molecular electrostatic potential (MEP) and complementary interactions in the 3-amino-4-iodo-5-methyisoxazole. | ||
Therefore, it is important for scientists to consider that interactions between electrostatic potential surfaces constitute a dynamic process. Each potential, when interacting with a neighboring electrostatic potential, induces a rearrangement of the entire potential surface of the two molecules involved, and the site of interaction tends toward neutrality of potential. This differentiation begins between the electrostatic potentials of individual molecules and those continuously modified by the addition of new molecules and/or molecular blocks. Here, the emergent property begins to direct the nucleation process until the formation of the proto-crystal, thereby marking the commencement of crystalline design.
One of the factors supporting the approach that considers intermolecular interactions based on the complementarity of electrostatic potential is the correlation between the contact area data between the molecules and the interaction energy (Fig. 4). The data showed that it is reasonable to assume that the magnitude of the stabilization energy between each neighboring molecule MN, and the central molecule M1 should be proportional to the contact surface area between the two molecules.62 To verify this assumption in our experiment, we analyzed the correlation between the normalized stabilization energy of the intermolecular interactions (NG) and the respective normalized contact areas (NC) for approximately 700 dimers.29–37,62–76 The use of normalized data takes into account the value of N (number of MN molecules neighboring M1) and is necessary to ensure that each molecule contributes proportionally to the formation of the crystal, allowing direct comparisons and consistent interpretations between different molecular systems. Considering the total interaction energy of the cluster (Gcluster) to be 1.0 and imagining the interaction energy to be homogeneous for the whole surface, the interaction energy between M1 and any of the neighboring molecules would be the same. In other words, each molecule in the first coordination sphere would contribute with a 1/N fraction of the total stabilization energy (Gcluster). But we understand almost all organic molecules do not have the same contact surface with every neighboring molecule and do not interact with the same energy with all of them. Therefore, to determine which molecules have the most important interactions for the stabilization of the cluster, we search for a set of parameters from the ideal contribution of 1/N of the energy and 1/N of the contact surface. Given that NGM1⋯MN and NCM1⋯MN are the normalized energy and normalized surface, eqn (2) and (3), respectively, in a supramolecular cluster with N molecules, the robustness of the interaction between M1 and any MN molecule can be estimated from the contributions of these normalized parameters. As a result, after observing the NC and NG correlation between the 700 dimers, we observed that 67% of the points demonstrate the correlation trend (Fig. 4, R2 = 0.670). It is worth mentioning that the NC and NG data used for the dimers were obtained from models previously reported by our own research group. The NC or NG values of a dimer can be higher than 1, since certain dimers have very high NC or NG contributions to the cluster formation. Thus normalized data can be greater than 1.0 because, as mentioned earlier, normalization is on the scale of N (e.g., N = 14, 16, etc.). We observed that some outliers may emerge from interactions in dimers that present, the first being (i) energies much higher than expected when compared with the contact surface between the molecules. Generally, this is associated with small areas possessing high electrostatic potential (e.g., interactions primarily resulting from hydrogen bonds).62 The second, (ii) contact surfaces larger than expected given the detected energy, which can be attributed to the cancellation of interaction energies due to electrostatic potentials of equal sign in the interaction regions of the same molecule.75 Thus, when we disregard 2% of the points (Fig. 4, marked in the blue circle) that have (NG − NC) > 1.75, the points demonstrating the NG vs. NC correlation trend increase to over 82%.
 | ||
| Fig. 4 Correlation between calculated normalized energy interaction (NGM1⋯MN) and normalized contact surface (NCM1⋯MN) in the supramolecular cluster. Each point corresponds to an intermolecular interaction between the central molecule (M1) and a neighboring molecule (MN), interactions in organic compounds that do not present intermolecular interactions of the interaction bond type. (NGM1⋯MN = 1.150 NC − 0.154; R2 = 0.670; r = 0.820; N = 638; NGM1⋯MN = 1.146 NC − 0.217; R2 = 0.820; r = 0.906; N = 624). | ||
5. Supramolecular cluster – a demarcation tool
In the 2010s, our research group demonstrated the necessity of demarcating a minimal portion of the crystal where it would be feasible to determine all pertinent information regarding intermolecular interactions within the crystal lattice. In the literature, several approaches to demarcation within the crystal system have been identified, more notably with: (i) unit cells; (ii) a set of unit cells; (iii) supramolecular synthons, motifs, among others.52–59Although the unit cell is a fundamental mathematical component for constructing the crystal structure, the cell parameters themselves do not constitute an adequate demarcation. This is because a unit cell does not contain enough molecules to fully describe the phenomenon under study: intermolecular interactions. Employing a set of unit cells can be a suitable demarcation for lattice energy calculations, proving useful in determining the average stabilization of a molecule when immersed in the crystal compared to the same molecule in vacuum. Conversely, supramolecular synthons are suitable for specific comparative studies of recurring intermolecular interactions in several crystal structures. However, synthons do not provide a sufficient demarcation for studying the phenomenon as they only include interactions deemed stronger by the observer, such as intermolecular interactions of an electrostatic nature.52–59
Conversely, it is important to remember, in accordance with the scientific method,80 that when delineating the phenomenon to be studied, one must ensure the representation of both the parts and the whole of the phenomenon. This includes a linear-Cartesian perspective (the part) and a systemic perspective (the whole); it should not be overlooked that the crystal structure constitutes a complex system, which, as such, presents emergent properties.1–12
Hence, we conclude that it is imperative to extend this demarcation: consider a molecule M1 and its first coordination sphere; this ensemble embodies a “supramolecular cluster”, representing the smallest segment that contains comprehensive information on the existing intermolecular interactions within the crystal.
