
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Toward dynamic crystal structure prediction: integrating thermodynamic and kinetic modeling
Prem K.
Reddy†
,
Anish V.
Dighe†
 ,
Rajan R.
Bhawnani
and
Meenesh R.
Singh
*
,
Rajan R.
Bhawnani
and
Meenesh R.
Singh
*
Department of Chemical Engineering, University of Illinois Chicago, 929 W Taylor St, Chicago, IL 60607, USA. E-mail: mrsingh@uic.edu; Tel: +1 (312) 413 7673
First published on 6th February 2026
Abstract
Precise control over the physical properties of organic molecular crystals is crucial for the design and manufacture of pharmaceutical compounds. Many organic molecules exhibit polymorphism, the ability to exist in multiple solid-state forms, each possessing distinct physical properties such as bioavailability, density, and dissolution rate. These variations are not merely academic; they can render a pharmaceutical compound ineffective or even hazardous if the undesired polymorph forms. Achieving reliable control over polymorphism therefore requires a fundamental understanding of the mechanisms of crystallization and molecular self-assembly spanning from the molecular to the macroscopic scale. Insights into self-assembly at multiple scales offer the opportunity to elucidate the dynamics of weak intermolecular interactions, advancing both crystal engineering and materials design. Existing computational approaches to crystal structure prediction (CSP) primarily focus on identifying dense, energy-minimized packings of molecules within rationally selected space groups. Although such thermodynamic methods can enumerate possible polymorphs, they often fail to consistently reproduce the experimentally observed crystal form under realistic kinetic and solvent conditions. This review aims to establish a conceptual framework for a multiscale model of crystallization, integrating both energetic and kinetic perspectives. By critically assessing the computational methods that model molecular self-assembly, nucleation, and growth, this work outlines a pathway toward dynamic crystal structure prediction, a unified approach capable of predicting not just feasible polymorphs but also the dominant crystal form that emerges under specific crystallization conditions.
 Prem K. Reddy | Prem K. Reddy received his PhD in Chemical Engineering from the University of Illinois Chicago, under the guidance of Dr. Meenesh Singh. His research focuses on exploring the crystallization of small molecules and metal–organic frameworks through theoretical approaches including molecular dynamics, population balance and microkinetic modeling. He is currently a postdoctoral associate at the Massachusetts Institute of Technology. |
 Anish V. Dighe | Anish V. Dighe is a Senior Scientist at Merck. He earned his PhD in Chemical Engineering from the University of Illinois Chicago, where he conducted his doctoral research under the supervision of Meenesh R. Singh. He subsequently completed postdoctoral training at the Massachusetts Institute of Technology. His research expertise lies in the mathematical modeling and simulation of crystallization processes for small molecules and metal–organic frameworks. |
 Rajan R. Bhawnani | Rajan R. Bhawnani received his Ph.D. in Chemical Engineering under the guidance of Dr. Meenesh R. Singh from the University of Illinois, Chicago (UIC) with a specialization in porous crystalline materials. He currently is a postdoctoral associate in the chemical engineering department at Massachusetts Institute of Technology (MIT) with Dr. Allan S. Myerson. |
 Meenesh R. Singh | Meenesh R. Singh is a Professor and Director of the Materials & Systems Engineering Lab at the University of Illinois Chicago. He also holds a Joint Appointment at Argonne National Laboratory and a visiting professorship at the Indian Institute of Technology Roorkee. He received his PhD in Chemical Engineering from Purdue University and then continued his career as a postdoctoral scholar at Lawrence Berkeley National Laboratory and at the University of California, Berkeley. His work is focused on all aspects of materials chemistry and electrochemical energy conversion and storage. |
1. Introduction
1.1. Crystals, characteristics, and properties
Self-assembly of atoms or molecules into periodic structures is central to the formation of a wide variety of inorganic and organic crystalline materials such as semiconductors, catalysts, pharmaceuticals, agrochemicals, biocrystals, adsorbents, inorganic–organic frameworks, food products, and specialty chemicals.1–6 The process of forming crystals begins with the self-assembly of molecules in solution to form small clusters, followed by layer-by-layer growth of clusters via self-assembly of molecules on their surface.7,8 These two stages of crystallization, namely nucleation and growth, are driven by the ordered self-assembly of molecules at different length scales, which collectively determine three primary characteristics – crystal structure (also referred to as polymorph or form), morphology (also referred to as shape), and size.5 The other characteristics such as defects, grain boundaries, compositional purity, and chirality, which arise from disordered self-assembly of atoms or molecules, can be important in fields ranging from electronic materials to pharmaceutical solids. The primary characteristics of crystalline materials can affect their physical properties. For example, the stability and bioavailability of pharmaceutical crystals are governed by the polymorph obtained; the activity and selectivity of catalytic reactions depend on the distribution of exposed facets on the catalyst; the band gap of semiconductor crystals varies with morphology and size; and downstream operations such as filtration, drying, and conveying of crystalline particles depend strongly on their aspect ratio. The relationship between crystallization conditions, crystal characteristics, and resulting physical properties is briefly depicted in Fig. 1. The relationship between crystal characteristics and physical properties can be determined from either theory (e.g., molecular simulations, density functional theory, or population balance modeling) or experiments (e.g., X-ray diffraction, atomic force microscopy, or spectroscopy) that are specific to the crystalline material and its application domain.9–12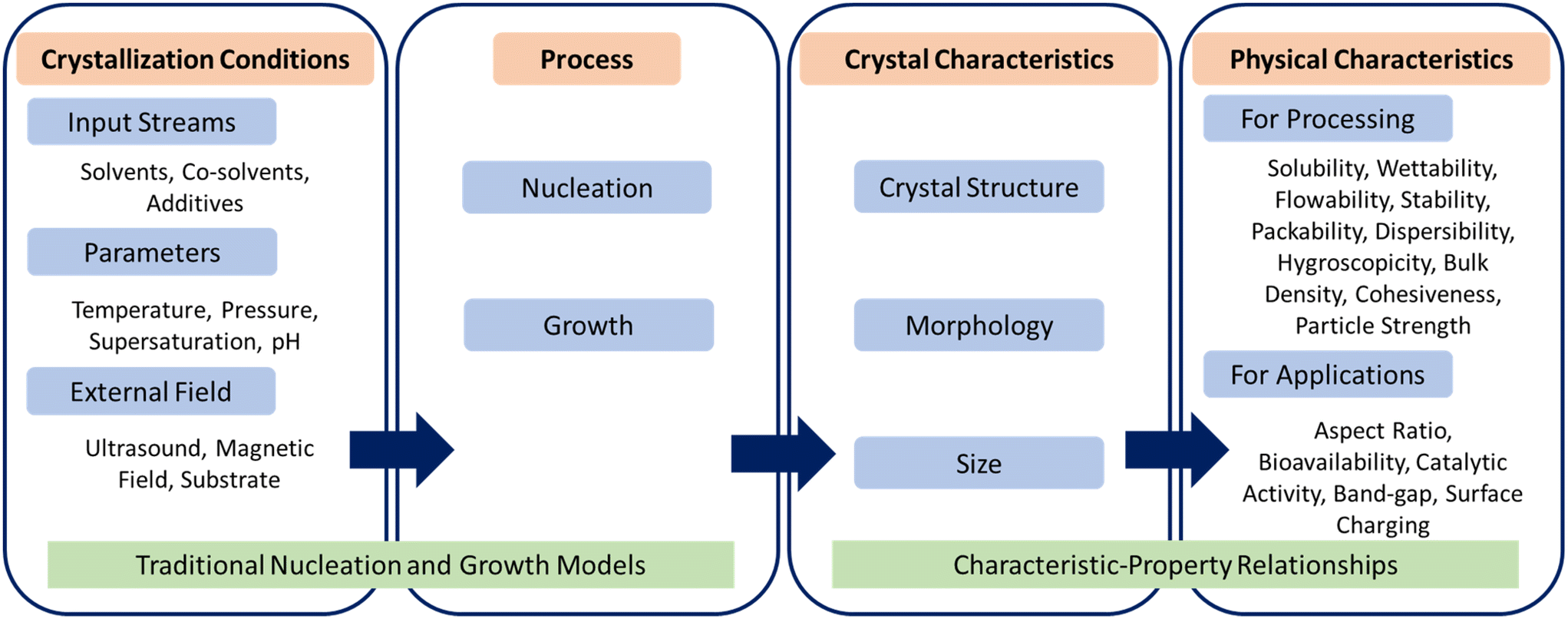 | ||
| Fig. 1 Relationship between crystallization conditions, primary characteristics and physical properties of crystalline materials. | ||
Several computational models have been developed to predict structure–property relationships for crystalline materials, such as the Hartman–Perdok theory for morphology prediction, population balance modeling for size distributions, and molecular dynamics or density functional theory (DFT)-based simulations for polymorph stability.13–18 Recent work has further integrated machine learning with these frameworks to predict crystal shapes and lattice energies directly from molecular descriptors, demonstrating rapid and accurate polymorph screening.19 The primary characteristics that yield desired physical properties can thus be identified from such computationally established characteristics–properties relationships. However, the most difficult task in crystallization remains to reproducibly grow crystals with targeted characteristics under realistic conditions, a challenge that has been central to crystallization research for several decades. This difficulty arises from the high sensitivity of nucleation and growth to supersaturation, solvent interactions, and impurities, making the control of polymorphism and morphology an ongoing pursuit across chemistry, materials science, and process engineering.20
1.2. Crystallization from the bird's-eye view
To tackle such a challenging task, it is of immense importance to understand the process of forming crystals – crystallization – and the physicochemical factors that govern it. Crystallization is one of the most widely employed separation and purification techniques in the chemical and pharmaceutical industries, used to isolate molecules or atoms in a pure solid form.21 The process of solution crystallization begins with nucleation (the birth of crystals) followed by crystal growth in a supersaturated solution consisting of solute molecules – the target species to crystallize – solvent molecules – which govern the kinetics of solute transport and attachment – and, in some cases, additives or impurities that modulate molecular interactions to favor or inhibit particular morphologies or polymorphs.21 The degree of supersaturation, defined as the chemical potential difference between solute molecules in the solution and those in equilibrium, serves as the primary driving force for crystallization.21 Supersaturation can be generated and controlled by adjusting either the temperature (cooling crystallization) or the solvent composition (antisolvent crystallization), based on the solid–liquid equilibrium (SLE) data (schematic is shown in Fig. 2a).22,23 At sufficiently high supersaturation, crystals may nucleate spontaneously within the homogeneous bulk of the solution (known as primary nucleation) or on foreign surfaces or existing particles (heterogeneous or secondary nucleation), as depicted in Fig. 2b.21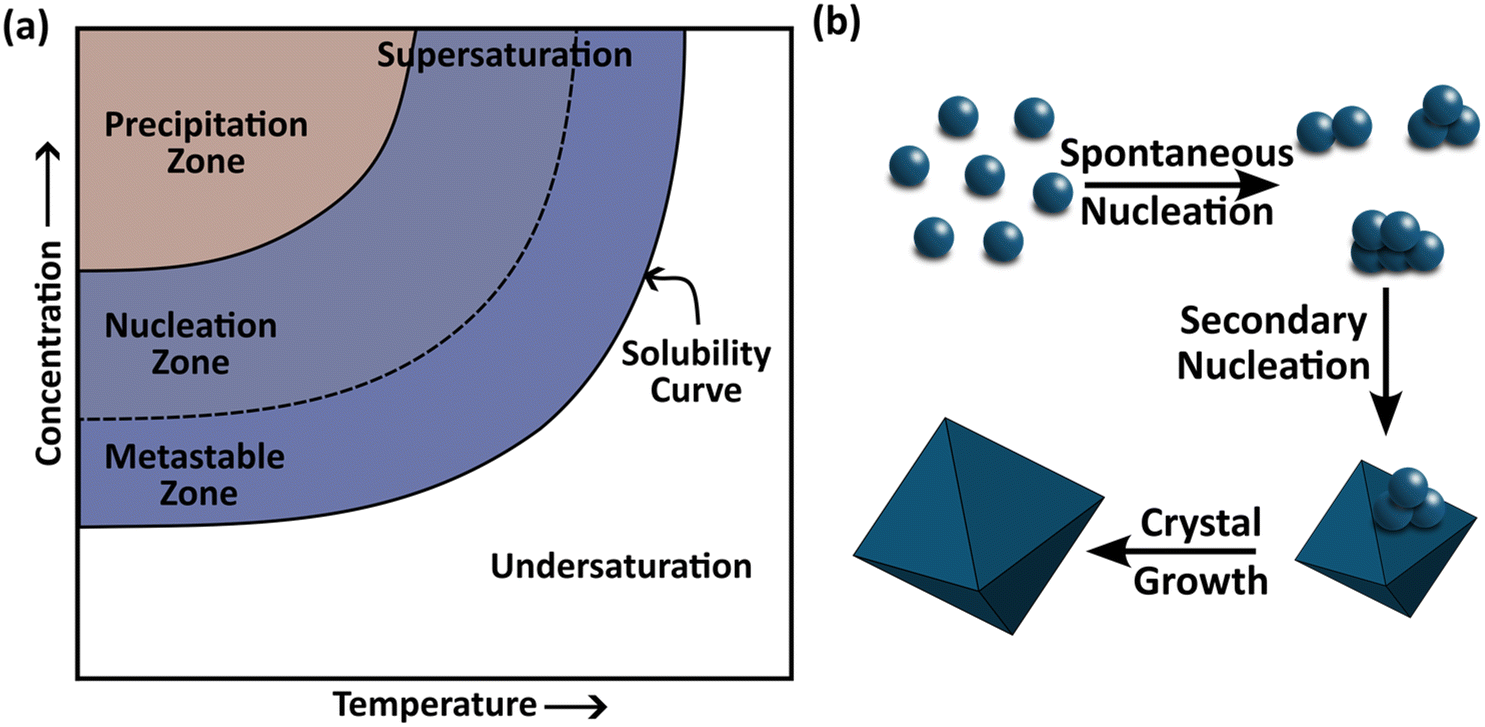 | ||
| Fig. 2 (a) Representative solid–liquid equilibrium curve showing the relationship between temperature, solute concentration, and the supersaturation region where primary and secondary nucleation occur. (b) Depiction of nucleation and crystal growth. | ||
The maintenance of supersaturation throughout the process ensures a continuous driving force for molecular assembly. As supersaturation increases, the chemical potential of solute molecules rises, enhancing molecular interactions within the solvation shell and promoting aggregation. When this driving force exceeds the stabilizing solvation interactions, solute molecules self-assemble into clusters.24 Initially, these small aggregates possess higher surface free energy than bulk free energy and are therefore thermodynamically unstable, favoring redissolution. As clusters grow, a critical size is reached where further molecular addition becomes energetically favorable; the formation of this cluster marks the onset of nucleation.24,25 Although the initial rate of cluster formation may be lower than its dissociation rate, the persistent supersaturation ensures that aggregation eventually dominates, leading to stable nuclei formation.
Nucleation establishes a structural template for subsequent crystal growth. During growth, the periodic arrangement of molecules evolves, and crystal faces with slower growth rates become dominant in the final morphology. Faces that grow rapidly disappear from the equilibrium shape (morphology, habit, and shape are used interchangeably in the literature). It is well established that the level of supersaturation determines the prevailing growth mechanism, such as spiral growth, two-dimensional (2D), or rough growth.26 Controlling crystal growth is crucial because the shape evolution directly influences material performance. For instance, in optical or electronic applications, macroscopic crystals with smooth, defect-free surfaces are desired, whereas in pharmaceutical manufacturing, microscopic crystals with a higher specific surface area provide enhanced dissolution rates and bioavailability.27 Thus, understanding the interplay between supersaturation, nucleation, and growth mechanisms is essential for tailoring crystals with targeted properties across disciplines from materials science to pharmaceutical engineering.
