
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of 2,7-dioctylbiphenylenodithiophenes for use as organic semiconducting materials in solution-processed organic thin-film transistors
Makoto
Watanabe
 *a,
Masato
Miyashita
a,
Takashi
Fukuda
b and
Shinya
Oku
a
*a,
Masato
Miyashita
a,
Takashi
Fukuda
b and
Shinya
Oku
a
aAdvanced Materials Research Laboratory, Tosoh Corporation, 2743-1 Hayakawa, Ayase-shi, Kanagawa 252-1123, Japan. E-mail: makoto-watanabe-zr@tosoh.co.jp
bResearch and Development Planning, Tosoh Corporation, 2-2-1 Yaesu, Tyuou-ku, Tokyo 104-8467, Japan
First published on 9th March 2026
Abstract
A synthetic route to 2,7-dialkylbiphenylenodithiophenes was developed and their potential as a new π-core system for organic semiconductors was investigated. The semiconductors 2,7-dioctylbiphenyleno[2,1-b:6,5-b′]dithiophene (C8-B1-BPDT), 2,7-dioctylbiphenyleno[1,2-b:5,6-b′]dithiophene (C8-B2-BPDT), and 2,7-dioctylbiphenyleno[2,3-b:6,7-b′]dithiophene (C8-L-BPDT) were synthesized from dihalobiphenylenes. Structural analysis by single-crystal X-ray crystallography revealed that C8-B1-BPDT adopts a herringbone motif whereas C8-B2-BPDT has an interdigitated structure. C8-B1-BPDT and C8-B2-BPDT displayed high solubilities of more than 0.80 wt% in toluene. In contrast, C8-L-BPDT exhibited a low solubility of 0.060 wt% in toluene at room temperature. Although C8-B2-BPDT formed noncontinuous films on a parylene-C dielectric layer by solution deposition, films of C8-B1-BPDT covered the whole area of the parylene-C layer. Bottom-contact bottom-gate thin-film transistors with a mobility of 0.58 cm2 V−1 s−1 were fabricated by drop casting of a solution of C8-B1-BPDT.
Introduction
Fused benzothiophene compounds have attracted considerable attention over the past few decades because of their use as semiconductor materials in organic thin-film transistors (OTFTs).1–12 Solution-processable OTFT materials are attractive for healthcare monitoring systems because they enable the use of printing technologies such as inkjet printing to fabricate the organic semiconducting layer of OTFTs.13,14 However, only a limited number of fused rigid benzothiophenes have yielded thin films with high mobility by solution deposition under ambient conditions.2,7,9 These materials also require high thermal stability to withstand the heat during the device manufacturing process. Producing low-molecular-weight fused benzothiophene organic semiconductors with both high thermal stability and high solubility is still challenging.Biphenylene is a fused rigid compound containing a four-membered ring and two benzene rings.15 Biphenylene compounds are expected to have higher solubility than the corresponding anthracene compounds because of the smaller ring of the former compared with that of the latter. Therefore, here we focused on biphenylenodithiophene, which consists of a biphenylene and two thiophene rings, as a new π-core system.
Compounds with a biphenylene unit have not been used as semiconductor active layers for OTFTs to date. Only a few thiophene-fused biphenylenes have been reported and their synthetic methods are quite limited.16,17 It is desirable to produce a wide range of biphenylenodithiophenes as potential semiconductor materials for OTFTs. Here we report the synthesis of three isomers of 2,7-dioctylbiphenylenodithiophene with centrosymmetry and their use in solution-processed OTFTs.