Based on these premises, in 2014,62 our research group proposed a demarcation utilizing the supramolecular cluster, consisting of an M1 molecule and the MN molecules that form the first molecular coordination sphere of M1. These are the molecules that maintain contact, as determined by the van der Waals radius, with this molecule. The first coordination sphere of a molecule had previously been delineated by Kitaigorodskii and others.81–83 Additionally, Russian researchers84–86 had already demonstrated that most organic molecules presented a first coordination sphere predominantly composed of 14 molecules (in over 50% of compounds), followed by 12 or 16 molecules.
Thus, we conceptualize this supramolecular cluster (M1 + MN neighboring molecules) as the smallest unit of the crystal that contains all topological and energetic information. With this in mind, we applied the Voronoi–Dirichlet polyhedron method (VDP, calculated with the ToposPro software85) to define the first coordination sphere of an M1 molecule (symmetry code x, y, z). The VDP concept was introduced by Fischer and Koch82 to determine the number of neighboring molecules in contact with a given central molecule (M1).
From constructing the supramolecular cluster, data on the contact area (CM1⋯MN, in Å2) and stabilization energy (GM1⋯MN, in kcal mol−1) were obtained for each dimer formed between the M1 molecules and the neighboring MN molecules (Fig. 5). The number of MN molecules determines the molecular coordination number (N). The VDP was used to obtain the contact area for each dimer, and this data was provided by the software used. The intermolecular stabilization energy of each dimer was determined following eqn (1), where GM1+MN is the total stabilization energy of the considered dimer, GM1 is the stabilization energy of the M1 molecule, and GMN is the stabilization energy of the neighboring MN molecule. To standardize all contact area and energy data on the same scale, thereby allowing the comparison of two or more compounds, it is necessary to normalize the data to obtain NCM1⋯MN and NGM1⋯MN, according to eqn (2) and (3), respectively. Lastly, from the contact area and interaction energy data, it is possible to determine the cluster contact area (CCluster) and the cluster interaction energy (GCluster) (eqn (4) and (5)). Importantly, the GCluster demonstrated a strong correlation with properties such as the experimental enthalpy of fusion and sublimation for some compounds studied in our laboratory.29,31,63
| GM1⋯MN = GM1+MN − (GM1 + GMN) | (1) |
 | (2) |
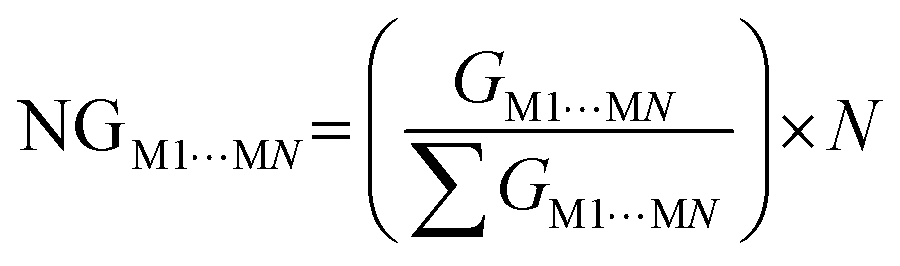 | (3) |
| Ccluster = ∑CM1⋯MN | (4) |
| Gcluster = ∑GM1⋯MN | (5) |
 | ||
| Fig. 5 Cluster of first coordination sphere around M1 is formed by MN molecules for a compound with N = 14: (a) M1 (red), six in-plane molecules (pink), four above-plane and four below-plane molecules (green); (b) supramolecular cluster for 3-amino-4-iodo-5-methyisoxazole (Mercury software, spacefill). | ||
With this approach, we have demonstrated in recent years29–37,62–76 that the demarcation of the supramolecular cluster is sufficient to obtain important information such as: (i) the total stabilization energy of the cluster, which provides valuable data for comparisons of polymorph stabilities,36,64,70,71,74 comparison with experimental data of fusion and sublimation enthalpy;29,31,63 (ii) the energies and contact areas of each M1⋯MN dimer, which form the supramolecular cluster, enabling a hierarchical analysis of the interactions from both energetic and topological perspectives. Standardizing these data allows comparison between compounds with different crystalline designs and varying coordination numbers (N).
These data are then organized in decreasing order of energy (i.e., from the dimers with the highest stabilizing energy contributions to those with the lowest), as illustrated in Fig. 6. This method enables one to identify the first blocks and stable nuclei in the crystallization process (Fig. 1), highlighting a possible crystallization pathway.29–37,65,67,69–76 To aid in proposing crystallization mechanisms, two parameters are used: NG/NC (eqn (6)) and NCG% (eqn (7)). The first indicates the dominant factor in crystal formation, with values above 1.0 suggesting energetic dominance, below 1.0 implying topological dominance; and values of 1.0 indicating a balance between the two factors. NCG% evaluates the energetic and topological contributions at each stage of crystallization, as defined in the proposed mechanisms.
 | (6) |
 | (7) |
 | ||
| Fig. 6 Retrocrystallization scheme (top) and the normalized energies showing the crystallization process of 3-amino-4-iodo-5-methyisoxazole (bottom). | ||
What is more, our studies have revealed that uncharged organic molecules present only stabilizing intermolecular interactions within all molecules of the supramolecular cluster.29–34,36,37,62,65,67,69–72,74,76 Recently, we applied this method to the study of crystals of di-, tri-, and tetraalkyl ammonium halides,73 mesoionic compounds,35 and ammonium carboxylates,75 making it possible to verify, in addition to stabilized intermolecular interactions, the existence of destabilizing intermolecular interactions.
With the aim of deepening the discussion on the crystallization process in light of the supramolecular cluster concept, this work limits our discussions to mainly single-component models, reported from 2014 to 2025. However, as mentioned earlier, we emphasize that the concept of supramolecular clusters is applicable to systems containing a series of organic molecules (see Table S1, ref. 29–37, 63–65, 67 and 69–76), which also included examples of molecular systems containing two molecules in the asymmetrical unit (see Table S1, ref. 30, 33, 37 and 65), including salts (see Table S1, ref. 73 and 75), cocrystals (see Table S1, ref. 34), mesoionic compounds (see Table S1, ref. 35), polymorphs (see Table S1, ref. 36, 64, 70, 71 and 74) and molecules containing mechanical bond, e.g., 2-rotaxanes (see Table S1, ref. 38 and 68–70). A detailed discussion of each of these systems would require a separate chapter, which cannot be included in this article. However, the reader can check our discussions on the crystallization results of each system in the cited references.