1.3. Molecular view of crystallization
Crystallization is a multiscale process that extends from the molecular level to the macroscopic scale, where the binding and organization of solute molecules underlie the formation of ordered solids. The fundamental step of crystallization is the repeated association of solute molecules through noncovalent interactions such as hydrogen bonding, π–π stacking, van der Waals forces, and electrostatic interactions.28 If we take a black-box perspective, as shown in Fig. 3, the crystallization process can be viewed as a continuous series of association and dissociation events of solute molecules occurring over different time and length scales. These elementary interactions collectively give rise to distinct stages of crystallization, including pre-nucleation clustering, nucleation, and subsequent crystal growth. The dynamic balance between molecular attachment and detachment governs both the rate and direction of crystal evolution, determining which polymorph or morphology ultimately emerges.29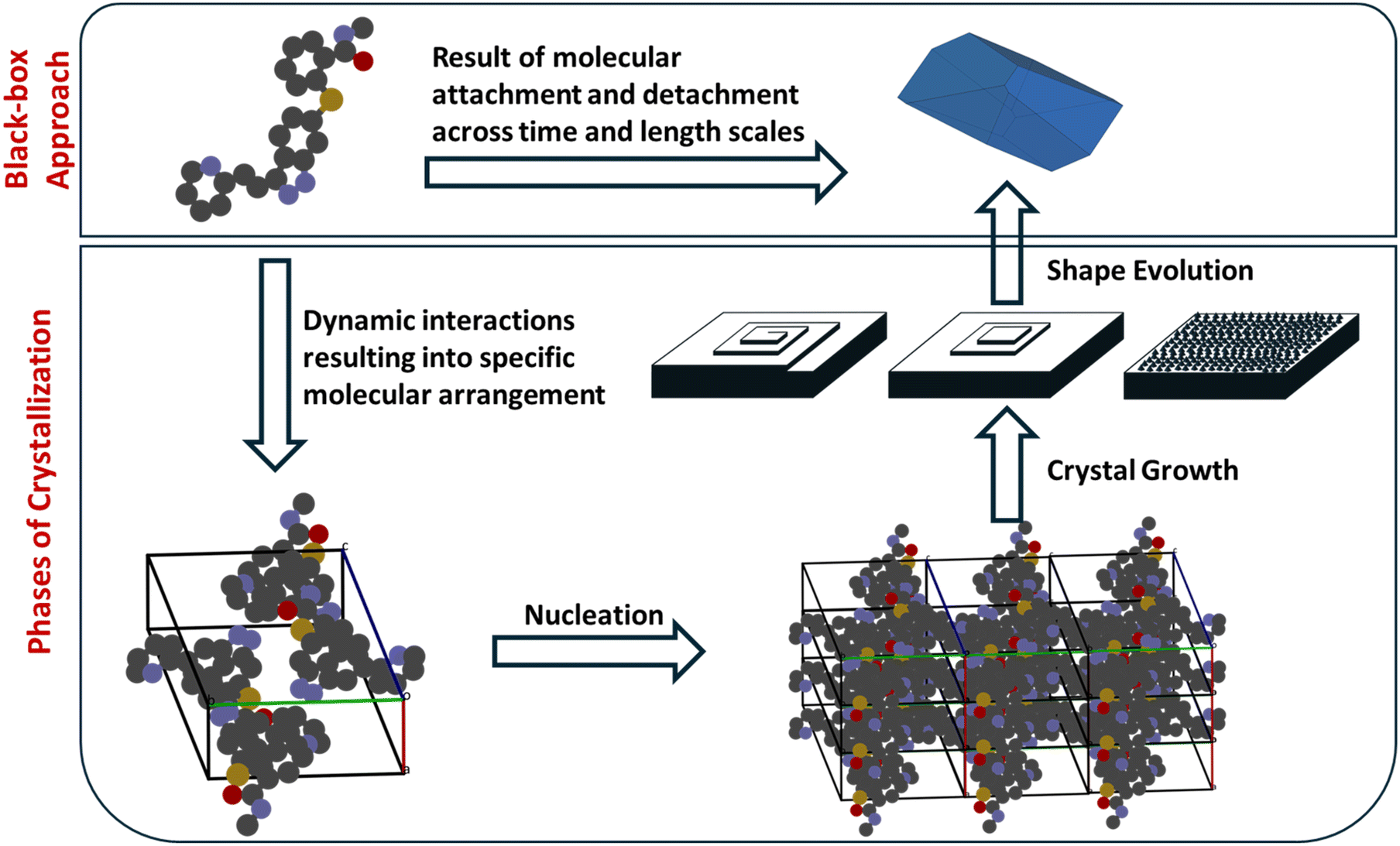 | ||
| Fig. 3 Two perspectives on the crystallization process. Top: The ‘black-box approach’ representing experimental reality, where often only the initial inputs (supersaturated solution) and final outputs (crystals) are observable, leaving the governing molecular mechanisms obscured. Bottom: The computational goal, which aims to ‘open’ the black box by resolving the process into distinct, modellable phases from molecular self-assembly and nucleation to macroscopic crystal growth, thereby enabling the mechanistic prediction of crystal structure and morphology. | ||
1.4. Scope of review
While the crystal structure is primarily determined during the early stage of nucleation, the morphology and size of crystals can continue to evolve throughout the growth phase. The evolution of these characteristics depends on several rate-governing processes controlled by the system's chemical composition, temperature, pressure, and interspecies interaction energies (see Fig. 1). For instance, the crystal structure depends on the rate of molecular self-assembly into specific lattices and symmetry groups, while the crystal morphology is determined by the relative growth rates of crystallographic faces, and the crystal size varies approximately linearly with the overall growth rate magnitude.30–32 Several theories have been proposed to quantitatively predict crystal-face growth rates.33,34 These include models based on lattice geometry, such as the Bravais–Friedel–Donnay–Harker (BFDH) method, surface energetics (the Hartman–Perdok theory), and mechanistic frameworks describing molecular attachment, including the two-dimensional (2D) nucleation model, the Burton–Cabrera–Frank (BCF) spiral growth model, the Chernov bulk-diffusion model, the Winn–Doherty shape-evolution model, MONTY Monte Carlo crystal-growth simulations, and rare-event sampling approaches.35–44 These growth models collectively enable prediction of dynamic and steady-state crystal morphology and size under different supersaturation or solvent conditions.31,43,45–47 Because most growth-rate models depend explicitly on intermolecular interactions specific to a given crystal lattice, the resulting morphology is strongly structure-dependent, as indicated schematically in Fig. 1. Therefore, crystal structure is the most fundamental characteristic governing not only morphology but also a broad range of physical properties. Since the dynamics of crystal-structure formation are difficult to probe experimentally – owing to nanoscopic time scales and transient ordering phenomena – computational models have become indispensable tools for identifying and predicting crystal structures under specific crystallization conditions. This review provides a critical assessment of computational frameworks that aim to predict crystal structure, emphasizing their implementation strategies, underlying assumptions, and key limitations.The occurrence of polymorphism arises from exceedingly small energy differences between alternative solid-state arrangements of the same molecule. The crystal structure of a solid is defined by the atomic geometry, molecular orientation, and space group symmetry within the unit cell. Even minute perturbations in molecular packing – such as subtle rotations, conformational changes, or hydrogen-bond rearrangements – can yield distinct polymorphs that differ in lattice energy by only a few kilojoules per mole.3 In many cases, metastable polymorphs exist within 5–10 kJ mol−1 of the most stable form, yet can dominate experimentally due to kinetic trapping or solvent-mediated stabilization.48 Capturing such fine energetic distinctions represents one of the greatest computational challenges in condensed-phase modeling. Ironically, as computational capabilities have grown, so too has the realization that polymorphism is governed by a complex interplay of thermodynamic and kinetic factors, prompting further research rather than closure. This persistent complexity underscores the need for multiscale approaches that integrate molecular energetics, nucleation kinetics, and growth dynamics – an overarching theme of this review.
In this article computational models used across the various phases of crystallization are critically reviewed, highlighting their underlying assumptions, predictive capabilities, and present limitations. It distinguishes aspects of crystallization that are well represented by current theory from those that remain poorly captured or oversimplified, thereby identifying opportunities for advancement. Particular attention is devoted to multiscale computational strategies that capture the fundamental molecular processes of self-assembly, nucleation, and growth, with an overarching goal of advancing the predictive modeling of polymorphism.
2. Crystal structure prediction up to a few lattices
2.1. Thermodynamic prediction of crystal structure using energy minimization techniques
In 1988, John Maddox pointed out that it was not yet possible to determine a crystal structure purely from first principles.4 Following this statement, there was a surge of interest in generating crystal structures directly from molecular formulae, without prior experimental input. This effort led to the emergence of the field now known as crystal structure prediction (CSP), which has since experienced an exponential increase in publications, as shown in Fig. 4. A summary of key review articles in this area is presented in Table 1, outlining milestones in CSP development and computational approaches toward addressing the problem of polymorphism.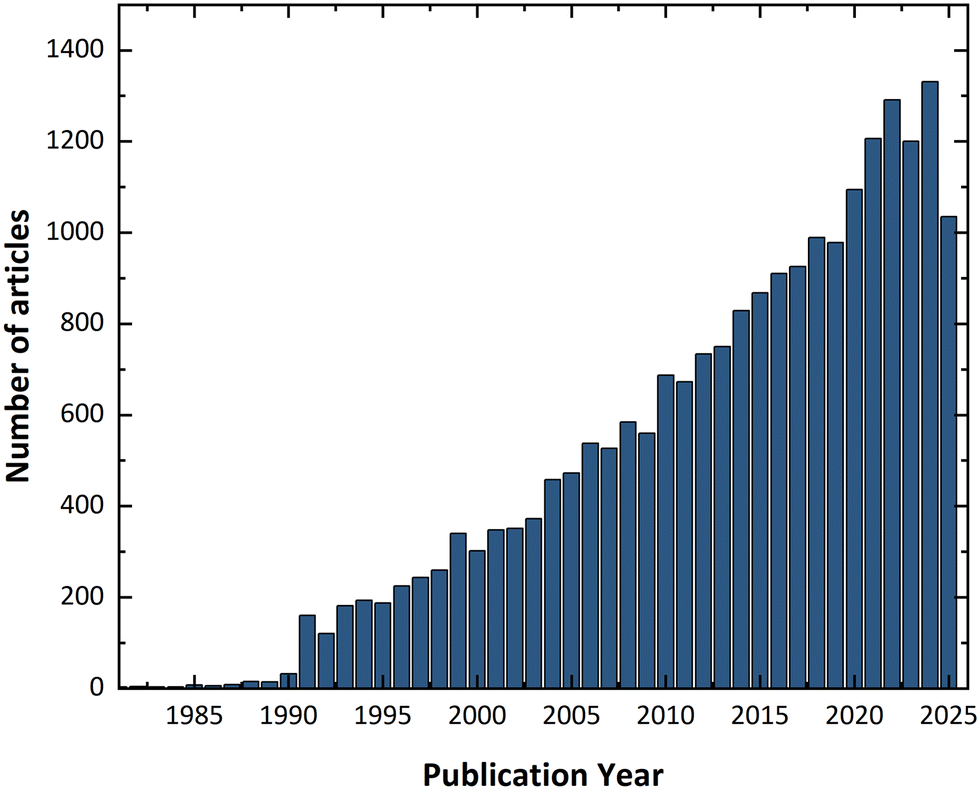 | ||
| Fig. 4 Number of articles published over the years. Data obtained by searching the Web of Science™ database for the keyword “crystal structure prediction” (accessed 2025). | ||
| Authors (year) | Title | Purpose of review |
|---|---|---|
| Moulton & Zaworotko47 (2001) | From molecules to crystal engineering: supramolecular isomerism and polymorphism in network solids | To outline challenges and prospects in crystal engineering with a focus on polymers and organic molecules |
| Vippagunta et al.53 (2001) | Crystalline solids | Overview of analytical and characterization techniques of polymorphism in pharmaceutical molecules, with a sub-section devoted to prediction of polymorphism |
| Price S. L.54 (2004) | The computational prediction of pharmaceutical crystal structures and polymorphism | Review of computational structure prediction methods and describing use of computational structure prediction methods to complement characterization of pharmaceutical solids |
| Datta & Grant55 (2004) | Crystal structures of drugs: advances in determination, prediction, and engineering | Critical review of analytical and computational methods for crystal structure determination |
| Desiraju G. R.56 (2007) | Crystal engineering: holistic view | Highlight challenges in crystal engineering with focus on crystallization |
| Woodley & Catlow57 (2008) | Crystal structure prediction from first principles | Rigorous review of computational techniques used in CSP and their application wide areas of chemistry |
| Price S. L.18 (2008) | Computational prediction of organic crystal structures and polymorphism | Review of CSP methodology and discussion on kinetic factors of crystallization |
| Thakur et al.58 (2015) | Crystal structure and prediction | Review of CSP and describing status of the field |
| Beran G. J. O.17 (2016) | Modeling polymorphic molecular crystals with electronic structure theory | Comprehensive review of use of electronic structure theory for modeling polymorphism with sections devoted to CSP techniques |
| Graser et al.59 (2018) | Machine learning and energy minimization approaches for crystal structure predictions: a review and new horizons | Computational methods such as DFT and ML are reviewed to CSP, along with the promise and challenges encountered in these techniques |
| Oganov et al.60 (2019) | Structure prediction drives materials discovery | CSP techniques are reviewed and their potential for the study of different materials systems and new material discoveries |
| Bowskill et al.61 (2021) | Crystal structure prediction methods for organic molecules: state of the art | Reviews methodologies for CSP focused on current research efforts to improve reliability and wide applicability for organic molecules |
| Yin et al.62 (2022) | Search methods for inorganic materials crystal structure prediction | Highlights the search methods for CSP such as empirical, guided-sampling algorithms, optimization-based search, and data-driven approaches |
| Clements et al.63 (2022) | Roles and opportunities for machine learning in organic molecular crystal structure prediction and its applications | Reviews the impact of machine learning CSP and its applications focused on evaluation energies, landscapes, and property-targeted molecule identification |
| Beran G. J. O.64 (2023) | Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials | Details high-level overview of the CSP methods, current challenges, and case studies for applications |
During their early years, lattice-energy-minimization (LEM) techniques were primarily used to refine incomplete or unreliable crystal structures derived from partial X-ray diffraction data. Identifying weakly bonded fragments in a crystal lattice was particularly challenging but could be handled effectively through energy-minimization schemes. Na2Ti9O19 and several organometallic compounds were among the first systems for which such computational refinements were successfully demonstrated.7,49 In these early applications, the space group symmetry of the crystal was known, and approximate molecular positions within the unit cell were available. The concept of a space group – defined by Bravais lattices and their associated symmetry operations – characterizes the internal periodicity of a crystal. Once the molecular arrangement within a lattice and its symmetry operations are established, the complete three-dimensional crystal structure can be generated by repeating this motif through those operations. Following Maddox's challenge, researchers began attempting CSP without using diffraction data.30,31 For relatively simple molecules, these methods could reproduce experimental structures; however, for more flexible or complex molecules, significant deviations were observed. These discrepancies were attributed to approximations in molecular-potential parameters, and it was hypothesized that improving the accuracy of these potentials would yield experimentally correct crystal structures across diverse systems. Since then, there has been intensive progress in this area. CSP using LEM has not only predicted known polymorphs but has also forecast new, previously unobserved crystal forms, many of which were later confirmed experimentally.32–36 This predictive capability has advanced structural chemistry by uncovering insights into conformational flexibility, molecular packing preferences, and the interplay between intramolecular and lattice energetics.37–46,50 Modern CSP approaches now routinely combine lattice-energy minimization with machine-learning-driven energy evaluations and dispersion-corrected quantum-mechanical methods, achieving near-DFT accuracy at a fraction of the computational cost.51,52
Methods for crystal structure prediction (CSP) are aimed at finding the densest and thermodynamically most stable packing arrangements of a molecule that result in minimized lattice energy. The lattice energy represents the sum of intramolecular and intermolecular interactions within the crystal lattice. It is generally hypothesized that, with an accurate description of these interactions, the experimentally observed polymorph will appear among the lowest-energy candidates in a CSP search.65Fig. 5 illustrates the overall workflow of the lattice-energy-minimization (LEM) approach. The process begins with a molecular formula, representing the compound whose crystal structure is to be predicted. The next step is to optimize the three-dimensional geometry of the molecule – typically by quantum-chemical methods (e.g., DFT). These initial stages are relatively easy owing to the wide availability of reliable computational tools (Gaussian, ORCA, TURBOMOLE, etc.).66–68
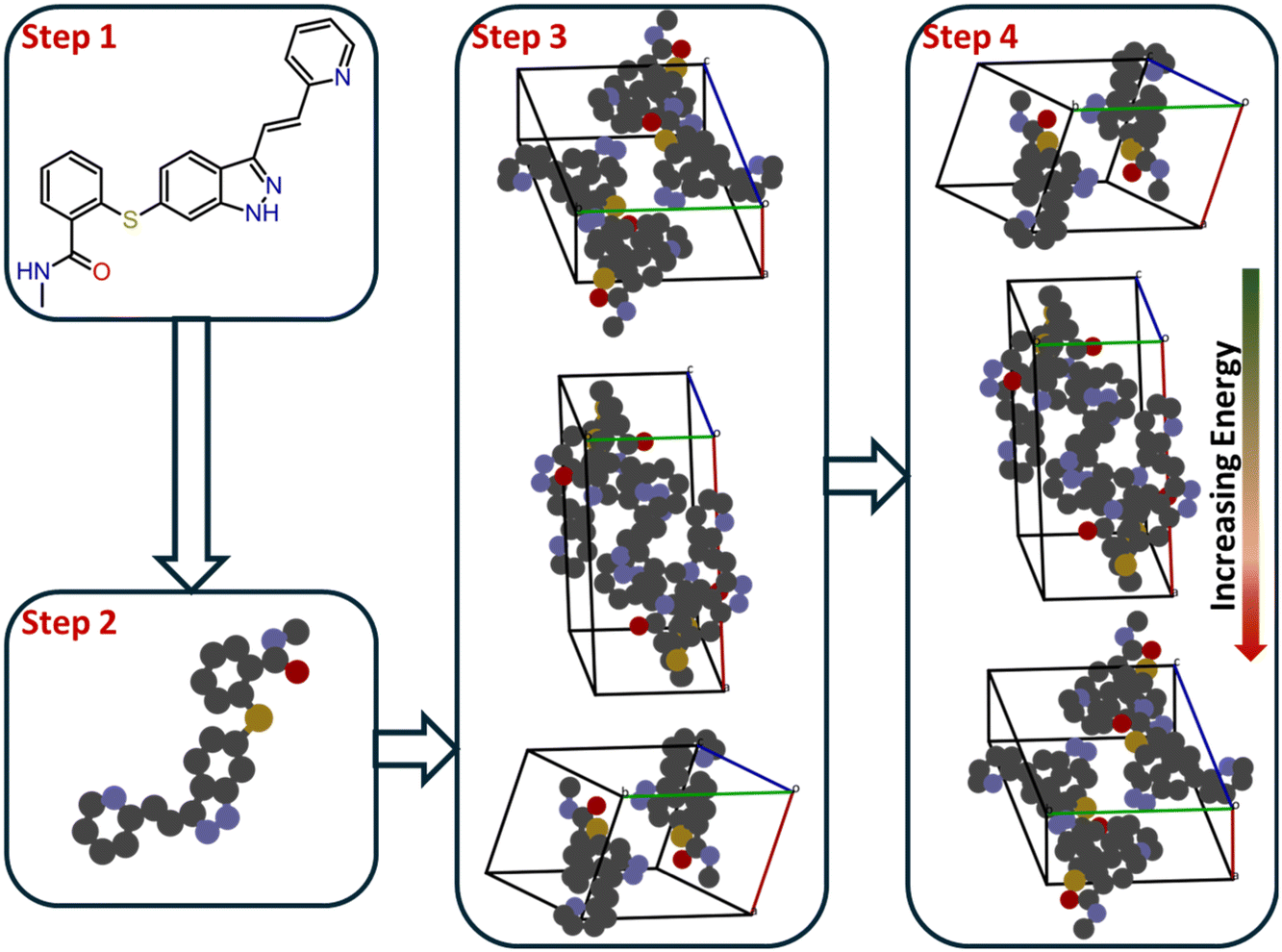 | ||