Results and discussion
Synthesis of biphenylenodithiophenes
The chemical structures of the three isomers are shown in Chart 1. The biphenylenodithiophene cores are denoted as BPDT with B- and L-indicating bent and linear cores, respectively. The selective syntheses of C8-B1-BPDT, C8-B2-BPDT, and C8-L-BPDT are shown in Scheme 1.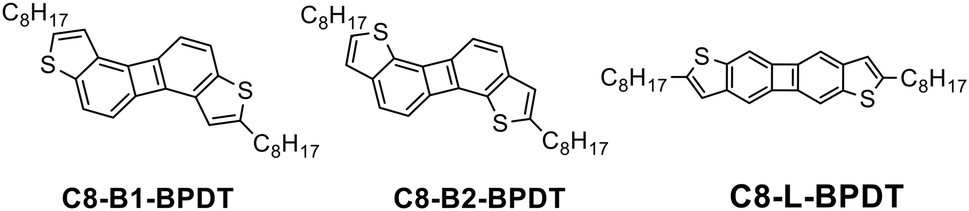 | ||
| Chart 1 Molecular structures of three biphenylenodithiophene isomers. | ||
 | ||
| Scheme 1 Syntheses of C8-B1-BPDT, C8-B2-BPDT, and C8-L-BPDT. | ||
The synthetic methodology was first planned to consist of the syntheses of 2,6-difluorobiphenylene (4) and 1,5-dichlorobiphenylene (10), regioselective dihalogenation, chemoselective Sonogashira coupling reactions at the iodo or bromo sites, and ring closure with sodium sulfide. Biphenylene 4 was prepared by Negishi coupling of 1-bromo-4-fluorophenyl-2-zinc chloride derived from 1 with 2-bromo-4-fluoro-1-iodobenzene (2) to give 3, followed by dilithiation of 3 with BuLi and CuCl2-mediated oxidative cyclization to give 4.184 is the intermediate for the synthesis of C8-B1-BPDT and C8-L-BPDT. We first carried out LDA19-mediated dilithiation of 4 followed by diiodination with iodine to afford a mixture of 2,6-difluoro-1,5-diiodobiphenylene (5) and an isomer 6 in a percentage ratio of 88% to 12%. It was found that the percentage ratio of 5 to 6 increased to 97% to 3% when lithium 2,2,6,6-tetramethylpiperidine (LTMP) was used instead of LDA. After purification by column chromatography and recrystallization, 5 was isolated. The symmetrical structure of 5 was confirmed by nuclear magnetic resonance (NMR) analyses (Fig. S15.5, SI). Sonogashira coupling of 5 with 1-decyne afforded 7, which was cyclized with sodium sulfide20,21 under mild reaction conditions to give 2,7-dioctylbiphenyleno[2,1-b:6,5-b′]dithiophene (C8-B1-BPDT) (Scheme 1).
The synthesis of the bent isomer 2,7-dioctylbiphenyleno[1,2-b:5,6-b′]dithiophene (C8-B2-BPDT) was also attempted by the same method starting with 10, which was prepared by lithium-mediated homocoupling22 of 8via an aryne species followed by the lithium-mediated intramolecular cyclization23 of 9 and quenching with hexachloroethane. Unfortunately, lithiation of 10 by lithium amide bases such as LDA followed by iodination gave only 1,5-dichloro-2-iodobiphenylene, and further lithiation and iodination resulted in the undesired formation of 1,5-difluoro-2,7-diiodobiphenylene. Therefore, a different route to synthesize C8-B2-BPDT was examined. In this alternative route, 10 was converted to 1,5-bis(2,2-dimethoxyethylthio)bipheneylene (11) in the presence of sodium sulfide and chloroacetaldehyde dimethyl acetal. Next, 11 was subjected to polyphosphoric acid-mediated cyclization24 to give unsubstituted biphenyleno[1,2-b:5,6-b′]dithiophene (12). Dilithiation by BuLi at the 2- and 7-positions of the thiophene rings and bromination using 1,2-dibromotetrachloroethane afforded 2,7-dibromo compound 13. Finally, C8-B2-BPDT was obtained by palladium-catalyzed coupling of 13 with octylzinc chloride.
The linear isomer C8-L-BPDT was synthesized following a similar process to that used to obtain C8-B1-BPDT. Electrophilic bromination of 4 using NBS selectively gave 2,6-dibromo-3,7-difluorobiphenylene (14) in moderate yield. The symmetry of 14 was also confirmed by NMR analysis (Fig. S15.23, SI). Sonogashira coupling of 14 with 1-decyne and cyclization using sodium sulfide gave 2,7-dioctylbiphenyleno[2,3-b:6,7-b′]dithiophene (C8-L-BPDT).
All the new materials were fully characterized by spectroscopic analyses including NMR and mass spectrometry (MS). Furthermore, single-crystal X-ray analyses of C8-B1-BPDT and C8-B2-BPDT confirmed the structure of the dialkylbiphenylenodithiophene framework (vide infra).