6. The concept of retrocrystallization
E. J. Corey87 established his concept of retrosynthetic analysis in the 1960s, revolutionizing both the practice and teaching of organic chemistry. The numerous synthetic methods developed by him and his co-workers have proven invaluable in organic synthesis. The application of the retrosynthesis concept remains current with the development of useful software for synthetic planning. In this method, the chemist seeks to identify bonds formed between preexisting synthesis blocks in complex molecules obtained through natural or artificial synthesis to pinpoint the types of reactions involved in each step. In this way, step by step, the chemist can trace back to the synthesis blocks used to obtain the target molecule.By comparing the synthesis of an organic compound to the “synthesis of a crystal”, one can observe that: (i) in organic synthesis, covalent bonds are broken and formed at each reaction step; (ii) in the synthesis of a crystal, molecules come together due to stabilization energies from intermolecular interactions as well as topological factors, leading to the formation of first blocks and stable nuclei during the nucleation process for crystal formation. Therefore, by considering crystal formation as a synthesis process, we can suggest its retrosynthesis, which, in the case of the crystal, can be called retrocrystallization. In this process, retrocrystallization involves breaking intermolecular interactions until reaching a molecule representing the first synthesis block.
The information obtained from the supramolecular cluster, defined previously, has facilitated the adaptation of Corey's retrosynthesis method87 into a retrocrystallization approach, where the main “synthesis” blocks of the crystal are determined. The retrocrystallization data have allowed proposals to be made regarding what we call the crystallization mechanism of compounds. These crystallization mechanisms have been corroborated by experimental data (e.g., 1H NMR and mass spectrometry)29–37 that allow information to be obtained about the first nucleation blocks of the crystal that were formed while still in solution.
The use of mass spectrometry (MS) has provided clear examples of the observation of initial crystallization nuclei, as in the case of mesoionic compounds.35 In one of our studies, we investigated a compound exhibiting a three-step crystallization mechanism, the first step being characterized by the formation of a dimeric nucleus. The dimer formed in this initial crystallization stage had a stabilization energy of approximately −17 kcal mol−1. The existence of this dimer throughout the crystallization process was experimentally confirmed by LC-MS/MS. In the LC-MS/MS experiment (positive mode, scan method), it was observed that the dimer has practically the same relative intensity as the molecular ion.35 In addition to this model, other models from the group have been observed and will be published soon.
Fig. 6 shows the retrocrystallization process of 3-amino-4-iodo-5-methylisoxazole: (a) the crystal or crystalline lattice, is a three-dimensional (3D) block; (b) supramolecular cluster, a sample of the crystal, is also a 3D block composed of a central molecule M1 (in red) surrounded by 14 molecules that form the first molecular coordination sphere; (c) the energy data allows deconstructing the supramolecular cluster into a one-dimensional (1D) block; (d) reducing further into a dimer; (e) and, finally, into the monomer M1. Additionally, Fig. 6 also shows the energy data of 3-amino-4-iodo-5-methylisoxazole,29 revealing a non-classical nucleation process that occurs in three stages: (i) in the first stage, dimer 1, possibly representing the first block, forms as a closed dimer involving hydrogen bonds; (ii) in the second stage, this dimer participates in stacking to constitute a stable 1D nucleus; (iii) in the third stage, stable 2D and 3D nuclei form, exhibiting small stability differences that obscure view of the 2D → 3D sequence.
The influence of concentration on the chemical shifts of the amino and methyl groups was evaluated29 in CDCl3 and CD3OD solvents. In CDCl3, the signal of the 3-NH2 hydrogens shows a large downfield shift as the concentration increases, indicating the formation of a hydrogen bond. The 5-CH3 hydrogens show an upfield shift as concentration increases, which is attributed to the anisotropic magnetic effect due to the proximity of the aromatic ring of another heterocyclic molecule. The same titration experiment was performed in CD3OD. The pattern of chemical shift changes in the signals due to the 5-CH3 group was similar to that observed in CDCl3. Nonetheless, the chemical shift changes in the 3-NH2 hydrogens signal were negligible. These results were expected, indicating a tendency for weak dimerization through the N–H⋯N (NH2) interaction in CD3OD. The protic solvent competes with the hydrogen bonds of the dimer, whereas the same solvent does not interfere with the π–π interactions responsible for the anisotropic magnetic effect on the 5-CH3 hydrogens.
7. Proposals for crystallization mechanisms
Once the energetic and topological parameters of the supramolecular cluster have been defined based on the retrocrystallization data of the cluster, mechanistic proposals for the crystallization of the substance under study can be inferred. Several crystallization mechanisms for different organic compounds have been reported by our research group30,32,63 (Fig. S1). These studies have demonstrated that the formation of crystal nuclei can occur through pathways alternative to classical nucleation and involving supramolecular intermediates. These mechanisms vary significantly depending on the predominant intermolecular interactions, including π–π stacking and, especially, hydrogen bonds, as well as the influence of specific functional groups. Identifying these alternative pathways broadens our understanding of crystallization processes and is fundamental for developing strategies to control crystal growth in complex systems.Our studies, involving over 200 crystalline substances, suggested the definition of nine main crystallization mechanisms (CR).29–37,63–65,67,69–76 All of them begin with the monomer (M), which can then form a stable 1D nucleus, a 2D nucleus, and reach the 3D proto-crystal. This crystallization process is called the CRM123 mechanism. Next, the routes can start from the monomer and go to the 1D nucleus and then directly to the 3D proto-crystal. This process appears to be the most common among the compounds studied and is referred to as the CRM13 mechanism. In the third type of mechanism, the monomer directly forms a 2D nucleus and then reaches the 3D proto-crystal (CRM23 mechanism). There is a fourth type of mechanism, in which crystallization starts from the monomer and directly forms the 3D proto-crystal, defined as CRM3. To illustrate each of these types of mechanisms mentioned, Fig. S1 represents the retrocrystallization scheme, showing the crystallization process for the compounds reported by our research group. Five other mechanisms follow similar paths to the mechanisms described above; however, they stop at first blocks such as closed dimers (e.g., CRMD123, CRMD13, CRMD23, and CRMD3) and closed tetramers (CRMTt123).