| Fig. 5 Steps in energy-minimization techniques, corresponding to the key stages of the approach. The first step is to obtain the molecular formula; the second is to optimize the molecular geometry. These are relatively straightforward compared with the third and fourth steps: (iii) generating all possible molecular packings in selected space groups, and (iv) evaluating and ranking based on their energies. | ||
The second step is critical because it defines the scope of the conformational search that will be required in subsequent steps. For flexible molecules, it is necessary to identify all possible low-energy conformations in the gas or solvated phase. Once these conformations are determined, one can infer the most probable space groups for crystallization, often guided by statistical analyses of the Cambridge Structural Database (CSD).69,70 At this point, the rigid-molecule approximation is commonly applied, assuming that the intramolecular geometry in the gas phase corresponds closely to that in the crystal lattice.71 This approximation remains valid for small and moderately flexible organic molecules, allowing a major reduction in computational cost without compromising accuracy. The third and fourth steps form the core of LEM-based CSP. After selecting the likely space groups, all possible trial crystal structures (packings) are generated within those symmetry constraints. This search phase may employ random or quasi-random sampling, Monte-Carlo exploration, simulated annealing, or evolutionary algorithms to explore the configurational space efficiently.72 The goal is to achieve broad coverage of configurational space while avoiding entrapment in local minima. Recent algorithms increasingly integrate machine-learning-guided or Bayesian optimization strategies to improve sampling efficiency and convergence.73,74 For example, a machine-learned force field within an evolutionary CSP framework was used to reproduce all known polymorphs of 66 test molecules with near-DFT accuracy.75 Once the candidate structures are generated, the lattice energy of each packing is computed—this being the most computationally expensive stage of CSP. Each molecule contributes six lattice degrees of freedom (three lattice lengths and three interaxial angles), and the complexity increases exponentially with the number of molecules in the asymmetric unit. Consequently, efficient search and ranking protocols are essential. The resulting structures are rank-ordered by lattice energy, with lower-energy packings representing the most stable polymorph candidates.
For search-space exploration, the third step, the most widely used algorithms include random or quasi-random searches, simulated annealing, Monte-Carlo sampling, evolutionary or genetic algorithms, and molecular-dynamics-based exploration.72,76,77 Random and quasi-random searches are undirected methods that often generate a large number of infeasible packings, most of which are quickly rejected due to unrealistic molecular overlaps or density constraints. Simulated-annealing and Monte-Carlo schemes employ stochastic acceptance rules (typically Boltzmann-weighted) that allow occasional uphill steps, helping the search escape local minima.78 Evolutionary and genetic algorithms apply biologically inspired operations – mutation, crossover, and selection – to evolve more stable structures iteratively.79,80 Hybrid methods combining evolutionary search with Bayesian optimization now represent the state of the art in balancing accuracy and efficiency.81
Simulated annealing (SA) is a stochastic global-optimization technique used to search for trial crystal structures and was among the first algorithms applied in CSP.30,31,82–84 SA is a computational analogue of the physical annealing process in metallurgy. In this method, the temperature of a system is progressively lowered from a high initial value, allowing the system to explore a wide energy landscape and gradually settle into low-energy configurations. At each cooling step, the system's equilibrium configuration is updated according to its Boltzmann-weighted free energy, and stochastic acceptance criteria prevent premature convergence to local minima. SA is typically coupled with Monte Carlo (MC) sampling, which generates random molecular displacements accepted or rejected using the Metropolis criterion. Together, these algorithms promote efficient exploration of configurational space and direct the system toward the global minimum of the lattice-energy surface.85 Evolutionary and genetic algorithms (EA & GA) form another class of stochastic search methods that have been successfully applied to CSP.86–90 Inspired by the principles of evolution and natural selection, these algorithms begin with a randomly generated population of crystal structures and evolve them via operations such as mutation, crossover, insertion, and deletion. Over successive generations, high-fitness structures (i.e., those with lower energies or denser packings) are preferentially retained, ensuring broad sample-space coverage while avoiding local minima. Modern EA implementations, such as GAtor, and CALYPSO, now integrate machine-learned energy surrogates and adaptive mutation schemes to accelerate convergence and improve energy ranking.91,92 Other stochastic approaches, independent of SA, use Monte-Carlo sampling guided by symmetry or molecular conformations. These methods generate trial structures based on the possible symmetry operations or feasible conformational degrees of freedom of the molecule.93,94 By restricting sampling to symmetry-allowed packings, such algorithms reduce computational expense while preserving diversity in candidate structures.
Molecular dynamics (MD) has also been employed to locate minimum-energy crystal structures and to investigate temperature-dependent polymorphic transitions.95,96 In this approach, the simulation box and number of molecules are chosen according to the selected space group or experimental cell parameters. MD is a force-balance technique that integrates Newton's equations of motion to propagate molecular positions and velocities over time, yielding trajectories that reflect the system's microscopic evolution. Randomized initial configurations evolve toward stable crystalline arrangements, and the ensemble-averaged energy provides a macroscopic measure of stability. However, the applicability of MD is often constrained by computational cost and limited time scales – typically a few picoseconds to nanoseconds, with femtosecond integration steps. Energy evaluations in MD rely on parameterized force fields, which define bonded and non-bonded interaction potentials; the quality of these parameters directly affects the accuracy of lattice-energy predictions.97,98
Energy evaluation represents one of the most critical and computationally intensive components of CSP. The accuracy of energy calculations directly dictates the predictive quality of a CSP workflow. Traditionally, two major computational routes are employed: (i) empirical or semi-empirical force fields and (ii) electronic-structure methods based on density functional theory (DFT).95,96,99–108 Force-field methods use predefined analytical functions to describe inter- and intramolecular interactions, which are broadly categorized as bonded (stretching, bending, torsion) and non-bonded (electrostatics, dispersion, hydrogen bonding) contributions. During crystallization, bonded interactions are often neglected, since molecules typically preserve their covalent connectivity upon crystallization. The challenge lies in accurate parameterization of non-bonded terms that govern molecular packing and dispersion. Different force fields – such as OPLS, GAFF, and tailor-made CSP-FFs – are optimized for specific chemical classes.109,110 DFT-based lattice-energy calculations, though computationally more demanding, often yield superior accuracy compared with traditional force fields. However, DFT simulations are limited by system-size scaling and convergence issues, especially when modeling flexible molecules or large unit cells.
To address the computational bottleneck of these energy evaluations, machine learning (ML) strategies are increasingly explored as intermediate screening steps in the energy minimization workflow. Recent approaches utilize graph neural networks and atom-centered symmetry functions to construct ML interatomic potentials (MLIPs) that aim to approximate DFT accuracy at a reduced computational cost.111,112 Recent efforts have introduced machine-learning-derived or fragment-based force fields, which significantly improve transferability and accuracy for molecular crystals.113 However, the reliability of these data-driven models for final polymorph ranking remains a critical question compared to established physics-based methods. Consequently, their most effective current application is as efficient surrogates for rapidly screening thousands of trial packings, allowing computationally expensive quantum mechanical calculations to be reserved for the final ranking of the most promising candidates. Furthermore, while generative ML models are being investigated to propose crystal structures directly, rigorous validation against standard stochastic search algorithms remains essential as the field evolves.
The CSP blind tests are collaborative benchmarking exercises organized by the Cambridge Crystallographic Data Centre (CCDC) that provide the necessary platform for such validation.114–119 In each test, participants are provided with molecular structures (without experimental crystal data) and a fixed time frame to submit their predicted crystal packings. To date, seven official blind tests (1999–2024) have been completed.114–121 The timeline of the blind tests and the evolution of CSP methodologies are depicted as shown in Fig. 6. With each new test, the molecular complexity has increased – typically measured by the number of molecules in the asymmetric unit (Z′) and conformational flexibility. While earlier CSP methods have successfully predicted many low-Z′ crystal forms, significant challenges remain for flexible or multi-component systems. For example, a metastable structure was predicted as the global minimum, while the experimentally stable form corresponded to a higher-energy structure (∼0.5 kJ mol−1 difference) in a different space group.122 Such discrepancies revealed that kinetic and entropic effects, often neglected in energy-minimization approaches, can alter polymorph stability. Subsequent blind tests exposed additional limitations – particularly the inadequacy of isotropic force fields in representing anisotropic electrostatics.119 Some participants who succeeded in earlier rounds failed to maintain accuracy in later tests, prompting the CCDC to increase the allowed number of submissions from three to one hundred. This change acknowledged that experimentally observed structures often correspond to kinetically accessible metastable forms, and ranking accuracy must be judged across a broader candidate set. Despite mixed results, the blind tests have driven major advances in methodology, leading to the adoption of dispersion-corrected DFT in modern CSP workflows.123
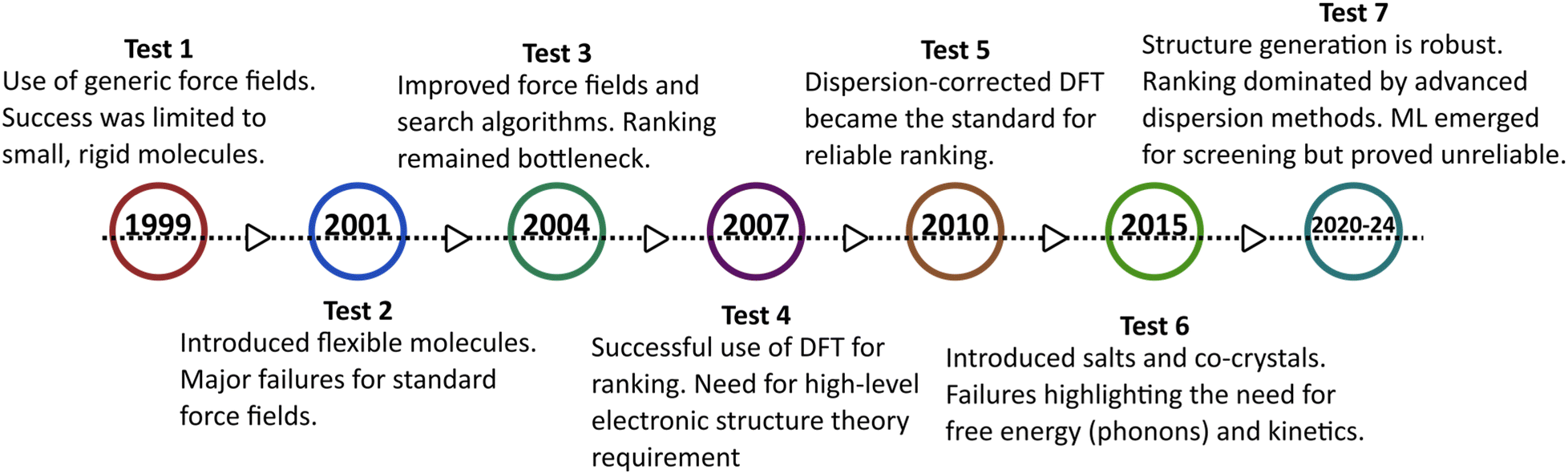 | ||
| Fig. 6 Timeline of the CCDC blind tests illustrating the evolution of CSP methodologies. The progression highlights the transition from empirical force fields (tests 1–2) to the adoption of DFT for energy ranking (tests 4–5), while the most recent tests (6–7) demonstrate the standardization of dispersion-corrected DFT as the benchmark for stability prediction. | ||
Accurately modeling weak van der Waals (vdW) interactions is a fundamental requirement for molecular CSP, as these forces frequently dominate the lattice energy. While early semi-local DFT functionals (e.g., PBE) failed to capture these long-range interactions, the development of robust dispersion corrections has effectively resolved this limitation for standard applications. Modern DFT approaches – such as DFT-D3, many-body dispersion (MBD), and hybrid functionals – have successfully integrated both pairwise and higher-order many-body effects into the electronic structure framework and substantially improved accuracy in ranking polymorphs.124–126 Fragment-based wave-function methods and ML-assisted DFT energy corrections now represent promising directions for balancing cost and precision.113 While earlier blind tests revealed challenges with flexible systems and anisotropic electrostatics, the results of the newly concluded seventh blind test (2024) provided a definitive assessment of modern capabilities.116,120,121 This test revealed that while generating candidate structures is now considered robust, accurately ranking them remains the primary challenge. The evaluation demonstrated that DFT incorporating specific dispersion corrections (such as D3, MBD, or XDM) yielded the most consistent results. Conversely, the test highlighted the limitations of ML potentials for final stability determination; while valuable for screening, they frequently struggled to discriminate the subtle energy differences required to identify the global minimum without subsequent DFT refinement. These findings have driven the field toward a consensus workflow: utilizing ML or empirical force fields for broad search, followed by dispersion-corrected DFT for the rigorous final ranking of stability.