Properties of biphenylenodithiophenes
The solubilities of bent C8-B1-BPDT and C8-B2-BPDT in toluene were as high as 1.6 and 0.80 wt%, respectively, at ambient temperature. Thus, drop casting of these solutions was able to be carried out under ambient conditions. High solubility is one of the requisite factors to realize printed electronics. Printing techniques such as inkjet printing are usually conducted under ambient conditions. A solution of C8-B1-BPDT in toluene formed a continuous film on a parylene-C layer by drop casting, whereas a crystalline precipitate with a dendrite-like morphology was obtained by drop casting of a solution of C8-B2-BPDT. Drop casting of a toluene solution of the linear isomer C8-L-BPDT resulted in precipitation of solid C8-L-BPDT because its solubility in toluene was only 0.060 wt% under ambient conditions. Thus, a thin film of C8-L-BPDT was fabricated by vacuum deposition. Fig. S1 (SI) exhibits laser microscope images of thin films of C8-B1-BPDT and C8-B2-BPDT obtained by drop casting a toluene solution on the surface of a parylene-C layer insulator film. The thin film morphology was also examined by scanning electron microscope (SEM) (Fig. S3 and S5, SI) and atomic force microscope (AFM) (Fig. S4 and S5, SI).Ultraviolet–visible (UV–vis) spectra of C8-B1-BPDT in dichloromethane solution and a thin film are presented in Fig. 1. UV–vis spectra of C8-B2-BPDT and C8-L1-BPDT in dichloromethane are shown in Fig. S6 (SI). The optical highest occupied molecular orbital (HOMO)–lowest unoccupied molecular orbital (LUMO) gap of the thin film of C8-B1-BPDT estimated from the absorption edge (λedge = 427 nm) was about 2.9 eV.
 | ||
| Fig. 1 UV–vis spectra of C8-B1-BPDT in dichloromethane solution and a thin film. | ||
The ionization potentials (IPs) of the biphenylenodithiophenes were estimated using photoelectron spectroscopy in air. Estimated IPs of thin films of C8-B1-BPDT and C8-L-BPDT were 5.2 and 5.7 eV, respectively (Fig. S7, SI). It is assumed that the smaller IP for C8-B1-BPDT than that of C8-L-BPDT may imply that the former has a more closely packed molecular arrangement than the latter, arising from molecular packing and intermolecular interactions.25 Judging from its HOMO–LUMO gap and IP, C8-B1-BPDT is expected to be electrochemically stable (Fig. S12, SI).
Differential scanning calorimetry (DSC) measurements revealed that C8-B1-BPDT had a melting point of 140 °C without any phase rearrangement (Fig. 2), whereas C8-B2-BPDT had a relatively low melting point of 107 °C (Fig. S8.1, SI) and linear C8-L-BPDT had a high melting point of 285 °C without any phase rearrangement (Fig. S8.2, SI). Thus, C8-B1-BPDT was found to have both high solubility in toluene and a high melting point.
 | ||
| Fig. 2 DSC curves of C8-B1-BPDT. The scan rate was 10 °C min−1. | ||
Fig. 3 and S9.1 and S9.2 (SI) show out-of-plane X-ray diffraction (XRD) patterns of drop-cast thin films of C8-B1-BPDT and C8-B2-BPDT and a vacuum-deposited thin film of C8-L-BPDT on parylene-C fabricated at ambient temperature, respectively. C8-B1-BPDT adopted a layered structure and exhibited a series of peaks assignable to multiple (00l) diffractions, meaning that the molecules were aligned perpendicular to the substrate. The interlayer distance (d-spacing) was calculated to be 26.6 Å from these reflections, which is almost half the crystallographic c-axis of its bulk single crystal and its molecular length (vide infra). In contrast, the diffraction pattern of C8-B2-BPDT displayed only a weak peak corresponding to 10.0 Å, meaning that the molecules were not oriented perpendicular to the substrate. The C8-L-BPDT thin film displayed weak peaks assignable to (001) and (004) diffractions and the interlayer distance (d-spacing) was calculated to be 31.8 Å.