Table S1 lists the crystallization mechanisms of crystalline compounds that were studied by our group from 2014 to 2025.29–37,63–65,67,70–76 The numbers of compounds in the table correspond to those in the original reference. In some instances, the mechanisms detailed in the table were revised and may differ from those proposed in the original references. Fig. 7 shows the distribution of crystallization mechanism types for the organic compounds studied. Table 1 summarizes the nine types of crystallization mechanisms previously described in the literature for the models studied, providing the symbol of each pathway and the number of associated compounds.
 | ||
| Fig. 7 Distribution of the types of crystallization mechanisms of the 213 organic compounds studied. | ||
| Path | Mechanism abbreviation | No. of compounds studied |
|---|---|---|
| Monomer → 1D → 2D → 3D | CRM123 | 37 |
| Monomer → 1D → 3D | CRM13 | 74 |
| Monomer → 2D → 3D | CRM23 | 49 |
| Monomer → 3D | CRM3 | 5 |
| Monomer → dimer → 1D → 2D → 3D | CRMD123 | 3 |
| Monomer → dimer → 1D → 3D | CRMD13 | 18 |
| Monomer → dimer → 2D → 3D | CRMD23 | 22 |
| Monomer → dimer → 3D | CRMD3 | 4 |
| Monomer → tetramer → 1D → 2D → 3D | CRMTt123 | 1 |
8. Conclusions and future perspectives
This highlight presents methods that aimed to systematize supramolecular structural data of crystalline compounds in order to better understand and interpret the design and crystallization mechanisms of crystalline supramolecular structures. Our research group has developed these studies since 2014,62 and there are no similar studies in the literature. Consequently, several concepts and parameters have been necessarily developed under the rigor of the scientific method, including (i) using the supramolecular cluster to define the smallest crystal unit that presents the structural and energetic information of the entire crystal; (ii) determining normalized interaction energies and normalized contact areas between molecules, which allows for comparisons between crystalline supramolecular structures of different compounds; (iii) developing proposed crystallization mechanisms based on energetic and topological data, enabling the establishment of stages in the crystal formation process, corroborated by NMR and MS data of molecular blocks detected in solution; and (iv) developing a specific nomenclature for the main crystallization mechanisms observed in a sample space of two hundred crystalline compounds.Future perspectives for this study include increasing the number of crystal structures studied; conducting a more detailed analysis of new structures to consolidate the number of possible crystallization mechanisms; seeking additional experimental data to corroborate these proposals; and introducing machine learning algorithms to learn from the systems already studied, thereby advancing automated processes using the available big data.
Conflicts of interest
There are no conflicts to declare.Data availability
We declare that the data described in the article is in the supplementary information (SI) and references cited in the article. Supplementary information is available. See DOI: https://doi.org/10.1039/d5ce01132a.Acknowledgements
The authors kindly acknowledge the Rio Grande do Sul Research Support Foundation (FAPERGS process no. 17/2551-0000981-9), the National Council for Scientific and Technological Development (CNPq process no. 405071/2016-7), and the Coordination for the Improvement of Higher Education Personnel (CAPES) for financial support. The fellowships from CNPq (M. A. P. M.) and CAPES (P. S. V. L., E. F. S., S. K. K.) are also acknowledged.References
- J.-M. Lehn, Perspectives in Chemistry—Steps towards Complex Matter, Angew. Chem., Int. Ed., 2013, 10(4), 2836–2850, DOI:10.1002/anie.201208397.
- J.-M. Lehn, From matter to life: Chemistry?!, Resonance, 1996, 39–53, DOI:10.1007/BF02835621.
- G. R. Desiraju, Crystal Engineering: A Holistic View, Angew. Chem., Int. Ed., 2007, 46(44), 8342–8356, DOI:10.1002/anie.200700534.
- G. R. Desiraju, Chemistry: The Middle Kingdom, Resonance, 2007, 44–60, DOI:10.1007/s12045-007-0072-8.
- R. F. Ludlow and S. Otto, Systems chemistry, Chem. Soc. Rev., 2008, 37, 101–108, 10.1039/B611921M.
- J. Vicens and Q. Vicens, Emergences of supramolecular chemistry: from supramolecular chemistry to supramolecular science, J. Inclusion Phenom. Macrocyclic Chem., 2011, 71, 251–274, DOI:10.1007/s10847-011-0001-z.
- J. Vicens and Q. Vicens, Origins and emergences of supramolecular chemistry, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65, 221–235, DOI:10.1007/s10847-009-9602-1.
- O. Pandoli and G. P. Spada, The supramolecular chemistry between eastern philosophy and the complexity theory, J. Inclusion Phenom. Macrocyclic Chem., 2009, 65, 205–219, DOI:10.1007/s10847-009-9643-5.
- A. Kurakin, The universal principles of self-organization and the unity of Nature and knowledge, in The SOFT, 2007, pp. 1–34, http://www.alexeikurakin.org/text/thesoft.pdf Search PubMed.
- B. Küppers, Towards an experimental analysis of molecular self-organization and precellular Darwinian evolution, Naturwissenschaften, 1979, 66(5), 228–243, DOI:10.1007/bf00571603.