Beyond dispersion, a more subtle but equally critical challenge in DFT-based ranking is the inherent ‘delocalization error’ (or self-interaction error) present in some semi-local functionals.127 This error can artificially stabilize specific electronic configurations, leading to incorrect energy rankings. This issue is particularly pronounced in conformational polymorphs, where the relative stability of different molecular geometries is sensitive to electron delocalization across conjugate π-systems. A notable example is the highly polymorphic system known as ROY (5-methyl-2-[(2-nitrophenyl)amino]-3-thiophenecarbonitrile). As demonstrated in previous work, standard DFT functionals often fail to reproduce the experimental stability order of ROY polymorphs largely due to errors in predicting the relative energies of the monomer conformations.128 Achieving reliable ranking in such challenging cases frequently requires moving beyond standard DFT to incorporating monomer energy corrections derived from second-order Møller–Plesset perturbation theory (MP2), which effectively mitigates the delocalization error.129 Thus, for molecular crystals with complex electronic structures, a hierarchical approach—refining DFT rankings with high-level MP2 calculations—is often suggested to ensure accuracy.
A further critical limitation of standard CSP workflows is the reliance on static lattice energies, which correspond to the crystal structure at 0 K without zero-point fluctuations.130 In reality, crystallization occurs at finite temperatures where vibrational entropy and thermal expansion play decisive roles in relative stability. Neglecting these thermal free-energy contributions can result in incorrect ranking, particularly for enantiotropic systems where stability order reverses with temperature. Incorporating these effects requires harmonic (or quasi-harmonic) phonon calculations to estimate the vibrational density of states. However, these calculations are roughly two orders of magnitude more expensive than static energy evaluations, making them computationally prohibitive for all but a few final candidates. Recent work has demonstrated that accurate ranking often requires coupling many-body dispersion effects with rigorous phonon calculations.131 Similarly, analysis of the 7th blind test highlighted that while dispersion-corrected DFT is robust for static lattices, the absence of thermal corrections remains a major source of uncertainty when predicting experimental forms under ambient conditions.120
Energy-minimization-based CSP approaches are limited by their neglect of nucleation kinetics and reliance on approximations to reduce computational expense. As molecular complexity increases, crystallization often becomes kinetically, rather than thermodynamically, controlled.132 Under such conditions, ranking polymorphs solely by lattice energy yields incomplete or misleading predictions. Numerous studies have emphasized that incorporating kinetic effects – such as molecular diffusion, solvent interactions, and metastable intermediate states – is essential for realistic CSP.48,133–138 Even if future algorithms successfully reproduce all experimentally known polymorphs within the top 100 predicted structures, identifying which polymorph actually forms experimentally will still demand coupling CSP with kinetic modeling and multiscale simulations.132,135 Although energy-minimization techniques may not yet fully capture the dynamics of polymorph selection, they remain invaluable for mapping the thermodynamic landscape and identifying potential metastable forms, thus guiding both experiment and process design.
However, despite these advances in ranking accuracy, the field remains challenged by the fundamental ‘over-prediction problem’. CSP landscapes invariably contain many more distinct crystal structures within a thermodynamically accessible energy window (typically <10 kJ mol−1 from the global minimum) than are ever observed experimentally.135 While these predicted structures represent valid local minima on the potential energy surface, the vast majority are never realized in the laboratory. Recent work has demonstrated that this discrepancy is partly due to the neglect of thermal effects; many distinct minima on the 0 K surface are separated by low energy barriers and effectively merge into single vibrational basins at finite temperatures.139 By applying threshold clustering algorithms to group these minima, the number of distinct predicted polymorphs can be significantly reduced, aligning predictions closer to experimental reality. However, even after accounting for these thermal basins, the number of energetically feasible structures often exceeds observation. This underscores that lattice energy minimization provides only a partial picture: it identifies what is thermodynamically possible, but not what is kinetically accessible. The selection of specific polymorphs from this crowded landscape is ultimately governed by the height of nucleation barriers and the rates of crystal growth, necessitating the transition from purely static energy calculations to the dynamic kinetic models discussed in the following sections.
2.2. Kinetic prediction of crystal structure
The ordered assembly of molecular building blocks is a ubiquitous phenomenon underlying diverse materials – from molecular crystals to supramolecular frameworks.140 Crystallization can be regarded as a self-assembly process operating across multiple time and length scales. The concept of self-assembly is broad, and whether the organization of organic molecules in solution under a strong driving force qualifies as spontaneous “self-assembly” or as a kinetically driven formation process remains debated. In this work, we treat the association of solute molecules in solution as a self-assembly event leading to crystal nucleation.Modeling molecular self-assembly is crucial because initial aggregation of solute molecules often gives rise to recurring structural motifs called “synthons”.56,141 Under specific thermodynamic conditions, only one dominant synthon may form, and this early motif is frequently retained in the crystal lattice – an idea known as the “link hypothesis”, recently validated experimentally for tetrolic acid crystals using in situ FTIR spectroscopy.142 In polymorphic systems, variations in the structure of the early aggregates can dictate which polymorph ultimately appears. Unfortunately, few studies have explicitly connected polymorph selection to early-stage self-assembly, though recent combined experimental–simulation analyses are beginning to close this gap.143
Modeling self-assembly involves computing the forces and potential-energy surfaces governing solute–solute and solute–solvent interactions, or mapping the temporal and spatial evolution of the system toward low-energy configurations. Such simulations are computationally intensive and thus limited to short time (10−9–10−6 s) and nanoscale dimensions. They are primarily aimed at capturing the dynamic structure of pre-nucleation aggregates under near-equilibrium conditions. Comprehensive reviews of self-assembly modeling approaches can be found elsewhere.144
To date, only a few studies have explicitly related polymorphism to initial molecular aggregation. Both employed MD simulations to compute radial distribution functions (RDFs) for solute–solute and solute–solvent pairs in various solvents.145 One examined 5-fluorouracil (5-FU) and the other tetrolic acid (TTA).146,147 Both reached a similar conclusion: polymorph selection is governed by the nature of early molecular aggregates and their solvent-mediated stability. The TTA study further demonstrated that analyzing these interactions could guide the rational design of solvent composition to steer crystallization toward desired polymorphs. Recent work using enhanced-sampling MD and machine-learning descriptors now allows quantifying the transition from disordered clusters to ordered nuclei in small molecules.148–150
Crucially, integrating explicit solvent interactions into these pre-nucleation models has proven successful in predicting metastable polymorphs in organic systems that standard thermodynamic CSP methods miss. Traditional lattice energy minimization, often performed under vacuum or with implicit solvation, tends to favor the densest packing. However, recent studies utilizing explicit solvent molecular dynamics have demonstrated that strong solvent–solute interactions can stabilize specific ‘solution-phase synthons’ that structurally resemble a metastable polymorph rather than the stable one. For instance, in the case of TTA, explicit solvent simulations revealed that specific solvents favored a carboxylic acid catemer motif in the liquid phase, directly steering nucleation toward the metastable catemeric polymorph despite the cyclic dimer form being thermodynamically more stable.147 More recently, advanced sampling techniques have shown that the desolvation penalty itself acts as a kinetic filter; solvent molecules that are tightly bound to specific functional groups can influence the formation of the most stable nucleus, thereby kinetically trapping the system into a metastable pathway.145 These findings underscore that for many polymorphic molecular crystals, the ‘prediction’ of structure is less about finding the global energy minimum and more about identifying which solvent-stabilized cluster is kinetically selected from solution.
Beyond MD simulations, Brownian dynamics (BD) and kinetic Monte Carlo (KMC) provide complementary means to probe self-assembly, as shown in Fig. 7. In BD, solvent molecules are not treated explicitly; instead, stochastic forces represent solvent collisions, enabling simulations over much longer time scales (micro- to milliseconds) at a fraction of MD's cost. KMC is a probabilistic technique where the system evolves through discrete events, each assigned a rate or probability. Events that reduce energy are accepted with probability 1, whereas those that raise energy are accepted with a probability proportional to exp(−ΔE/kT) – the Metropolis criterion. This formulation yields the most probable kinetic pathway connecting successive metastable states. BD and KMC methods have been widely used to investigate protein folding, protein-structure prediction, polymer assembly, and nanoparticle self-organization in solution.151–156 Their increasing application to small-molecule crystallization suggests a growing role for multiscale kinetic models linking early molecular assembly with observable polymorph outcomes.
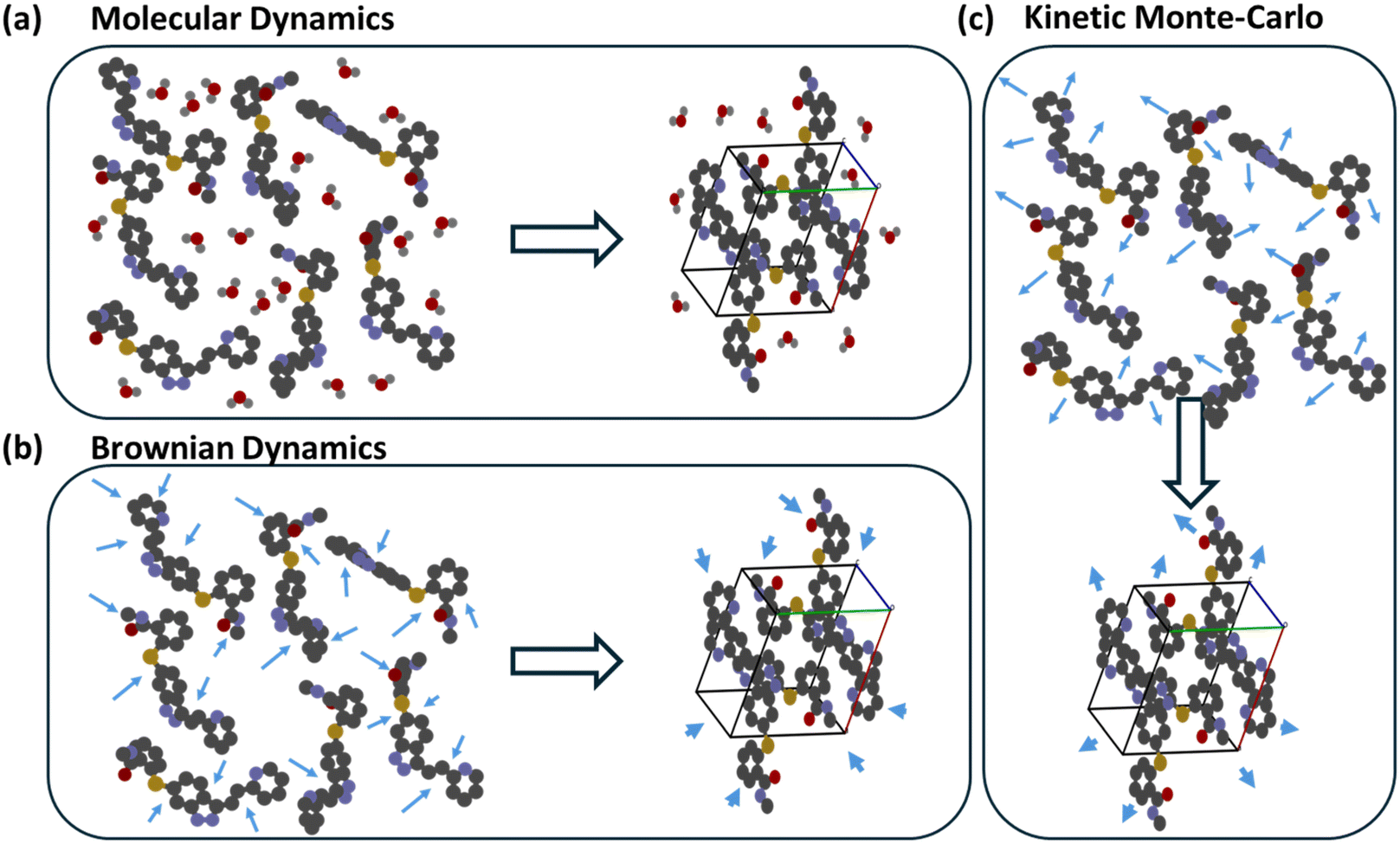 | ||
| Fig. 7 Modeling techniques for self-assembly. (a) Molecular dynamics (MD) explicitly represents solute and solvent molecules and integrates Newton's equations of motion, calculating forces and energies at each time step – demanding substantial computational resources. (b) Brownian dynamics (BD) treats solvent effects statistically via stochastic force terms, shown as arrows, reducing cost and extending accessible time scales. (c) Kinetic Monte Carlo (kMC) is a probabilistic method that tracks the system's most likely energetic evolution, shown as arrows, though energy evaluation at each step limits its scalability. | ||
2.3. Challenges of disappearing and late-appearing polymorphs
A persistent challenge in pharmaceutical crystallization is the phenomenon of “disappearing polymorphs”, where a long-manufactured metastable form is unexpectedly replaced by a more stable, late-appearing polymorph that was previously unknown.157 This occurrence, famously observed in the case of ritonavir, is driven by the Ostwald rule of stages, where the system initially crystallizes into a kinetically accessible metastable form before eventually converting to the thermodynamic global minimum.158,159 Standard nucleation models often fail to predict these events because they focus on the kinetics of the first emerging nucleus rather than the long-term competitive dynamics between forms.Recent work has highlighted that “late-appearing” polymorphs are often those that are thermodynamically stable but kinetically hindered. A conceptual framework was proposed that differentiates between the energy landscape (thermodynamic stability) and the crystallizability landscape (kinetic accessibility).157 They suggest that late-appearing stable forms often possess high molecular strain energy in their crystal conformation, which imposes a high kinetic barrier to nucleation even if the lattice energy is favorable. Consequently, a complete CSP workflow must not only rank structures by lattice energy but also assess their “crystallizability”, predicting not just what is stable, but when (or if) it will appear.
From a modeling perspective, capturing these dynamics presents significant computational hurdles. Solution-mediated phase transformation (SMPT)—the primary mechanism for late-appearing polymorphs—occurs over timescales ranging from hours to years, far beyond the nanosecond reach of standard MD. Simulating the transition requires rare-event sampling techniques, such as forward flux sampling (FFS) or metadynamics, to model the dissolution of the metastable form and the concomitant nucleation of the stable phase. Furthermore, the reaction coordinate is often complex, involving not just the order parameter of the new crystal but the explicit interfacial interactions between the dissolving metastable surface and the emerging stable nucleus (cross-nucleation). Without accurate models for these specific interfacial energies and transformation rates, identifying cases where a stable form exists but is kinetically “silent” remains a frontier challenge for the field.