 | ||
| Fig. 3 Out-of-plane XRD profile of a drop-cast thin film of C8-B1-BPDT. | ||
In-plane reciprocal lattice mapping measurements were carried out for the drop-cast thin film of C8-B1-BPDT (Fig. 4a). The XRD profiles collected by scanning the φ angle (0–360°) were integrated and converted to a linear 2θχ scan pattern (Fig. 4b). The peak at 16.2° (d-spacing of 5.5 Å) was assigned to the (200) diffraction of the single-crystal lattice, which is almost half the crystallographic a-axis (10.9 Å) of its bulk single crystal (vide infra).
 | ||
| Fig. 4 In-plane XRD analysis of a drop cast thin film of C8-B1-BPDT (incident angle, 0.20°). (a) In-plane reciprocal lattice mapping and (b) in-plane XRD profile. | ||
Single crystals suitable for X-ray structural analysis were obtained for C8-B1-BPDT and C8-B2-BPDT. The single crystal of C8-B1-BPDT belonged to the monoclinic I2/a space group with two molecules along the c-axis in the unit cell. The molecules were arranged with herringbone packing, which enables two-dimensional carrier transport, and the alkyl moieties were located above and below the core plane (Fig. 5). In contrast, C8-B2-BPDT crystallized in the monoclinic P1 space group. The XRD analysis unveiled an interdigitated arrangement, which is unfavorable for carrier transport, and the alkyl moieties were located on the same plane as the core (Fig. S10, SI). Crystallographic parameters were summarized in Table S1 (SI).
 | ||
| Fig. 5 Crystal structure of C8-B1-BPDT. (a) b-Axis projection showing a layer-by-layer arrangement and (b) c-axis projection. | ||
OTFT devices with biphenylenodithiophenes
Bottom-contact bottom-gate OTFT devices were fabricated using a solution process to form semiconducting layers of C8-B1-BPDT and C8-B2-BPDT. Silver was thermally deposited as the gate electrodes on a glass substrate, and then a parylene-C dielectric layer was deposited by chemical vapor deposition. Gold was vacuum deposited through a shadow mask as the source–drain electrodes, which were treated with PFBT to give a channel length and width of 100 and 500 μm, respectively. Finally, a toluene solution of organic semiconductor material was drop cast to complete fabrication of OTFT devices. C8-B1-BPDT covered the source–drain electrodes to give a crystalline thin film, whereas C8-B2-BPDT formed a noncontinuous film that partially covered the source–drain electrodes (Fig. S1, SI). Because of its poor solubility, C8-L-BPDT was vacuum deposited on the source–drain electrodes at ambient substrate temperature. These devices were evaluated under ambient conditions and showed typical p-channel responses. The field-effect mobilities were extracted from the saturation regime. The characteristics of the OTFTs are summarized in Table 1. C8-B1-BPDT-based devices exhibited a mobility of 0.21 cm2 V−1 s−1 (Fig. S13, SI). Their mobility was increased up to 0.58 cm2 V−1 s−1 with an Ion/Ioff ratio of 4.6 × 107 using a polystyrene (PS) blend formulation (Fig. 6). It is assumed that vertical phase separation26 between the organic semiconductor and insulating polymer gave rise to this mobility increase. That is, PS could fill voids in the interface between C8-B1-BPDT, the dielectric and the source–drain electrodes in a bottom-gate bottom-contact device and afford smoother underlayer to C8-B1-BPDT than the absence of PS. In the presence of PS, interface trap density (Dit) values were found to decrease (Table S2, SI). The morphology of a drop-cast thin film of C8-B1-BPDT with PS was observed by SEM and AFM (Fig. S5, SI).| Compound | Deposition method | Mobilitya (cm2 V−1 s−1) | I on/Ioff | V th (V) |
|---|---|---|---|---|
a Extracted from the saturation regime (VDS = −20 V).
b 0.2 wt% of the compound in toluene was used.
c 0.05 wt% of polystyrene (MW = 280![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) was included.
d The substrate was maintained at room temperature.
e 0.4 wt% of the compound in toluene was spin-coated at 2000 rpm for 40 s. 000) was included.
d The substrate was maintained at room temperature.
e 0.4 wt% of the compound in toluene was spin-coated at 2000 rpm for 40 s.