- F. Heylighen, Complexity and Self-organization, prepared for the Encyclopedia of Library and Information Sciences, ed. M. J. Bates and M. Niles Maack, Taylor & Francis, 2008 Search PubMed.
- E. Morin, La Methode, Du Seuil, Paris, France, 1977–1980, vol. 1–2 Search PubMed.
- J. W. Gibbs, The Collected Works, Thermodinamics, Longmans, Green and Co., New York, 1928, vol. I Search PubMed.
- M. W. Anderson, M. Bennett, R. Cedeno, H. Coelfen, S. J. Cox, A. J. Cruz-Cabeza, J. J. De Yoreo, R. Drummond-Brydson, M. K. Dudek, K. A. Fichthorn, A. R. Finney, I. Ford, J. M. Galloway, D. Gebauer, R. Grossier, J. H. Harding, A. Hare, D. Horvath, L. Hunter, J. Kim, Y. Kimura, C. E. A. Kirschhock, A. A. Kiselev, W. Kras, C. Kuttner, A. Y. Lee, Z. Liao, L. Maini, S. O. N. Lill, N. Pellens, S. L. Price, I. B. Rietveld, J. D. Rimer, K. J. Roberts, J. Rogal, M. Salvalaglio, I. Sandei, G. Schuszter, J. Sefcik, W. Sun, J. H. ter Horst, M. Ukrainczyk, A. E. S. Van Driessche, S. Veesler, P. G. Vekilov, V. Verma, T. Whale, H. P. Wheatcroft and J. Zeglinski, Understanding crystal nucleation mechanisms: where do we stand? General discussion, Faraday Discuss., 2022, 235, 219–272, 10.1039/d2fd90021a.
- S. Karthika, T. K. Radhakrishnan and P. Kalaichelvi, A Review of Classical and Nonclassical Nucleation Theories, Cryst. Growth Des., 2016, 16(11), 6663–6681, DOI:10.1021/acs.cgd.6b00794.
- K. Gorman, Experimental Investigation of Nucleation Phenomena on the Nanoscale: Classical or Nonclassical? Literature Seminar November, 2019, pp. 1–3.
- J. De Yoreo, A Perspective on Multistep Pathways of Nucleation, Crystallization via Nonclassical Pathways Volume 1: Nucleation, Assembly, Observation & Application ACS Symposium Series, ed. X. Zhang, American Chemical Society, Washington, DC, 2020, ch. 1, pp. 1–17, DOI:10.1021/bk-2020-1358.ch001.
- M. Svärd, Mesoscale clusters of organic solutes in solution and their role in crystal nucleation, CrystEngComm, 2022, 24, 5182–5193, 10.1039/d2ce00718e.
- Y. Tsarfati, S. Rosenne, H. Weissman, L. J. W. Shimon, D. Gur, B. A. Palmer and B. Rybtchinski, Crystallization of Organic Molecules: Nonclassical Mechanism Revealed by Direct Imaging, ACS Cent. Sci., 2018, 4, 1031–1036, DOI:10.1021/acscentsci.8b00289.
- D. Gebauer, P. Raiteri, J. D. Gale and H. Cölfen, On Classical And Non-Classical Views On Nucleation, Am. J. Sci., 2018, 318, 969–988, DOI:10.2475/09.2018.05.
- H. Chen, M. Li, Z. Lu, X. Wang, J. Yang, Z. Wang, F. Zhang, C. Gu, W. Zhang, Y. Sun, J. Sun, W. Zhu and X. Guo, Multistep nucleation and growth mechanisms of organic crystals from amorphous solid states, Nat. Commun., 2019, 10, 3872–3882, DOI:10.1038/s41467-019-11887-2.
- R. J. Davey, S. L. M. Schroeder and J. H. Horst, Nucleation of Organic Crystals - A Molecular Perspective, Angew. Chem., Int. Ed., 2013, 52, 2166–2179, DOI:10.1002/anie.201204824.
- J. H. Harding, C. L. Freemana and D. M. Duffy, Oriented crystal growth on organic monolayers, CrystEngComm, 2014, 16, 1430–1438, 10.1039/c3ce41677a.
- R. J. Davey, K. Allen, N. Blagden, W. I. Cross, H. F. Lieberman, M. J. Quayle, S. Righini, L. Seton and G. J. T. Tiddy, Crystal engineering – nucleation, the key step, CrystEngComm, 2002, 4, 257–264, 10.1039/B203521A.
- A. Spitaleri, C. A. Hunter, J. F. McCabe, M. J. Packer and S. L. Cockroft, A 1H NMR study of crystal nucleation in solution, CrystEngComm, 2004, 6, 489–493, 10.1039/b407163h.
- C. S. Towler and L. S. Taylor, Spectroscopic Characterization of Intermolecular Interactions in Solution and Their Influence on Crystallization Outcome, Cryst. Growth Des., 2007, 7(4), 633–638, DOI:10.1021/cg0602358.
- M. Kaissaratos, L. Filobelo and P. G. Vekilov, Cryst. Growth Des., 2021, 21, 5394–5402 Search PubMed.
- P. G. Vekilov, in Crystallization via Nonclassical Pathways Volume 1: Nucleation, Assembly, Observation and Application, American Chemical Society, 2020, ch. 2, vol. 1358, pp. 19–46 Search PubMed.
- M. A. P. Martins, A. R. Meyer, A. Z. Tier, K. Longhi, L. C. Ducati, H. G. Bonacorso, N. Zanatta and C. P. Frizzo, Proposal for Crystallization of 3-Amino-4-Halo-5-Methylisoxazoles: An Energetic and Topological Approach, CrystEngComm, 2015, 17(38), 7381–7391, 10.1039/c5ce01295c.