3. Crystal structure prediction beyond a few lattices
3.1. Kinetic models for nucleation
The nucleus of a crystal is characterized by a cluster of molecules that tends to grow rather than dissociate into solution. The process by which these nuclei form is termed nucleation. Nucleation is traditionally described using classical nucleation theory (CNT), which assumes that (i) the structure of the nucleus is identical to that of the bulk crystal, (ii) the surface tension is independent of curvature and temperature, (iii) the nucleus is incompressible, and (iv) growth occurs via sequential addition of single “growth units” or monomers.160 CNT describes the change in Gibbs free energy (ΔG) as the balance between bulk volumetric and interfacial surface energies. Once a cluster exceeds a critical radius, further addition of molecules reduces its free energy, making the cluster stable – this marks the formation of a nucleus, as depicted in Fig. 8. It is essential to draw a sharp distinction between the ‘self-assembled’ clusters discussed in section 2.2 and the ‘critical nucleus’ defined here. Pre-nucleation clusters generally refer to stable or metastable solute aggregates—potentially amorphous or possessing liquid-like order—that exist in dynamic equilibrium with the solution. In contrast, the critical nucleus represents a thermodynamically unstable transition state at the peak of the free energy barrier (ΔG*). While CNT assumes the nucleus forms directly with the bulk crystal structure from monomers, the self-assembly perspective aligns more closely with non-classical mechanisms, where stable pre-nucleation clusters serve as precursors that must undergo subsequent densification or structural reordering to reach the critical size and structure required for crystal growth.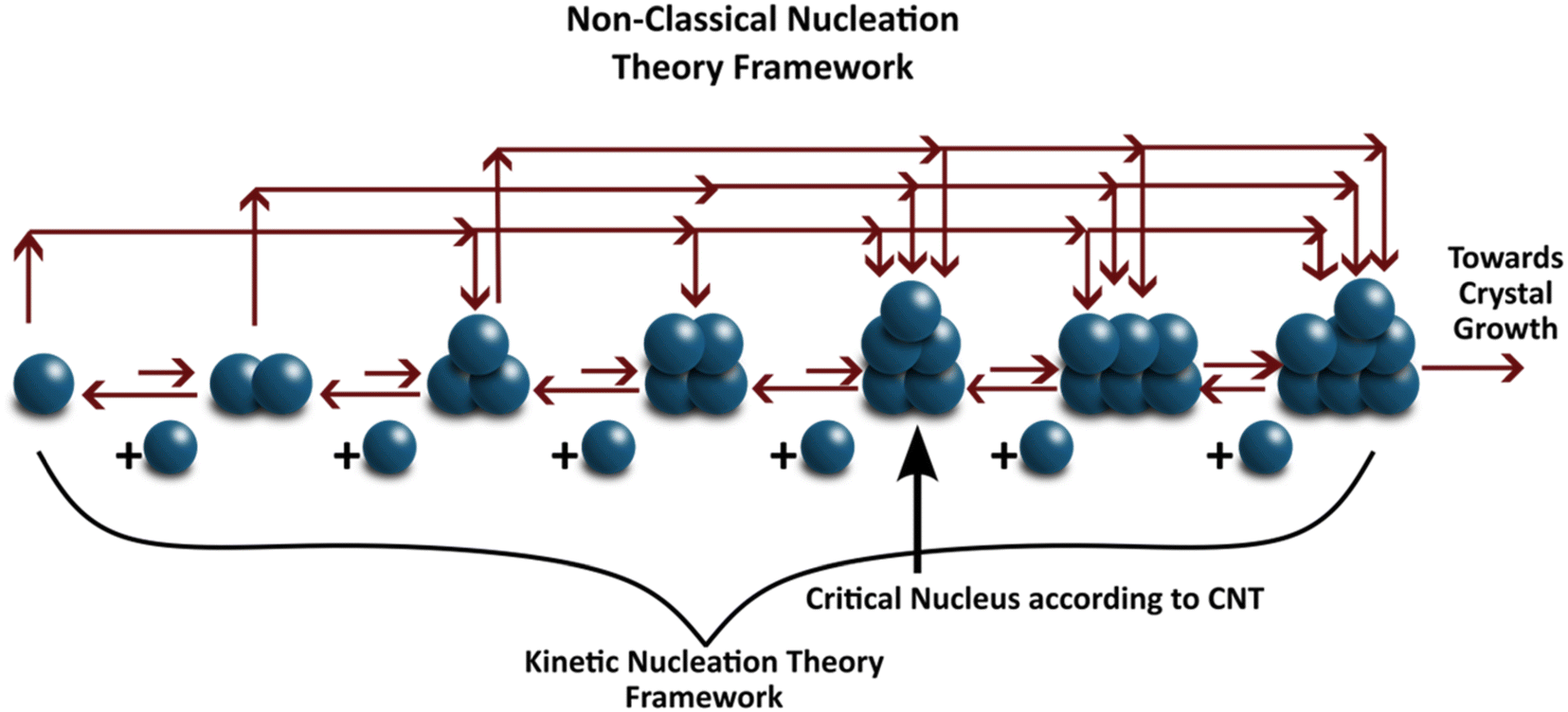 | ||
| Fig. 8 Frameworks of different nucleation theories. The upper path indicate two-step (nonclassical) nucleation, where small amorphous clusters densify into larger crystalline nuclei. The lower path represents monomer-by-monomer addition, characteristic of classical nucleation theory. | ||
To account for the discrete kinetics of molecular attachment, CNT has been extended to the kinetic nucleation theory (KNT) framework, where nucleation is treated as a series of elementary association reactions between growth units.8,161–163 In this work, we adopt KNT to describe the temporal evolution of cluster formation. One approach to model the kinetics of nucleation involves solving the unsteady-state differential equation for cluster population dynamics.164,165 The analytical solution of this equation, developed in the 20th century, gives the nucleation rate (J) as:
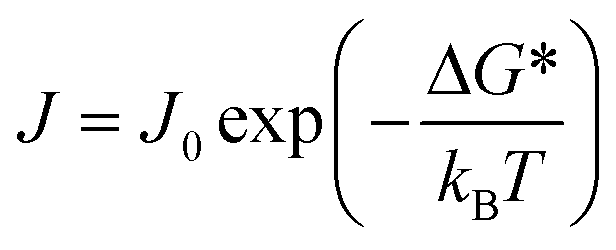 | (1) |
| J0 = zρsAkin | (2) |
Beyond CNT and KNT, extensive theoretical and experimental evidence now supports two-step or nonclassical nucleation mechanisms, where solute molecules first aggregate into dense, amorphous clusters, followed by structural ordering (densification) into crystalline nuclei.168,169 The corresponding free-energy landscape exhibits two barriers – one for cluster formation and another for ordering – each influencing the overall nucleation rate.170 These models better describe the multi-step kinetics observed in proteins, colloids, and small organic molecules. Recent molecular-dynamics and metadynamics simulations have revealed that such multi-step pathways are prevalent even in simple molecular systems.145,171
3.2. Probabilistic models for nucleation
Probabilistic formulations of nucleation employ the Fokker–Planck equation (FPE) to describe the time evolution of the probability density of observing a cluster of a given size.167,172 The FPE can be derived from the master equation for birth–death processes, which balances the rates of cluster growth (birth) and dissolution (death). Expanding this master equation in a Taylor series and truncating at the second order yields a Fokker–Planck-type equation, linking deterministic drift (growth) and stochastic diffusion (fluctuations) in cluster size space.173 The solution to the FPE can be obtained numerically using Ito's stochastic differential equations (SDEs), where an ensemble of simulated trajectories converges to the time-dependent cluster-size distribution.174 These stochastic frameworks bridge molecular-level kinetics and continuum models by explicitly incorporating fluctuations and non-Markovian effects neglected in classical rate theories. Recent studies combine the Fokker–Planck approach with machine-learning algorithms, allowing efficient and accurate solutions.1753.3. Population balance models
Population balance equations (PBEs) are another important class of models applied to study the process of nucleation and growth in crystallization.176,177 PBEs originated from Smoluchowski's coagulation equation, which describes the time evolution of particle populations under aggregation and breakage events.178 PBEs are analogous to traditional transport equations in that they express the change in an internal coordinate (e.g., particle size, volume, or composition) with respect to time and position. In the case of nucleation, this internal coordinate is usually particle size. The solution of PBEs yields the particle-size distribution (PSD) of nuclei or crystals in a crystallizer. Recent studies comparing PBE-based predictions with kinetic rate equations report that both approaches produce comparable accuracy, but PBEs require fewer empirical parameters. Moreover, stochastic extensions of PBEs have been developed to describe fluctuations in cluster populations, enabling the study of the probabilistic nature of nucleation in solutions.MD simulations, which operate on much shorter time scales, has also been extensively applied to study nucleation mechanisms. Comprehensive reviews of molecular simulations for nucleation are available throughout the literature.20,179,180 Because nucleation events often occur within the time scales accessible to MD (typically nanoseconds to microseconds), enhanced-sampling techniques such as metadynamics, umbrella sampling, or forward-flux sampling are required to access rare-event kinetics efficiently.181 These methods allow selectively focusing on key collective variables while disregarding irrelevant motions. In one such study, MD simulations revealed that the mechanism of nucleation changes with cluster size, transitioning from diffuse aggregation to ordered crystal growth as the system crosses the critical nucleus size.182 The authors identified a non-intuitive reaction coordinate that better captured the nucleation pathway, underscoring the need for data-driven and machine-learning-assisted descriptors to interpret nucleation dynamics.
Modeling nucleation during crystallization primarily aims to predict nucleation rates. If we assume that molecular self-assembly and potential polymorphs are well characterized, then differences in polymorph nucleation rates dictate which crystal form will ultimately dominate. Polymorphs with higher nucleation rates are kinetically favored under given conditions. The essential input in these models is the attachment frequency or attachment free energy barrier for a growth unit joining the nucleus. All models discussed above – including CNT, KNT, and PBEs – require this attachment frequency, which depends on molecular interactions and supersaturation. PBEs, in particular, require the explicit input of the nucleation rate to compute the resulting particle-size distribution in a crystallizer. Therefore, coupling PBEs with kinetic rate equations has become an active area of research to close the multiscale modeling gap.183 Such integrated frameworks enable simulation of entire crystallization processes, from nucleation to growth, under realistic process conditions.
3.4. Crystal growth
The shape and size of crystals profoundly influence their physical and functional properties. In many applications, such as pharmaceuticals, optoelectronics, or catalysis, it is essential to control the final crystal morphology. Shape evolution depends on the growth rate of specific crystal faces: faces with higher growth rates disappear quickly, producing elongated or needle-like morphologies, while slow-growing faces become dominant facets. Controlling these growth rates enables targeted shape design. The crystal-growth phase begins once nuclei have formed and molecules attach to the crystal surface. Historically, models of crystal growth began with geometric and equilibrium approaches. The earliest geometric framework was the Bravais–Friedel–Donnay–Harker (BFDH) model, which proposed that the growth rate of a crystal face is inversely proportional to its interplanar spacing.184–186 In contrast, equilibrium models sought the morphology that minimizes total crystal free energy – an assumption valid only when growth kinetics are sufficiently fast to approximate equilibrium. Despite simplifications, the BFDH and equilibrium models laid the foundation for modern crystal growth modeling.187The Hartman–Perdok theory, which extended these models, introduced the concept of periodic bond chains (PBCs) to classify crystal faces as flat (F), stepped (S), or kinked (K) depending on the number of PBCs intersecting the face.188–190 Faces containing multiple PBCs have higher bonding periodicity and thus lower surface energy and greater stability. With time, mechanistic models emerged – such as two-dimensional (2D) nucleation, the Burton–Cabrera–Frank (BCF) model, the Chernov bulk-diffusion model, the Winn–Doherty model, and the Monte Carlo-based MONTY algorithm.191–194 These frameworks clarified that crystal growth typically proceeds through three primary mechanisms: spiral growth, 2D layer-by-layer nucleation, and rough (continuous) growth, as shown in Fig. 9, depending on the supersaturation regime. The Winn–Doherty model in particular advanced the field by defining the growth-mechanism regimes as functions of supersaturation and interfacial free energy. It predicts that: at low supersaturation, spiral growth dominates; at moderate supersaturation, 2D nucleation prevails; and at high supersaturation, rapid rough growth occurs.195 Building upon this foundation, subsequent studies have formulated shape-evolution and morphology-prediction models, continuously refined with experimental validation and computational integration.196
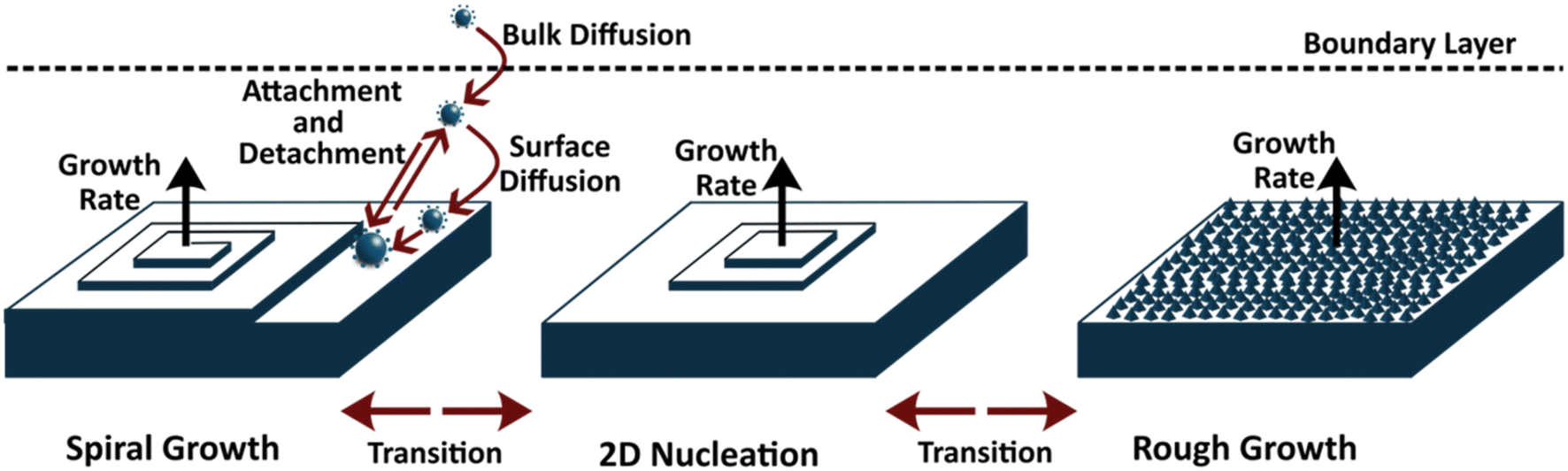 | ||
| Fig. 9 Processes occurring on the surface during crystal growth. Solvated molecules from the bulk solution may attach directly to kink sites or first adsorb and diffuse along the surface. Depending on the level of supersaturation, crystal growth proceeds via spiral, 2D nucleation, or rough growth. Transitions between these mechanisms can occur due to surface defects or solvent-mediated effects. | ||
The fundamental mechanisms of crystal growth – particle addition, aggregation, or breakage – are naturally captured by PBEs.178,197 The generalized stochastic form of PBEs for particulate systems was extended to describe nucleation and growth processes.198 Solving PBEs in more than one dimension (because real crystal growth is inherently three-dimensional), however, remains computationally challenging. Common approaches include dimensionality reduction (via symmetry or shape assumptions) or variable transformation to measurable quantities, though these can reduce predictive accuracy.199 Recent advances employ principal component analysis (PCA) and morphological PBEs, which enable solving multidimensional problems by projecting high-dimensional data onto a reduced vector space without losing shape information.200 The method of characteristics has been particularly effective for solving time-dependent PBEs describing facet-specific growth rates. Modern implementations construct morphology sets, graphs, and domains, which are then solved using the MorphologyDomain software package.16
Despite continuous progress, crystal growth modeling remains incomplete. Current models often neglect the thickness and energetics of the solvation shell surrounding the solute molecule. This solvation layer strongly influences the binding energy of growth units on crystal surfaces, particularly under low supersaturation conditions where attachment kinetics are rate-limiting. The Winn–Doherty model, for instance, assumes isotropic binding energy and computes only relative growth rates. Moreover, a single crystal face may host multiple spirals, pits, and surface defects, which are typically neglected in continuum models. Growth mechanisms can also transition between regimes, as shown in Fig. 9, (e.g., from 2D nucleation to rough growth) depending on interfacial or solvent effects, yet the conditions governing such transitions remain poorly understood. Developing a model that can quantitatively capture growth-unit binding and growth-mechanism transitions will require incorporating molecular-scale free-energy calculations – similar to those used in nucleation modeling – into mesoscale and process-scale frameworks. A more comprehensive review and a new model for crystal growth will be the topic for another article.