|
||||
| C8-B1-BPDT | Drop castingb | 0.21 | 1.0 × 10−7 | −1.0 |
| C8-B1-BPDT | Drop castingb | 0.58 | 4.6 × 10−7 | −2.9 |
| C8-B2-BPDT | Drop castingb | 0.0002 | 1.0 × 10−3 | −0.9 |
| C8-L-BPDT | Vacuum depositiond | 0.004 | 1.0 × 10−4 | −10.3 |
| C8-B1-BPDT | Spin coatinge | 0.073 | 1.0 × 10−5 | −1.2 |
| C8-B1-BPDT | Spin coatinge | 0.12 | 2.4 × 10−5 | −2.6 |
 | ||
| Fig. 6 Typical characteristics of OTFTs with drop-cast C8-B1-BPDT layers: output characteristics (left) and transfer characteristics at Vd = −20 V (right). | ||
Structurally comparable NDT3,27 and ADT5,28–33 compounds without a triethylsilylethynyl moiety did not show field-effect activity after solution deposition except for linear dihexylADT34 and bent monosubstituted ADT compounds.35 The bent isomer C8-B2-BPDT and linear C8-L-BPDT showed lower mobilities of 10−4 and 10−3 cm2 V−1 s−1, respectively (Fig. S11, SI). Spin coating methods were also examined for those three compounds. A solution of C8-B1-BPDT gave mobilities of 0.073 cm2 V−1 s−1 without PS and 0.12 cm2 V−1 s−1 with PS, respectively (Table 1 and Fig. S14, SI). On the other hand, continuous films that covered the source–drain electrodes were obtained from C8-B2-BPDT and C8-L-BPDT but both showed no transistor performance with or without PS. Fig. S2 (SI) exhibits a laser microscope image of thin films of these three compounds with PS obtained by spin coating.
Conclusions
A series of dialkylbiphenylenodithiophene compounds was synthesized and their use as organic semiconductor materials in OTFTs was explored. The straightforward synthetic route enabled the synthesis of a variety of bent and linear isomers of biphenylenodithiophene with different substituents. The bent core systems with biphenyleno[2,1-b:6,5-b′]dithiophene and biphenyleno[1,2-b:5,6-b′]dithiophene exhibited high solubility in toluene at ambient temperature, making them promising core systems for device fabrication using printing technology. In particular, C8-B1-BPDT formed continuous films by drop casting that showed a carrier mobility as high as 0.58 cm2 V−1 s−1. We will investigate this core system with different substituents as semiconductor materials for OTFTs in the future.Experimental
Synthesis of compounds
:heptane (2:1) to afford 2,6-difluoro-1,5-diiodobiphenylene (5, 2.30 g, 5.22 mmol, 69%) as a pale yellow solid. Mp 197.0–198.5 °C; 1H NMR (400 MHz, CDCl3): δ 6.43 (dd, J = 9.6 Hz, J = 7.3 Hz, 2H), 6.68 (dd, J = 7.2 Hz, J = 4.0 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 71.70 (d, J = 31.1 Hz, Cquat), 113.26 (d, J = 26.8 Hz, CH), 117.41 (d, J = 7.9 Hz, CH), 144.98 (Cquat), 156.57 (d, J = 1.8 Hz, Cquat), 162.24 (d, J = 249.1 Hz, Cquat).
:ethyl acetate = 20:1) to afford 1,5-bis((2,2-dimethoxyethy)thio)biphenylene (11) (886 mg, 2.26 mmol, 71%) as an amber viscous oil; 1H NMR (400 MHz, CDCl3): δ 3.10 (d, J = 5.8 Hz, 4H), 3.38 (s, 12H), 4.53 (t, J = 5.6 Hz, 2H), 6.54 (dd, J = 5.8 Hz, J = 1.8 Hz, 2H), 6.66–6.74 (m, 4H); 13C NMR (100 MHz, CDCl3): δ 35.37, 53.91, 103.29, 115.95, 126.65, 129.53, 148.64, 150.76.
:heptane (1:1) to afford 2,6-dibromo-3,7-difluorobiphenylene (14, 353 mg, 1.02 mmol, 61%) as pale yellow needles. Mp 230.1–231.0 °C; 1H NMR (400 MHz, CDCl3): δ 6.49 (d, J = 6.5 Hz, 2H), 6.82 (d, J = 5.7 Hz, 2H); 13C NMR (100 MHz, CDCl3): δ 107.42 (d, J = 24.1 Hz, Cquat), 109.31 (d, J = 28.1 Hz, CH), 123.44 (CH), 144.12 (Cquat), 150.33 (d, J = 6.6 Hz, Cquat), 159.94 (d, J = 250.0 Hz, Cquat).