- A. B. Pagliari, T. Orlando, P. R. S. Salbego, G. C. Zimmer, M. Hörner, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Supramolecular Packing of a Series of N-Phenylamides and the Role of NH⋯O=C Interactions, ACS Omega, 2018, 3(10), 13850–13861, DOI:10.1021/acsomega.8b01801.
- M. A. P. Martins, A. B. Pagliari, A. L. Belladona, A. Z. Tier, A. R. Meyer, L. V. Rodrigues, M. Hoerner, C. P. Frizzo, H. G. Bonacorso and N. Zanatta, Supramolecular self-assembly and thermodynamic properties of 5-aryl-1-(1,1-dimethylethyl)-1H-pyrazoles in the crystalline state, J. Mol. Struct., 2019, 1195, 570–581, DOI:10.1016/j.molstruc.2019.06.009.
- J. P. P. Copetti, P. R. S. Salbego, T. Orlando, J. M. L. Rosa, G. F. Fiss, J. P. G. de Oliveira, M. L. A. A. Vasconcellos, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Substituent effects on the crystallization mechanisms of 7-chloro-4-substitutedquinolines, CrystEngComm, 2020, 22, 4094–4107, 10.1039/d0ce00214c.
- L. C. Lopes, T. Orlando, P. H. B. Simões, F. F. S. Farias, H. G. Bonacorso, N. Zanatta, P. R. S. Salbego and M. A. P. Martins, Persistence of N-H⋯O=C Interactions in the Crystallization Mechanisms of Trisubstituted Bis-Ureas with Bulky Substituents, Cryst. Growth Des., 2021, 21(10), 5740–5751, DOI:10.1021/acs.cgd.1c00600.
- A. B. Pagliari, A. R. Meyer, V. B. Solner, J. M. L. Rosa, M. Hoerner, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Effect of hydrogen bonds and π-π interactions on the crystallization of phenyl-perfluorophenyl amides: understanding the self-organization of a cocrystal, CrystEngComm, 2022, 24, 5348–5363, 10.1039/D2CE00231K.
- P. S. V. Lima, G. H. Weimer, L. P. Oliveira, H. D. S. Souza, G. F. Fiss, H. G. Bonacorso and M. A. P. Martins, Mesoionic compounds: the role of intermolecular interactions in their crystalline design, CrystEngComm, 2023, 25, 4976–4991, 10.1039/d3ce00554b.
- J. M. L. Rosa, G. H. Weimer, P. R. S. Salbego, G. F. Fiss, M. Hoerner, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Polymorphism in Pyridine-2,6-dicarboxamides: The Role of 2 Molecular Conformation in Hydrate Formation, Cryst. Growth Des., 2023, 23(8), 5548–5563, DOI:10.1021/acs.cgd.3c00184.
- F. F. S. Farias, M. Mittersteiner, A. M. Kieling, P. S. V. Lima, G. H. Weimer, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, The Persistence of Hydrogen Bonds in Pyrimidinones: From Solution to Crystal, ACS Org. Inorg. Au, 2024, 4(5), 557–570, DOI:10.1021/acsorginorgau.4c00057.
- T. Orlando, J. M. Perez, F. F. S. Farias, P. R. S. Salbego, A. Martinez-Cuezva, M. A. P. Martins and J. Berna, Mechanical Bonding as a Promoter of Crystalline Diversity in Halogenated [2]Rotaxanes, Cryst. Growth Des., 2023, 23(5), 3794–3804, DOI:10.1021/acs.cgd.3c00194.
- S. J. Grabowski, What Is the Covalency of Hydrogen Bonding?, Chem. Rev., 2011, 111(4), 2597–2625, DOI:10.1021/cr800346f.
- P. Hobza and Z. Havlas, Blue-Shifting Hydrogen Bonds, Chem. Rev., 2000, 100(11), 4253–4264, DOI:10.1021/cr990050q.
- T. Steiner and G. R. Desiraju, Distinction between the weak hydrogen bond and the van der Waals interaction, Chem. Commun., 1998, 891–892, 10.1039/A708099I.
- C. A. Hunter and J. K. M. Sanders, The nature of π-π interactions, J. Am. Chem. Soc., 1990, 112(14), 5525–5534, DOI:10.1021/ja00170a016.
- L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Halogen Bonding in Supramolecular Chemistry, Chem. Rev., 2015, 115(15), 7118–7195, DOI:10.1021/cr500674c.
- G. Cavallo, P. Metrangolo, R. Milani, T. Pilati, A. Priimagi, G. Resnati and G. Terraneo, The Halogen Bond, Chem. Rev., 2016, 116(4), 2478–2601, DOI:10.1021/acs.chemrev.5b00484.
- M. H. Kolář and P. Hobza, Computer Modeling of Halogen Bonds and Other σ-Hole Interactions, Chem. Rev., 2016, 116(9), 5155–5187, DOI:10.1021/acs.chemrev.5b00560.
- H. Wang, W. Wang and W. J. Jin, σ-Hole Bond vs π-Hole Bond: A Comparison Based on Halogen Bond, Chem. Rev., 2016, 116(9), 5072–5104, DOI:10.1021/acs.chemrev.5b00527.
- C. J. Setter, J. J. Whittaker, A. J. Brock, K. S. A. Arachchige, J. C. McMurtrie, J. K. Clegg and M. C. Pfrunder, Straightening out halogen bonds, CrystEngComm, 2020, 22, 1687–1690, 10.1039/d0ce00176g.
- R. Gleiter, G. Haberhauer, D. B. Werz, F. Rominger and C. Bleiholder, From Noncovalent Chalcogen–Chalcogen Interactions to Supramolecular Aggregates: Experiments and Calculations, Chem. Rev., 2018, 118(4), 2010–2041, DOI:10.1021/acs.chemrev.7b00449.
- D. J. Pascoe, K. B. Ling and S. L. Cockroft, The Origin of Chalcogen-Bonding Interactions, J. Am. Chem. Soc., 2017, 139(42), 15160–15167, DOI:10.1021/jacs.7b08511.