4. Perspective – multiscale modeling for dynamic crystal structure prediction
Currently, multiple computational approaches can individually model distinct phases of crystallization – from molecular self-assembly to crystal growth. However, predicting the dynamic evolution of crystal structure requires an integrated multiscale framework capable of coupling these phases, as depicted in Fig. 10, under consistent thermodynamic and kinetic formalisms. This proposed framework functions as an information pipeline: it begins with molecular self-assembly, where simulations identify the dominant ‘synthons’ or pre-nucleation clusters that define the available polymorphic pathways. These structural motifs serve as the reaction coordinates for the nucleation phase, where stochastic equations determine the probability flux of specific polymorphs emerging from solution. Finally, these nucleation rates are passed as birth terms to the crystal growth phase, modeled via PBEs to predict macroscopic properties. The following paragraphs detail how these disparate scales could be mathematically bridged to achieve dynamic prediction.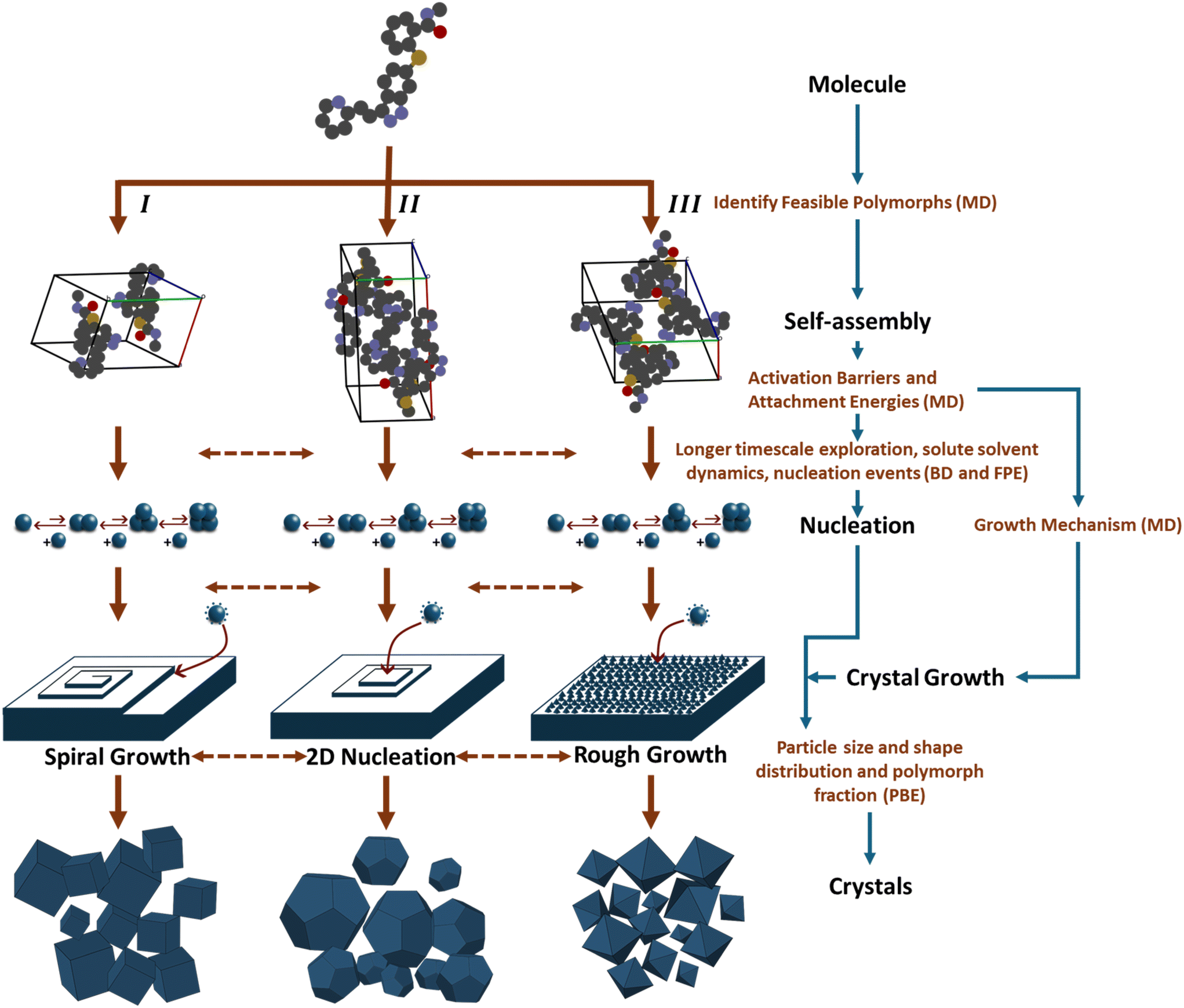 | ||
| Fig. 10 Schematic representation of the multiscale computational framework for modeling crystallization. The workflow illustrates the transition from individual molecules to bulk crystals for three distinct polymorphs (I, II, and III). The process integrates molecular dynamics (to identify feasible polymorphs, activation barriers, and attachment energies), Brownian dynamics and Fokker–Planck equations (for longer timescale exploration, capture nucleation events and solute–solvent dynamics), and population balance equations (to predict macroscopic crystal properties). Information flow from one state to another is represented with blue arrows. | ||
The self-assembly phase of crystallization focuses on identifying all feasible polymorphs of a solute molecule. In a specific solvent environment, solute–solute and solute–solvent interactions constrain the molecules to a limited number of low-energy configurations. This was confirmed through MD simulations for systems such as 5-fluorouracil and TTA, where distinct aggregation motifs or synthons were observed to dictate polymorph outcomes.146,147 Each synthon represents the fundamental unit of crystal growth, and its structural identity determines the feasible crystal forms accessible under given operating conditions. Although reliable force-field parameters and diffusivities remain a bottleneck and are rarely available for new molecules, machine learning models are a key research direction. Additional complexity arises from kinetic limitations: some molecules exhibit slow crystal growth due to sluggish diffusion or solvation-shell restructuring. To overcome MD's limited timescale (typically up to microseconds), BD provides an efficient alternative by solving the Langevin equation of motion rather than Newton's second law.201 BD models the molecular motion as a balance of drift (deterministic) and diffusive (stochastic) forces:
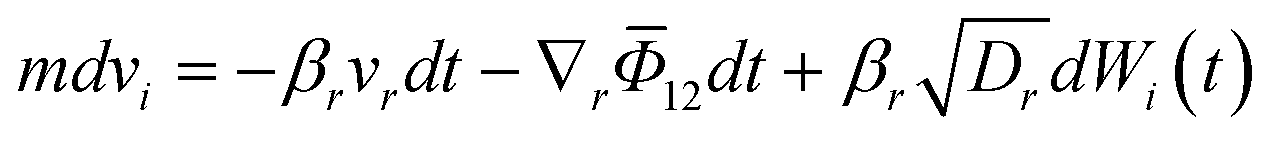 | (3) |
![[capital Phi, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c1.gif) is the mean field potential. For molecules where rotational motion cannot be neglected, the Langevin equation takes the form:
is the mean field potential. For molecules where rotational motion cannot be neglected, the Langevin equation takes the form: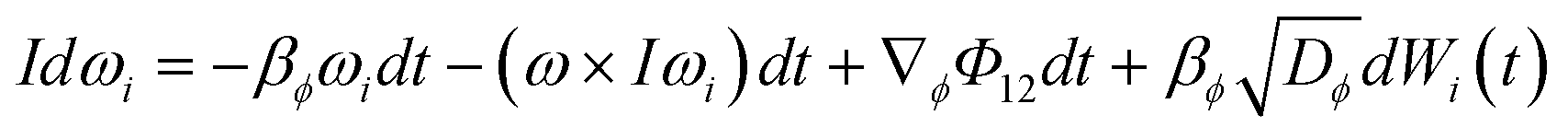 | (4) |
BD simulations thus enable longer-timescale exploration of solute-cluster dynamics, capturing key features of diffusive aggregation relevant to pre-nucleation clusters. To characterize orientation and packing differences between polymorphs, a molecular frame of reference must be defined. While radial distribution functions (RDFs) quantify solute–solute interactions, they cannot distinguish between distinct polymorphs. For small organic molecules, a rigid-body approximation is generally valid. A rigid molecule containing M atoms can be described by its internal atomic coordinates ai relative to its center of mass r. The position of a molecule at any time is given by the center of mass in a fixed Cartesian frame and the relative positions of constituting atoms can be obtained from the polar angle and the azimuthal angle such that:
| ai = T[θ(t), φ(t)]ai(0) | (5) |
Accurate force-field parameterization remains critical for describing both intra- and intermolecular forces during this phase.
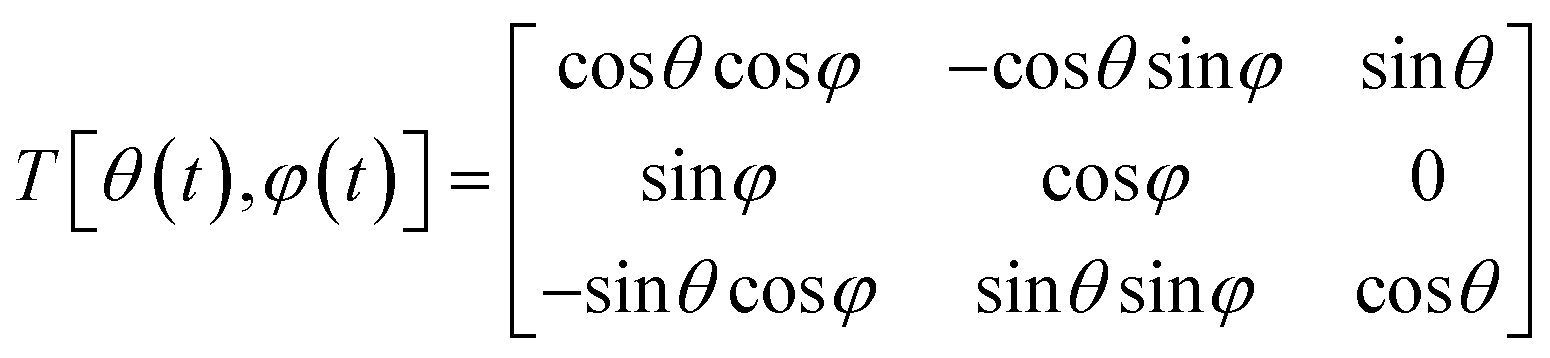
Modeling nucleation is essential for calculating nucleation rates and polymorph selection. This requires evaluation of the free energy of attachment between solute molecules, obtainable from potential-well analyses of solvated and desolvated species.22 Most CSP frameworks treat solvent implicitly or neglect competitive adsorption. Yet solvation dynamics, hydrogen-bonding rearrangements, and impurity incorporation strongly influence nucleation kinetics and facet-specific growth rates. Although detailed protocols for calculating attachment energies will be the topic of another article, once these energy barriers are known, nucleation can be treated stochastically using FPE coupled with the Langevin drift terms. During crystallization under supersaturation, drift terms dominate over diffusive terms in the Langevin equation, indicating that deterministic molecular attachment prevails over random motion. The probability density function of molecular configurations P(n,t) can thus be described by the FPE, allowing computation of time-dependent distributions of pre-nucleation synthons. Each polymorph corresponds to a distinct set of radial and angular coordinates. For a fixed number of solute molecules within the simulation volume, this approach can be used to estimate the population distribution of competing polymorphic clusters in solution.
To analyze which self-assembled structures lead to nucleation, it is necessary to define an internal coordinate that connects the molecular-level cluster evolution with the macroscopic nucleation process. This internal coordinate can be chosen as the particle number (n) or cluster size, which evolves according to:
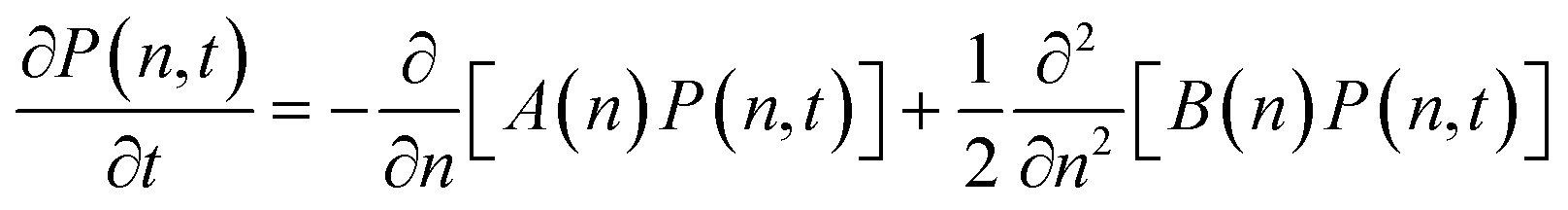 | (6) |
Building on the Fokker–Planck results, PBEs describe how these nuclei evolve into macroscopic crystal populations. PBEs express the number density of crystals n(V, t) as a function of crystal volume (V) and time:
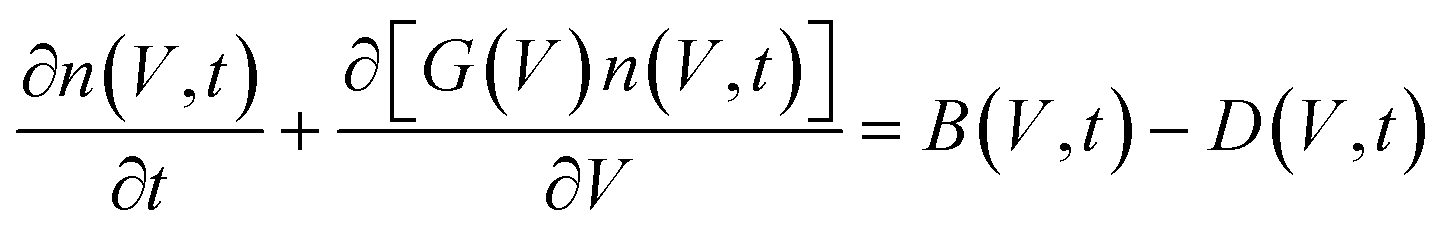 | (7) |
The structure exhibiting the highest nucleation rate or smallest ΔG* will correspond to the dominant polymorph. In practice, this framework allows one to: use MD/BD simulations to identify feasible synthons; compute attachment free energies for each synthon; incorporate these as kinetic parameters into the Fokker–Planck-based probability density model; predict which polymorph will form under given supersaturation and solvent conditions; calculate the particle size distribution and polymorph fraction of the crystals. Such a hierarchical stochastic framework – combining MD, BD, FPE, and PBE – constitutes a dynamic crystal-structure prediction approach that captures both thermodynamic stability and kinetic accessibility of polymorphs.
In the proposed multiscale framework (Fig. 10), machine learning has the potential to serve not merely as an auxiliary tool, but as the critical “bridge” necessary to unify these disparate computational scales. Rather than treating molecular dynamics and population balances as isolated steps, ML algorithms can derive kinetic rate constants and reaction coordinates from high-fidelity molecular simulations and seamlessly pass these parameters to mesoscale Fokker–Planck equations.203 This data-driven connectivity creates a continuous information pipeline: molecular-level insights directly update macroscopic process predictions without the need for prohibitive on-the-fly computations. While the reliability of such ML-integrated workflows is still being established compared to standard physics-based models, this approach offers a promising pathway to transform static individual methods into a dynamic, consolidated prediction engine with a good training data set.
5. Conclusion
Crystallization is an inherently multiscale and multiphase phenomenon, extending from molecular interactions to macroscopic structure evolution. Over the past three decades, research in crystal structure prediction (CSP) has advanced from purely thermodynamic frameworks, centered on lattice-energy minimization, to more sophisticated kinetic and stochastic models that capture the dynamic nature of molecular self-assembly, nucleation, and growth. While energy-minimization techniques have achieved remarkable success in reproducing known crystal structures and discovering new polymorphs, their neglect of nucleation kinetics and solvent-mediated effects continues to limit predictive reliability, especially for flexible molecules and complex crystallization environments.Recent progress in molecular dynamics (MD), Brownian dynamics (BD), and stochastic modeling has significantly broadened our ability to probe early aggregation and self-assembly events. These approaches have confirmed that molecular synthons, the building blocks of crystalline order, often determine the polymorph that ultimately emerges. The inclusion of two-step nucleation mechanisms and probabilistic population-balance equations (PBEs) has enabled more accurate predictions of nucleation rates and particle-size distributions across diverse systems. In parallel, mechanistic crystal-growth models, from the Hartman–Perdok to Winn–Doherty frameworks, have elucidated how supersaturation and interfacial free energy govern the evolution of crystal morphology and growth regimes.
Despite these advances, the field remains fragmented across scales: molecular-level models provide microscopic insight but limited time and length coverage, while continuum and population-balance models capture bulk phenomena but lack molecular specificity. Bridging this gap requires a multiscale, dynamically coupled modeling paradigm capable of linking intermolecular forces, stochastic kinetics, and macroscopic observables. The multiscale model proposed in this review provides a conceptual framework for such integration, beginning with MD-based synthon identification, extending through BD and Fokker–Planck-based stochastic modeling for nucleation, and culminating in PBE-based predictions of crystal growth and morphology.
The future of CSP will depend on the synergistic integration of thermodynamic and kinetic models with data-driven and machine-learning-assisted methods. Emerging hybrid approaches, combining quantum mechanical accuracy, enhanced-sampling kinetics, and morphology-resolved population balances, hold promise for transforming CSP from a static predictive tool into a dynamic, process-aware modeling framework. Such unified methodologies will not only improve the reliability of polymorph prediction but also enable rational control of crystallization pathways – a goal central to materials design, pharmaceutical manufacturing, and sustainable chemical engineering.
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This material is based on the work performed at the Materials and Systems Engineering Laboratory at the University of Illinois Chicago (UIC). M. R. S. acknowledges funding support from UIC and the US National Science Foundation (NSF) – EFRI Program (award no. 2132022), and CMMI (award no. 2326714).References
- A. Y. Lee, D. Erdemir and A. S. Myerson, Crystal Polymorphism in Chemical Process Development, Annu. Rev. Chem. Biomol. Eng., 2011, 2(1), 259–280, DOI:10.1146/annurev-chembioeng-061010-114224.
- R. Adhiyaman and S. K. Basu, Crystal modification of dipyridamole using different solvents and crystallization conditions, Int. J. Pharm., 2006, 321(1–2), 27–34, DOI:10.1016/j.ijpharm.2006.04.021.