Characterization of compounds
| Id = (WCi/2L)μFET(Vg − Vth)2, | (1) |
Interface trap density (Dit) was estimated from the values of subthreshold swing (SS) in the transfer characteristics using the following equation:36
| SS = (kT/q)ln10(1 + q2Dit/Ci) | (2) |
Author contributions
M. Watanabe: synthesis and characterization of synthetic products, writing the manuscript. M. Miyashita: synthesis and characterization of synthetic products. T. Fukuda and S. Oku: OTFT device fabrication, electric data analysis.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI).Supplementary information including NMR spectra of products is available. See DOI: https://doi.org/10.1039/d5ce01050k.
CCDC 2477388 (C8-B1-BPDT) and 2477349 (C8-B2-BPDT) contain the supplementary crystallographic data for this paper.37a,b
Acknowledgements
We thank Dr R. Tanaka (Sagami Chemical Research Institute) for measurement and analysis of single-crystal X-ray crystallography data.Notes and references
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- H. Ebata, T. Izawa, E. Miyazaki, K. Takimiya, M. Ikeda, H. Kuwabara and T. Yui, J. Am. Chem. Soc., 2007, 129, 15732–15733 CrossRef CAS PubMed.
- S. Shinamura, I. Osaka, E. Miyazaki, A. Nakao, M. Yamagishi, J. Takeya and K. Takimiya, J. Am. Chem. Soc., 2011, 133, 5024–5035 CrossRef CAS PubMed.
- K. Takimiya, S. Shinamura, I. Osaka and E. Miyazaki, Adv. Mater., 2011, 23, 4347–4370 CrossRef CAS PubMed.
- M. Nakano, K. Niimi, E. Miyazaki, I. Osaka and K. Takimiya, J. Org. Chem., 2012, 77, 8099–8111 CrossRef CAS PubMed.
- P. Gao, D. Beckmann, H. N. Tsao, X. Feng, V. Enkelmann, M. Baumgarten, W. Pisula and K. Müllen, Adv. Mater., 2009, 21, 213–216 CrossRef CAS.
- T. Okamoto, C. Mitsui, M. Yamagishi, K. Nakahara, J. Soeda, Y. Hirose, K. Miwa, H. Sato, A. Yamano, T. Matsushita, T. Uemura and J. Takeya, Adv. Mater., 2013, 25, 6392–6397 CrossRef CAS PubMed.
- C. Mitsui, T. Okamoto, M. Yamagishi, J. Tsurumi, K. Yoshimoto, K. Nakahara, J. Soeda, Y. Hirose, H. Sato, A. Yamano, T. Uemura and J. Takeya, Adv. Mater., 2014, 26, 4546–4551 CrossRef CAS PubMed.
- K. Fukuda, T. Minamiki, T. Minami, M. Watanabe, T. Fukuda, D. Kumaki and S. Tokito, Adv. Electron. Mater., 2015, 1, 1400052 CrossRef.
- H. Iino, T. Usui and J. Hanna, Nat. Commun., 2015, 6, 6828 CrossRef CAS PubMed.
- S. Inoue, T. Higashino, S. Arai, R. Kumai, H. Matsui, S. Tsuzuki, S. Horiuchi and T. Hasegawa, Chem. Sci., 2020, 11, 12493–12505 RSC.
- T. Higashino, S. Inoue, S. Arai, H. Matsui, N. Toda, S. Horiuchi, R. Azumi and T. Hasegawa, Chem. Mater., 2021, 33, 7379–7385 CrossRef CAS.
- S. Allard, M. Forster, B. Souharce, H. Thiem and U. Scherf, Angew. Chem., Int. Ed., 2008, 47, 4070–4098 CrossRef CAS PubMed.
- N. S. Yusof, M. F. P. Mohamed, N. A. Ghazali, M. F. A. J. Khan, S. Shaari and M. N. Mohtar, Alexandria Eng. J., 2022, 61, 11405–11431 CrossRef.
- H. Takano, T. Ito, K. S. Kanyiva and T. Shibata, Eur. J. Org. Chem., 2019, 2871–2883 CrossRef CAS.