- J. C. Ma and D. A. Dougherty, The Cation−π Interaction, Chem. Rev., 1997, 97(5), 1303–1324, DOI:10.1021/cr9603744.
- M. Giese, M. Albrecht and K. Rissanen, Anion−π Interactions with Fluoroarenes, Chem. Rev., 2015, 115(16), 8867–8895, DOI:10.1021/acs.chemrev.5b00156.
- P. Vishweshwar, A. Nangia and V. M. Lynch, Recurrence of Carboxylic Acid-Pyridine Supramolecular Synthon in the Crystal Structures of Some Pyrazinecarboxylic Acids, J. Org. Chem., 2002, 67, 556–565, DOI:10.1021/jo0162484.
- A. Dey, M. T. Kirchner, V. R. Vangala, G. R. Desiraju, R. Mondal and J. A. K. Howard, Crystal Structure Prediction of Aminols: Advantages of a Supramolecular Synthon Approach with Experimental Structures, J. Am. Chem. Soc., 2005, 127, 10545–10559, DOI:10.1021/ja042738c.
- E. Nahua, E. Kolehmainen and M. Nissinen, Packing incentives and a reliable N–H⋯N–pyridine synthon in co-crystallization of bipyridines with two agrochemical actives, CrystEngComm, 2011, 13, 6531, 10.1039/C1CE05730H.
- K. Shivakumar, A. Vidyasagar, A. Naidu, R. G. Gonnade and K. M. Sureshan, Strength from weakness: The role of CH…N hydrogen bond in the formation of wave-like topology in crystals of aza-heterocycles, CrystEngComm, 2012, 14, 519–524, 10.1039/C1CE05997A.
- J. D. Dunitz and A. Gavezzotti, Supramolecular Synthons: Validation and Ranking of Intermolecular Interaction Energies, Cryst. Growth Des., 2012, 12, 5873–5877, DOI:10.1021/cg301293r.
- E. Bosch, E. Speetzen and N. P. Bowling, Halogen-Bonded Supramolecular Parallelograms: From Self-Complementary Iodoalkyne Halogen-Bonded Dimers to 1:1 and 2:2 Iodoalkyne Halogen-Bonded Cocrystals, Cryst. Growth Des., 2024, 24(4), 1674–1681, DOI:10.1021/acs.cgd.3c01325.
- C. Esterhuysen, Hydrogen vs Halogen-Bonded R22(8) Rings in Organic Crystal Structures, Cryst. Growth Des., 2024, 24, 859–870, DOI:10.1021/acs.cgd.3c01343.
- D. Ejarque, T. Calvet, M. Font-Bardia and J. Pons, Structural Landscape of α-Acetamidocinnamic Acid Cocrystals with Bipyridine-Based Coformers: Influence of Crystal Packing on Their Thermal and Photophysical Properties, Cryst. Growth Des., 2024, 24(4), 1746–1765, DOI:10.1021/acs.cgd.3c01374.
- J. D. Dunitz and A. Gavezzotti, How molecules stick together in organic crystals: weak intermolecular interactions, Chem. Soc. Rev., 2009, 38, 2622–2633, 10.1039/B822963P.
- J. P. M. Lommerse, A. J. Stone, R. Taylor and F. H. Allen, The Nature and Geometry of Intermolecular Interactions between Halogens and Oxygen or Nitrogen, J. Am. Chem. Soc., 1996, 118, 3108–3116, DOI:10.1021/ja953281x.
- M. A. P. Martins, C. P. Frizzo, A. C. L. Martins, A. Z. Tier, I. M. Gindri, A. R. Meyer, H. G. Bonacorso and N. Zanatta, Energetic and Topological Approach for Characterization of Supramolecular Clusters in Organic Crystals, RSC Adv., 2014, 4, 44337–44349, 10.1039/c4ra06040g.
- C. P. Frizzo, A. Z. Tier, I. M. Gindri, A. R. Meyer, G. Black, A. L. Belladona and M. A. P. Martins, Thermodynamic, energetic, and topological properties of crystal packing of pyrazolo[1,5-a]pyrimidines governed by weak electrostatic intermolecular interactions, CrystEngComm, 2015, 17, 4325–4333, 10.1039/c5ce00613a.
- M. A. P. Martins, M. Hoerner, J. Beck, A. Z. Tier, A. L. Belladona, A. R. Meyer, N. Zanatta, H. G. Bonacorso and C. P. Frizzo, Polymorphism in an 18-membered macrocycle: an energetic and topological approach to understand the supramolecular structure, CrystEngComm, 2016, 18, 3866–3876, 10.1039/c5ce02123e.
- G. C. Zimmer, A. B. Pagliari, C. R. Bender, P. R. S. Salbego, T. Orlando, M. Hörner, N. Zanatta, H. G. Bonacorso and M. A. P. Martins, Insights on conformation in the solid state: a case study – s-cis and/or s-trans crystallization of 5(3)-aryl-3(5)-carboxyethyl-1-tert-butylpyrazoles, CrystEngComm, 2018, 20, 5154–5168, 10.1039/c8ce00984h.
- P. R. S. Salbego, C. R. Bender, M. Hörner, N. Zanatta, C. P. Frizzo, H. G. Bonacorso and M. A. P. Martins, Insights on the Similarity of Supramolecular Structures in Organic Crystals Using Quantitative Indexes, ACS Omega, 2018, 3(3), 2569–2578, DOI:10.1021/acsomega.7b02057.
- M. A. P. Martins, P. R. S. Salbego, G. A. Moraes, C. R. Bender, P. J. Zambiazi, T. Orlando, A. B. Pagliari, C. P. Frizzo and M. Hoerner, Understanding the crystalline formation of triazene N-oxides and the role of halogen⋯π interactions, CrystEngComm, 2018, 20, 96–112, 10.1039/c7ce02015e.