- G. M. Day, Advances in Crystal Structure Prediction and Applications to Pharmaceutical Materials, in Computational Pharmaceutical Solid State Chemistry, Wiley, 2016, pp. 87–115 Search PubMed.
- J. Maddox, Crystals from first principles, Nature, 1988, 335(6187), 201–201, DOI:10.1038/335201a0.
- R. R. Bhawnani, N. K. Dandu, P. K. R. Podupu, A. T. Ngo and M. R. Singh, Sequential Hydrolysis of Metal Oxo Clusters Drives Polymorphism in Electrodeposited Zirconium Metal–Organic Frameworks, Chem. Mater., 2024, 36(5), 2402–2411, DOI:10.1021/acs.chemmater.3c03065.
- R. R. Bhawnani, M. L. Barsoum, A. Dhakal, V. V. Gande, R. Y. Nemade, P. K. R. Podupu, R. dos Reis, G. Giri, V. Berry, V. Dravid and M. R. Singh, Dynamic structural and morphological transformations in high-entropy metal-organic frameworks, Mater. Today Chem., 2025, 49, 103009, DOI:10.1016/j.mtchem.2025.103009.
- J. Sanz-Aparicio, S. Martínez-Carrera, S. García-Blanco and A. Conde, Lattice-energy calculations on organometallic compounds, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1988, 44(3), 259–262, DOI:10.1107/s0108768188001053.
- P. K. Reddy, P. Verma, A. Dhakal, R. R. Bhawnani, M. Phister, A. V. Dighe, K. H. Stone, G. Giri and M. R. Singh, Mechanistic insights into metal-organic framework thin film growth from microkinetic analysis of in situ X-ray scattering data, Matter, 2025, 9(1), 102430, DOI:10.1016/j.matt.2025.102430.
- G. J. Beran, Modeling polymorphic molecular crystals with electronic structure theory, Chem. Rev., 2016, 116(9), 5567–5613 CrossRef CAS PubMed.
- R. F. Stewart, Valence Structure from X-Ray Diffraction Data: Physical Properties, J. Chem. Phys., 1972, 57(4), 1664–1668 CrossRef CAS.
- M. Iggland and M. Mazzotti, Population balance modeling with size-dependent solubility: Ostwald ripening, Cryst. Growth Des., 2012, 12(3), 1489–1500 CrossRef CAS.
- E. H. Chow, D.-K. Bučar and W. Jones, New opportunities in crystal engineering–the role of atomic force microscopy in studies of molecular crystals, Chem. Commun., 2012, 48(74), 9210–9226 RSC.
- P. Hartman and W. G. Perdok, On the relations between structure and morphology of crystals. I, Acta Crystallogr., 1955, 8, 49 CrossRef.
- R. F. P. Grimbergen, H. Meekes, P. Bennema, C. S. Strom and L. J. P. Vogels, On the prediction of crystal morphology. I. The Hartman–Perdok theory revisited, Acta Crystallogr., Sect. A: Found. Adv., 1998, 54(4), 491–500, DOI:10.1107/S0108767397019806.
- A. V. Dighe, R. Y. Nemade and M. R. Singh, Modeling and Simulation of Crystallization of Metal–Organic Frameworks, Processes, 2019, 7(8), 11, DOI:10.3390/pr7080527.
- M. R. Singh and D. Ramkrishna, A Comprehensive Approach to Predicting Crystal Morphology Distributions with Population Balances, in Crystal Growth & Design, 2013, vol. 13, pp. 1397–1411 Search PubMed.
- G. J. O. Beran, Modeling Polymorphic Molecular Crystals with Electronic Structure Theory, Chem. Rev., 2016, 116(9), 5567–5613, DOI:10.1021/acs.chemrev.5b00648.
- S. L. Price, Computational prediction of organic crystal structures and polymorphism, Int. Rev. Phys. Chem., 2008, 27(3), 541–568, DOI:10.1080/01442350802102387.
- C. H. Chan, M. Sun and B. Huang, Application of machine learning for advanced material prediction and design, EcoMat, 2022, 4(4), e12194 Search PubMed.
- G. C. Sosso, J. Chen, S. J. Cox, M. Fitzner, P. Pedevilla, A. Zen and A. Michaelides, Crystal Nucleation in Liquids: Open Questions and Future Challenges in Molecular Dynamics Simulations, Chem. Rev., 2016, 116(12), 7078–7116, DOI:10.1021/acs.chemrev.5b00744.
- A. S. Myerson, Handbook of Industrial Crystallization, 2002 Search PubMed.
- A. V. Dighe, P. K. R. Podupu, P. Coliaie and M. R. Singh, Three-Step Mechanism of Antisolvent Crystallization, Cryst. Growth Des., 2022, 22(5), 3119–3127, DOI:10.1021/acs.cgd.2c00014.
- A. V. Dighe and M. R. Singh, Solvent fluctuations in the solvation shell determine the activation barrier for crystal growth rates, Proc. Natl. Acad. Sci. U. S. A., 2019, 116, 23954 CrossRef CAS PubMed.
- M. Larson and J. Garside, Solute clustering in supersaturated solutions, Chem. Eng. Sci., 1986, 41(5), 1285–1289 CrossRef CAS.
- A. V. Dighe, L. Huelsenbeck, R. R. Bhawnani, P. Verma, K. H. Stone, M. R. Singh and G. Giri, Autocatalysis and Oriented Attachment Direct the Synthesis of a Metal–Organic Framework, JACS Au, 2022, 2(2), 453–462, DOI:10.1021/jacsau.1c00494.
- M. A. Lovette and M. F. Doherty, Predictive modeling of supersaturation-dependent crystal shapes, Cryst. Growth Des., 2012, 12(2), 656–669 CrossRef CAS.
- F. Artusio and R. Pisano, Surface-induced crystallization of pharmaceuticals and biopharmaceuticals: A review, Int. J. Pharm., 2018, 547(1–2), 190–208 CrossRef CAS PubMed.
- L. H. Chagas, M. C. De Souza, W. R. Do Carmo, H. A. De Abreu and R. Diniz, Structure characterization of materials by association of the raman spectra and x-ray diffraction data, in Vibrational Spectroscopy, IntechOpen, 2012 Search PubMed.
- D. S. Coombes, C. R. A. Catlow, J. D. Gale, A. L. Rohl and S. L. Price, Calculation of attachment energies and relative volume growth rates as an aid to polymorph prediction, Cryst. Growth Des., 2005, 5(3), 879–885 CrossRef CAS.
- J. Pannetier, J. Bassas-Alsina, J. Rodriguez-Carvajal and V. Caignaert, Prediction of crystal structures from crystal chemistry rules by simulated annealing, Nature, 1990, 346(6282), 343–345, DOI:10.1038/346343a0.
- R. J. Gdanitz, Prediction of molecular crystal structures by Monte Carlo simulated annealing without reference to diffraction data, Chem. Phys. Lett., 1992, 190(3–4), 391–396, DOI:10.1016/0009-2614(92)85357-g.
- J. A. Foster, K. K. Damodaran, A. Maurin, G. M. Day, H. P. G. Thompson, G. J. Cameron, J. C. Bernal and J. W. Steed, Pharmaceutical polymorph control in a drug-mimetic supramolecular gel, Chem. Sci., 2017, 8(1), 78–84, 10.1039/c6sc04126d.
- J.-B. Arlin, L. S. Price, S. L. Price and A. J. Florence, A strategy for producing predicted polymorphs: catemeric carbamazepine form V, Chem. Commun., 2011, 47(25), 7074, 10.1039/c1cc11634g.
- C. Noguez and F. Hidalgo, Ab Initio Electronic Circular Dichroism of Fullerenes, Single-Walled Carbon Nanotubes, and Ligand-Protected Metal Nanoparticles, Chirality, 2014, 26(9), 553–562, DOI:10.1002/chir.22348.
- V. K. Srirambhatla, R. Guo, S. L. Price and A. J. Florence, Isomorphous template induced crystallisation: a robust method for the targeted crystallisation of computationally predicted metastable polymorphs, Chem. Commun., 2016, 52(46), 7384–7386, 10.1039/c6cc01710j.
- R. W. Lancaster, P. G. Karamertzanis, A. T. Hulme, D. A. Tocher, T. C. Lewis and S. L. Price, The polymorphism of progesterone: Stabilization of a ‘disappearing’ polymorph by co-crystallization, J. Pharm. Sci., 2007, 96(12), 3419–3431, DOI:10.1002/jps.20983.
- M. Baias, J.-N. Dumez, P. H. Svensson, S. Schantz, G. M. Day and L. Emsley, De Novo Determination of the Crystal Structure of a Large Drug Molecule by Crystal Structure Prediction-Based Powder NMR Crystallography, J. Am. Chem. Soc., 2013, 135(46), 17501–17507, DOI:10.1021/ja4088874.
- O. G. Uzoh, A. J. Cruz-Cabeza and S. L. Price, Is the Fenamate Group a Polymorphophore? Contrasting the Crystal Energy Landscapes of Fenamic and Tolfenamic Acids, Cryst. Growth Des., 2012, 12(8), 4230–4239, DOI:10.1021/cg3007348.
- D. E. Braun, R. M. Bhardwaj, J.-B. Arlin, A. J. Florence, V. Kahlenberg, U. J. Griesser, D. A. Tocher and S. L. Price, Absorbing a Little Water: The Structural, Thermodynamic, and Kinetic Relationship between Pyrogallol and Its Tetarto-Hydrate, Cryst. Growth Des., 2013, 13(9), 4071–4083, DOI:10.1021/cg4009015.
- M. Baias, C. M. Widdifield, J.-N. Dumez, H. P. G. Thompson, T. G. Cooper, E. Salager, S. Bassil, R. S. Stein, A. Lesage, G. M. Day and L. Emsley, Powder crystallography of pharmaceutical materials by combined crystal structure prediction and solid-state 1H NMR spectroscopy, Phys. Chem. Chem. Phys., 2013, 15(21), 8069, 10.1039/c3cp41095a.
- M. D. Eddleston, K. E. Hejczyk, E. G. Bithell, G. M. Day and W. Jones, Polymorph Identification and Crystal Structure Determination by a Combined Crystal Structure Prediction and Transmission Electron Microscopy Approach, Chem. – Eur. J., 2013, 19(24), 7874–7882, DOI:10.1002/chem.201204368.
- L. S. Price, J. A. McMahon, S. R. Lingireddy, S.-F. Lau, B. A. Diseroad, S. L. Price and S. M. Reutzel-Edens, A molecular picture of the problems in ensuring structural purity of tazofelone, J. Mol. Struct., 2014, 1078, 26–42, DOI:10.1016/j.molstruc.2014.01.014.
- D. E. Braun, L. H. Koztecki, J. A. McMahon, S. L. Price and S. M. Reutzel-Edens, Navigating the Waters of Unconventional Crystalline Hydrates, Mol. Pharmaceutics, 2015, 12(8), 3069–3088, DOI:10.1021/acs.molpharmaceut.5b00357.
- M. Selent, J. Nyman, J. Roukala, M. Ilczyszyn, R. Oilunkaniemi, P. J. Bygrave, R. Laitinen, J. Jokisaari, G. M. Day and P. Lantto, Inside Back Cover: Clathrate Structure Determination by Combining Crystal Structure Prediction with Computational and Experimental 129Xe NMR Spectroscopy (Chem. Eur. J. 22/2017), Chem. – Eur. J., 2017, 23(22), 5386–5386, DOI:10.1002/chem.201700348.
- M. Arhangelskis, M. D. Eddleston, D. G. Reid, G. M. Day, D. K. Bučar, A. J. Morris and W. Jones, Rationalization of the Color Properties of Fluorescein in the Solid State: A Combined Computational and Experimental Study, Chem. – Eur. J., 2016, 22(29), 10065–10073, DOI:10.1002/chem.201601340.
- M. Habgood, Form II Caffeine: A Case Study for Confirming and Predicting Disorder in Organic Crystals, Cryst. Growth Des., 2011, 11(8), 3600–3608, DOI:10.1021/cg2005612.
- B. Moulton and M. J. Zaworotko, From Molecules to Crystal Engineering: Supramolecular Isomerism and Polymorphism in Network Solids, Chem. Rev., 2001, 101(6), 1629–1658, DOI:10.1021/cr9900432.
- S. L. Price, D. E. Braun and S. M. Reutzel-Edens, Can computed crystal energy landscapes help understand pharmaceutical solids?, Chem. Commun., 2016, 52(44), 7065–7077, 10.1039/c6cc00721j.
- F. R. Theobald, C. Catlow and A. Cormack, Lattice energy minimization as a complementary technique to refine structures obtained by high-resolution electron microscopy, J. Solid State Chem., 1984, 52(1), 80–90 CrossRef CAS.
- D. E. Braun, J. A. McMahon, L. H. Koztecki, S. L. Price and S. M. Reutzel-Edens, Contrasting Polymorphism of Related Small Molecule Drugs Correlated and Guided by the Computed Crystal Energy Landscape, Cryst. Growth Des., 2014, 14(4), 2056–2072, DOI:10.1021/cg500185h.
- L. M. LeBlanc, A. Otero-de-la-Roza and E. R. Johnson, Composite and Low-Cost Approaches for Molecular Crystal Structure Prediction, J. Chem. Theory Comput., 2018, 14(4), 2265–2276 CrossRef CAS PubMed.
- J. Ropers, M. M. Mosca, O. Anosova, V. Kurlin and A. I. Cooper, Fast predictions of lattice energies by continuous isometry invariants of crystal structures, in International conference on data analytics and management in data intensive domains, 2021, Springer, pp. 178–192 Search PubMed.
- S. R. Vippagunta, H. G. Brittain and D. J. W. Grant, Crystalline solids, Adv. Drug Delivery Rev., 2001, 48(1), 3–26, DOI:10.1016/s0169-409x(01)00097-7.
- S. L. Price, The computational prediction of pharmaceutical crystal structures and polymorphism, Adv. Drug Delivery Rev., 2004, 56(3), 301–319, DOI:10.1016/j.addr.2003.10.006.
- S. Datta and D. J. W. Grant, Crystal structures of drugs: advances in determination, prediction and engineering, Nat. Rev. Drug Discovery, 2004, 3(1), 42–57, DOI:10.1038/nrd1280.
- G. R. Desiraju, Crystal Engineering: A Holistic View, Angew. Chem., Int. Ed., 2007, 46(44), 8342–8356, DOI:10.1002/anie.200700534.
- S. M. Woodley and R. Catlow, Crystal structure prediction from first principles, Nat. Mater., 2008, 7(12), 937–946, DOI:10.1038/nmat2321.
- T. S. Thakur, R. Dubey and G. R. Desiraju, Crystal Structure and Prediction, Annu. Rev. Phys. Chem., 2015, 66(1), 21–42, DOI:10.1146/annurev-physchem-040214-121452.
- J. Graser, S. K. Kauwe and T. D. Sparks, Machine Learning and Energy Minimization Approaches for Crystal Structure Predictions: A Review and New Horizons, Chem. Mater., 2018, 30(11), 3601–3612, DOI:10.1021/acs.chemmater.7b05304.
- A. R. Oganov, C. J. Pickard, Q. Zhu and R. J. Needs, Structure prediction drives materials discovery, Nat. Rev. Mater., 2019, 4(5), 331–348, DOI:10.1038/s41578-019-0101-8.
- D. H. Bowskill, I. J. Sugden, S. Konstantinopoulos, C. S. Adjiman and C. C. Pantelides, Crystal Structure Prediction Methods for Organic Molecules: State of the Art, Annu. Rev. Chem. Biomol. Eng., 2021, 12, 593–623, DOI:10.1146/annurev-chembioeng-060718-030256.
- X. Yin and C. E. Gounaris, Search methods for inorganic materials crystal structure prediction, Curr. Opin. Chem. Eng., 2022, 35, 100726, DOI:10.1016/j.coche.2021.100726.
- R. J. Clements, J. Dickman, J. Johal, J. Martin, J. Glover and G. M. Day, Roles and opportunities for machine learning in organic molecular crystal structure prediction and its applications, MRS Bull., 2022, 47(10), 1054–1062, DOI:10.1557/s43577-022-00434-y.
- G. J. O. Beran, Frontiers of molecular crystal structure prediction for pharmaceuticals and functional organic materials, Chem. Sci., 2023, 14(46), 13290–13312, 10.1039/d3sc03903j.