- D. N. Nicolaides, Synthesis, 1977, 127–129 CrossRef CAS.
- A. Fukazawa, H. Oshima, Y. Shiota, S. Takahashi, K. Yoshizawa and S. Yamaguchi, J. Am. Chem. Soc., 2013, 135, 1731–1734 CrossRef CAS PubMed.
- V. G. Wittig and G. Klar, Liebigs Ann. Chem., 1967, 704, 91–108 CrossRef.
- P. I. Dosa, Z. Gu, D. Hager, W. L. Karney and K. P. C. Vollhardt, Chem. Commun., 2009, 1967–1969 RSC.
- T. Kashiki, S. Shinamura, M. Kohara, E. Miyazaki, K. Takimiya, M. Ikeda and H. Kuwabara, Org. Lett., 2009, 11, 2473–2475 CrossRef CAS PubMed.
- Y. Li, L. Cheng, B. Li, S. Jiang, L. Chen and Y. Shao, ChemistrySelect, 2016, 1, 1092–1095 CrossRef CAS.
- F. R. Leroux, L. Bonnafoux, C. Heiss, F. Colobert and D. A. Lanfranchi, Adv. Synth. Catal., 2007, 349, 2705–2713 CrossRef CAS.
- S. L. Wang, M. L. Pan, W. S. Su and Y. T. Wu, Angew. Chem., Int. Ed., 2017, 56, 14694–14697 CrossRef CAS PubMed.
- P. A. Ple and L. Marnett, J. Heterocyclic Chem., 1988, 25, 1271–1272 CrossRef CAS.
- H. Y. Chen, G. Schweicher, M. Planells, S. M. Ryno, K. Boch, A. J. P. White, D. Simatos, M. Little, C. Jellett, S. J. Cryer, A. Marks, M. Hurhangee, J. L. Bredas, H. Sirringhaus and I. McCulloch, Chem. Mater., 2018, 30, 7587–7592 CrossRef CAS.
- J. Smith, R. Hamilton, I. McCulloch, N. Stingelin-Stutzmann, M. Heeney, D. D. C. Bradley and T. D. Anthopoulos, J. Mater. Chem., 2010, 20, 2562–2574 RSC.
- S. Shinamura, E. Miyazaki and K. Takimiya, J. Org. Chem., 2010, 75, 1228–1234 CrossRef CAS PubMed.
- A. Pietrangelo, M. J. MacLachlan, M. O. Wolf and B. O. Patrick, Org. Lett., 2007, 9, 3571–3573 CrossRef CAS PubMed.
- A. Pietrangelo, B. O. Patrick, M. J. MacLachlan and M. O. Wolf, J. Org. Chem., 2009, 74, 4918–4926 CrossRef CAS PubMed.
- B. Tylleman, C. M. L. V. Velde, J. Y. Balandier, S. Stas, S. Sergeyev and Y. H. Geerts, Org. Lett., 2011, 13, 5208–5211 CrossRef CAS PubMed.
- M. Mamada, T. Minamiki, H. Katagiri and S. Tokito, Org. Lett., 2012, 14, 4062–4065 CrossRef CAS PubMed.
- Y. Yi, L. Zhu and J. L. Brédas, J. Phys. Chem. C, 2012, 116, 5215–5224 CrossRef CAS.
- C. Quinton, M. Suzuki, Y. Kaneshige, Y. Tatenaka, C. Katagiri, Y. Yamaguchi, D. Kuzuhara, N. Aratani, K. Nakayama and H. Yamada, J. Mater. Chem. C, 2015, 3, 5995–6005 RSC.
- J. G. Laquindanum, H. E. Katz and A. J. Lovinger, J. Am. Chem. Soc., 1998, 120, 664–672 CrossRef CAS.
- W. L. Liau, T. H. Lee, J. T. Chen and C. S. Hsu, J. Mater. Chem. C, 2016, 4, 2284–2288 RSC.
- M. Yoon, K. R. Ko, S. W. Min and S. Im, RSC Adv., 2018, 8, 2837–2843 RSC.
- (a) CCDC 2477388: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p4xrj; (b) CCDC 2477349: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2p4wh7.
| This journal is © The Royal Society of Chemistry 2026 |