- P. R. S. Salbego, C. R. Bender, T. Orlando, G. A. Moraes, J. P. P. Copetti, G. H. Weimer, H. G. Bonacorso, N. Zanatta, M. Hoerner and M. A. P. Martins, Supramolecular Similarity in Polymorphs: Use of Similarity Indices (IX), ACS Omega, 2019, 4, 9697–9709, DOI:10.1021/acsomega.8b03660.
- T. Orlando, P. R. S. Salbego, F. F. S. Farias, G. H. Weimer, J. P. P. Copetti, H. G. Bonacorso, N. Zanatta, M. Hoerner, J. Berná and M. A. P. Martins, Crystallization Mechanisms Applied To Understand The Crystal Formation of Rotaxanes, Eur. J. Org. Chem., 2019, 3451–3463, DOI:10.1002/ejoc.201801870.
- T. Orlando, P. R. S. Salbego, C. L. R. Taschetto, H. G. Bonacorso, N. Zanatta, M. Horner and M. A. P. Martins, Polymorphism in A Rotaxane Molecule: Intra- And Intermolecular Understanding, Cryst. Growth Des., 2019, 19, 1021–1030, DOI:10.1021/acs.cgd.8b01560.
- G. C. Zimmer, A. B. Pagliari, V. B. Solner, M. Hoerner, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Packing and Conformational Polymorphism in 1,2-Bis(aminocarbonyl(1-tert-butyl 1H pyrazol-(3)5-yl))ethanes: Illuminating Examples of Highly Flexible Molecules, Cryst. Growth Des., 2021, 21(8), 4690–4706, DOI:10.1021/acs.cgd.1c00541.
- T. Orlando, L. C. Lopes, D. A. M. Neumann, V. P. Andrade, M. Mittersteiner, C. Q. Rocha, N. Zanatta, H. G. Bonacorso, M. A. P. Martins and P. R. S. Salbego, Uncovering the origins of supramolecular similarity in a series of benzimidazole structures, CrystEngComm, 2022, 24, 6600–6610, 10.1039/D2CE00909A.
- G. H. Weimer, J. P. P. Copetti, T. Orlando, N. Zanatta, H. G. Bonacorso, P. R. S. Salbego and M. A. P. Martins, The role of attractive and repulsive interactions in the stabilization of ammonium salts structures, CrystEngComm, 2022, 24, 7039–7048, 10.1039/D2CE00891B.
- A. B. Pagliari, J. M. L. Rosa, P. S. V. Lima, G. C. Zimmer, M. E. C. da Silva, E. G. de Oliveira, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Polymorphism in N-(5-methylisoxazol-3-yl) malonamide: understanding the supramolecular structure and the crystallization mechanism, CrystEngComm, 2023, 25, 5118–5132, 10.1039/d3ce00643c.
- J. M. L. Rosa, P. S. V. Lima, H. G. Bonacorso, N. Zanatta and M. A. P. Martins, Ammonium carboxylate salts: the additivity of intermolecular interaction energies in charged organic compounds, CrystEngComm, 2024, 26, 796–808, 10.1039/d3ce01083j.
- F. F. S. Farias, G. H. Weimer, S. K. Kunz, R. R. Prates, T. Orlando and M. A. P. Martins, [2]Rotaxane Solvates: Insights into the Presence of Solvents in the Crystal Lattice, Cryst. Growth Des., 2024, 24, 4030–4044, DOI:10.1021/acs.cgd.3c01179.
- J. E. Lennard-Jones, Cohesion, Proc. Phys. Soc., 1931, 43, 461–482, DOI:10.1088/0959-5309/43/5/301.
- M. A. Spackman, J. J. McKinnon and D. Jayatilaka, Electrostatic potentials mapped on Hirshfeld surfaces provide direct insight into intermolecular interactions in crystals, CrystEngComm, 2008, 10, 377–388, 10.1039/b715227b.
- M. A. Spackman and D. Jayatilaka, Hirshfeld surface analysis, CrystEngComm, 2009, 11, 19–32, 10.1039/B818330A.
- K. Popper, The Logic Of Scientific Discovery, Routledge, Taylor & Francis, 2002 Search PubMed.
- A. I. Kitaigorodskii, Molecular Crystals and Molecules, Academic Press, New York, 1973 Search PubMed.
- W. Fischer and E. Koch, Geometrical packing analysis of molecular compounds, Z. Kristallogr. - Cryst. Mater., 1979, 150, 245–260, DOI:10.1524/zkri.1979.150.14.245.
- O. V. Shishkin, R. I. Zubatyuk, A. V. Maleev and R. Boese, Investigation of topology of intermolecular interactions in the benzene–acetylene co-crystal by different theoretical methods, Struct. Chem., 2014, 25, 1547–1552, DOI:10.1007/s11224-014-0413-7.
- I. A. Baburin and V. A. Blatov, Sizes of molecules in organic crystals: the Voronoi-Dirichlet approach, Acta Crystallogr., Sect. B: Struct. Sci., 2004, 60, 447–452, DOI:10.1107/S0108768104012698.
- V. A. Blatov, Voronoi–dirichlet polyhedra in crystal chemistry: theory and applications, Crystallogr. Rev., 2004, 10, 249–318, DOI:10.1080/08893110412331323170.
- T. G. Mitina and V. A. Blatov, Topology of 2-Periodic Coordination Networks: Toward Expert Systems in Crystal Design, Cryst. Growth Des., 2013, 13, 1655–1664, DOI:10.1021/cg301873m.
- E. J. Corey, The Logic of Chemical Synthesis: Multistep Synthesis of Complex Carbogenic Molecules (Nobel Lecture), Angew. Chem., Int. Ed. Engl., 1991, 30(5), 455–465, DOI:10.1002/anie.199104553.
| This journal is © The Royal Society of Chemistry 2026 |