- G. M. Day, J. Chisholm, N. Shan, W. S. Motherwell and W. Jones, An assessment of lattice energy minimization for the prediction of molecular organic crystal structures, Cryst. Growth Des., 2004, 4(6), 1327–1340 CrossRef CAS.
- M. e. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- F. Neese, Software update: The ORCA program system–Version 5.0, WIREs Comput. Mol. Sci., 2022, 12, e1606 CrossRef.
- S. G. Balasubramani, G. P. Chen, S. Coriani, M. Diedenhofen, M. S. Frank, Y. J. Franzke, F. Furche, R. Grotjahn, M. E. Harding, C. Hättig, A. Hellweg, B. Helmich-Paris, C. Holzer, U. Huniar, M. Kaupp, A. Marefat Khah, S. Karbalaei Khani, T. Müller, F. Mack, B. D. Nguyen, S. M. Parker, E. Perlt, D. Rappoport, K. Reiter, S. Roy, M. Rückert, G. Schmitz, M. Sierka, E. Tapavicza, D. P. Tew, C. van Wüllen, V. K. Voora, F. Weigend, A. Wodyński and J. M. Yu, TURBOMOLE: Modular program suite for ab initio quantum-chemical and condensed-matter simulations, J. Chem. Phys., 2020, 152(18), 184107, DOI:10.1063/5.0004635.
- T. G. Cooper, W. Jones, W. D. S. Motherwell and G. M. Day, Database guided conformation selection in crystal structure prediction of alanine, CrystEngComm, 2007, 9(7), 595, 10.1039/b702136d.
- H. P. G. Thompson and G. M. Day, Which conformations make stable crystal structures? Mapping crystalline molecular geometries to the conformational energy landscape, Chem. Sci., 2014, 5(8), 3173–3182, 10.1039/c4sc01132e.
- S. W. H. Bragg, The Structure of Organic Crystals, Proc. Phys. Soc., London, 1921, 34(1), 33–50, DOI:10.1088/1478-7814/34/1/306.
- R. S. Judson, M. Colvin, J. Meza, A. Huffer and D. Gutierrez, Do intelligent configuration search techniques outperform random search for large molecules?, Int. J. Quantum Chem., 1992, 44(2), 277–290 CrossRef CAS.
- P. W. Butler, R. Hafizi and G. M. Day, Machine-learned potentials by active learning from organic crystal structure prediction landscapes, J. Phys. Chem. A, 2024, 128(5), 945–957 CrossRef CAS PubMed.
- Y. Zuo, M. Qin, C. Chen, W. Ye, X. Li, J. Luo and S. P. Ong, Accelerating materials discovery with Bayesian optimization and graph deep learning, Mater. Today, 2021, 51, 126–135 CrossRef CAS.
- D. Zhou, I. Bier, B. Santra, L. D. Jacobson, C. Wu, A. Garaizar Suarez, B. R. Almaguer, H. Yu, R. Abel and R. A. Friesner, A robust crystal structure prediction method to support small molecule drug development with large scale validation and blind study, Nat. Commun., 2025, 16(1), 2210 CrossRef CAS PubMed.
- S. Yang and G. M. Day, Exploration and optimization in crystal structure prediction: Combining basin hopping with quasi-random sampling, J. Chem. Theory Comput., 2021, 17(3), 1988–1999 CrossRef CAS PubMed.
- P. Hajela, Stochastic search in structural optimization-genetic algorithms and simulated annealing, in Structural optimization: Status and promise, 1993, pp. 611–635 Search PubMed.
- D. Henderson, S. H. Jacobson and A. W. Johnson, The theory and practice of simulated annealing, in Handbook of metaheuristics, Springer, 2003, pp. 287–319 Search PubMed.
- A. R. Oganov, A. O. Lyakhov and M. Valle, How Evolutionary Crystal Structure Prediction Works and Why, Acc. Chem. Res., 2011, 44(3), 227–237 CrossRef CAS PubMed.
- N. L. Abraham and M. I. Probert, A periodic genetic algorithm with real-space representation for crystal structure and polymorph prediction, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 73(22), 224104 CrossRef.
- T. Yamashita, H. Kino, K. Tsuda, T. Miyake and T. Oguchi, Hybrid algorithm of Bayesian optimization and evolutionary algorithm in crystal structure prediction, Sci. Technol. Adv. Mater.: Methods, 2022, 2(1), 67–74 CAS.
- D. E. Akporiaye, H. Fjellvåg, E. N. Halvorsen, J. Hustveit, A. Karlsson and K. P. Lillerud, UiO-7: A New Aluminophosphate Phase Solved by Simulated Annealing and High-Resolution Powder Diffraction, J. Phys. Chem., 1996, 100(41), 16641–16646, DOI:10.1021/jp961046a.
- M. W. Deem and J. M. Newsam, Determination of 4-connected framework crystal structures by simulated annealing, Nature, 1989, 342(6247), 260–262, DOI:10.1038/342260a0.
- M. W. Deem and J. M. Newsam, Framework crystal structure solution by simulated annealing: test application to known zeolite structures, J. Am. Chem. Soc., 1992, 114(18), 7189–7198, DOI:10.1021/ja00044a035.
- B. Tidor, Simulated annealing on free energy surfaces by a combined molecular dynamics and Monte Carlo approach, J. Phys. Chem., 1993, 97(5), 1069–1073 CrossRef CAS.
- A. R. Oganov and C. W. Glass, Crystal structure prediction using ab initio evolutionary techniques: Principles and applications, J. Chem. Phys., 2006, 124 (24), DOI:10.1063/1.2210932.
- S. Bahmann and J. Kortus, EVO—Evolutionary algorithm for crystal structure prediction, Comput. Phys. Commun., 2013, 184(6), 1618–1625, DOI:10.1016/j.cpc.2013.02.007.
- V. E. Bazterra, M. B. Ferraro and J. C. Facelli, Modified genetic algorithm to model crystal structures: III. Determination of crystal structures allowing simultaneous molecular geometry relaxation, Int. J. Quantum Chem., 2003, 96(4), 312–320, DOI:10.1002/qua.10726.
- V. E. Bazterra, M. B. Ferraro and J. C. Facelli, Modified genetic algorithm to model crystal structures. I. Benzene, naphthalene and anthracene, J. Chem. Phys., 2002, 116(14), 5984–5991, DOI:10.1063/1.1458547.
- V. E. Bazterra, M. B. Ferraro and J. C. Facelli, Modified genetic algorithm to model crystal structures. II. Determination of a polymorphic structure of benzene using enthalpy minimization, J. Chem. Phys., 2002, 116(14), 5992–5995, DOI:10.1063/1.1458548.
- Q. Tong, L. Xue, J. Lv, Y. Wang and Y. Ma, Accelerating CALYPSO structure prediction by data-driven learning of a potential energy surface, Faraday Discuss., 2018, 211, 31–43 RSC.
- F. Curtis, X. Li, T. Rose, A. Vazquez-Mayagoitia, S. Bhattacharya, L. M. Ghiringhelli and N. Marom, GAtor: a first-principles genetic algorithm for molecular crystal structure prediction, J. Chem. Theory Comput., 2018, 14(4), 2246–2264 CrossRef CAS PubMed.
- K. J. Michel and C. Wolverton, Symmetry building Monte Carlo-based crystal structure prediction, Comput. Phys. Commun., 2014, 185(5), 1389–1393, DOI:10.1016/j.cpc.2014.01.015.
- J. Pillardy, Y. A. Arnautova, C. Czaplewski, K. D. Gibson and H. A. Scheraga, Conformation-family Monte Carlo: A new method for crystal structure prediction, Proc. Natl. Acad. Sci. U. S. A., 2001, 98(22), 12351–12356, DOI:10.1073/pnas.231479298.
- H. Kooijman, B. P. van Eijck and J. Kroon, Molecular dynamics simulations of crystal structure containing charged molecules, J. Mol. Struct., 1992, 268(1–3), 283–292, DOI:10.1016/0022-2860(92)85077-t.
- E. Schneider, L. Vogt and M. E. Tuckerman, Exploring polymorphism of benzene and naphthalene with free energy based enhanced molecular dynamics, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(4), 542–550, DOI:10.1107/s2052520616007873.
- R. L. Marchese Robinson, D. Geatches, C. Morris, R. Mackenzie, A. G. Maloney, K. J. Roberts, A. Moldovan, E. Chow, K. Pencheva and D. R. M. Vatvani, Evaluation of force-field calculations of lattice energies on a large public dataset, assessment of pharmaceutical relevance, and comparison to density functional theory, J. Chem. Inf. Model., 2019, 59(11), 4778–4792 CrossRef CAS PubMed.
- J. Nyman, O. S. Pundyke and G. M. Day, Accurate force fields and methods for modelling organic molecular crystals at finite temperatures, Phys. Chem. Chem. Phys., 2016, 18(23), 15828–15837 Search PubMed.
- J. R. Holden, Z. Du and H. L. Ammon, Prediction of possible crystal structures for C-, H-, N-, O-, and F-containing organic compounds, J. Comput. Chem., 1993, 14(4), 422–437, DOI:10.1002/jcc.540140406.
- A. M. Chaka, R. Zaniewski, W. Youngs, C. Tessier and G. Klopman, Predicting the crystal structure of organic molecular materials, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 1996, 52(1), 165–183, DOI:10.1107/s0108768195006987.
- B. P. Van Eijck, Crystal structure predictions using five space groups with two independent molecules. The case of small organic acids, J. Comput. Chem., 2002, 23(4), 456–462, DOI:10.1002/jcc.10042.
- B. P. van Eijck and J. Kroon, Upack program package for crystal structure prediction: Force fields and crystal structure generation for small carbohydrate molecules, J. Comput. Chem., 1999, 20(8), 799–812, DOI:10.1002/(sici)1096-987x(199906)20:8<799::aid-jcc6>3.0.co;2-z.
- M. A. Neumann, F. J. J. Leusen and J. Kendrick, A Major Advance in Crystal Structure Prediction, Angew. Chem., Int. Ed., 2008, 47(13), 2427–2430, DOI:10.1002/anie.200704247.
- D. M. Elking, L. Fusti-Molnar and A. Nichols, Crystal structure prediction of rigid molecules, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(4), 488–501, DOI:10.1107/s2052520616010118.
- C. C. Fischer, K. J. Tibbetts, D. Morgan and G. Ceder, Predicting crystal structure by merging data mining with quantum mechanics, Nat. Mater., 2006, 5(8), 641–646, DOI:10.1038/nmat1691.
- G. M. Day, W. D. S. Motherwell and W. Jones, A strategy for predicting the crystal structures of flexible molecules: the polymorphism of phenobarbital, Phys. Chem. Chem. Phys., 2007, 9(14), 1693–1704, 10.1039/b612190j.
- P. G. Karamertzanis and C. C. Pantelides, Ab initio crystal structure prediction. II. Flexible molecules, Mol. Phys., 2007, 105(2–3), 273–291, DOI:10.1080/00268970601143317.
- J. Yang, W. Hu, D. Usvyat, D. Matthews, M. Schütz and G. K.-L. Chan, Ab initio determination of the crystalline benzene lattice energy to sub-kilojoule/mole accuracy, Science, 2014, 345(6197), 640–643, DOI:10.1126/science.1254419.
- M. A. Neumann, Tailor-made force fields for crystal-structure prediction, J. Phys. Chem. B, 2008, 112(32), 9810–9829 CrossRef CAS PubMed.
- S. Anker, D. McKechnie, P. Mulheran, J. Sefcik and K. Johnston, Assessment of GAFF and OPLS force fields for urea: crystal and aqueous solution properties, Cryst. Growth Des., 2023, 24(1), 143–158 CrossRef PubMed.
- C. J. Nickerson and E. R. Johnson, Assessment of a foundational machine-learned potential for energy ranking of molecular crystal polymorphs, Phys. Chem. Chem. Phys., 2025, 27(22), 11930–11940, 10.1039/D5CP00593K.
- K. S. Nayal, D. O'Connor, R. Zubatyuk, D. M. Anstine, Y. Yang, R. Tom, W. Deng, K. Tang, N. Marom and O. Isayev, Efficient Molecular Crystal Structure Prediction and Stability Assessment with AIMNet2 Neural Network Potentials, Cryst. Growth Des., 2025, 25(21), 9092–9106 CrossRef CAS PubMed.
- D. McDonagh, C.-K. Skylaris and G. M. Day, Machine-learned fragment-based energies for crystal structure prediction, J. Chem. Theory Comput., 2019, 15(4), 2743–2758 CrossRef CAS PubMed.
- J. P. M. Lommerse, W. D. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Gavezzotti, D. W. M. Hofmann, F. J. J. Leusen, W. T. M. Mooij, S. L. Price, B. Schweizer, M. U. Schmidt, B. P. van Eijck, P. Verwer and D. E. Williams, A test of crystal structure prediction of small organic molecules, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2000, 56(4), 697–714, DOI:10.1107/s0108768100004584.
- W. D. S. Motherwell, H. L. Ammon, J. D. Dunitz, A. Dzyabchenko, P. Erk, A. Gavezzotti, D. W. M. Hofmann, F. J. J. Leusen, J. P. M. Lommerse, W. T. M. Mooij, S. L. Price, H. Scheraga, B. Schweizer, M. U. Schmidt, B. P. van Eijck, P. Verwer and D. E. Williams, Crystal structure prediction of small organic molecules: a second blind test, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2002, 58(4), 647–661, DOI:10.1107/s0108768102005669.
- G. M. Day, W. D. S. Motherwell, H. L. Ammon, S. X. M. Boerrigter, R. G. Della Valle, E. Venuti, A. Dzyabchenko, J. D. Dunitz, B. Schweizer and B. P. van Eijck, et al., A third blind test of crystal structure prediction, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2005, 61(5), 511–527, DOI:10.1107/s0108768105016563.
- G. M. Day, T. G. Cooper, A. J. Cruz-Cabeza, K. E. Hejczyk, H. L. Ammon, S. X. M. Boerrigter, J. S. Tan, R. G. Della Valle, E. Venuti and J. Jose, et al., Significant progress in predicting the crystal structures of small organic molecules – a report on the fourth blind test, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2009, 65(2), 107–125, DOI:10.1107/s0108768109004066.
- D. A. Bardwell, C. S. Adjiman, Y. A. Arnautova, E. Bartashevich, S. X. M. Boerrigter, D. E. Braun, A. J. Cruz-Cabeza, G. M. Day, R. G. Della Valle and G. R. Desiraju, et al., Towards crystal structure prediction of complex organic compounds – a report on the fifth blind test, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2011, 67(6), 535–551, DOI:10.1107/s0108768111042868.
- A. M. Reilly, R. I. Cooper, C. S. Adjiman, S. Bhattacharya, A. D. Boese, J. G. Brandenburg, P. J. Bygrave, R. Bylsma, J. E. Campbell and R. Car, et al., Report on the sixth blind test of organic crystal structure prediction methods, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2016, 72(4), 439–459, DOI:10.1107/s2052520616007447.
- L. M. Hunnisett, N. Francia, J. Nyman, N. S. Abraham, S. Aitipamula, T. Alkhidir, M. Almehairbi, A. Anelli, D. M. Anstine and J. E. Anthony, et al., The seventh blind test of crystal structure prediction: structure ranking methods, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2024, 80(6), 548–574, DOI:10.1107/S2052520624008679.
- L. M. Hunnisett, J. Nyman, N. Francia, N. S. Abraham, C. S. Adjiman, S. Aitipamula, T. Alkhidir, M. Almehairbi, A. Anelli and D. M. Anstine, et al., The seventh blind test of crystal structure prediction: structure generation methods, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2024, 80(6), 517–547, DOI:10.1107/S2052520624007492.
- G. M. Day, Current approaches to predicting molecular organic crystal structures, Crystallogr. Rev., 2011, 17(1), 3–52 CrossRef.
- R. A. Mayo, A. J. Price, A. Otero-de-la-Roza and E. R. Johnson, Assessment of the exchange-hole dipole moment dispersion correction for the energy ranking stage of the seventh crystal structure prediction blind test, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2024, 80(6), 595–605, DOI:10.1107/S2052520624002774.
- J. Hoja and A. Tkatchenko, First-principles stability ranking of molecular crystal polymorphs with the DFT+ MBD approach, Faraday Discuss., 2018, 211, 253–274 RSC.
- S. Wen and G. J. Beran, Crystal polymorphism in oxalyl dihydrazide: Is empirical DFT-D accurate enough?, J. Chem. Theory Comput., 2012, 8(8), 2698–2705 Search PubMed.
- S. Grimme, A. Hansen, J. G. Brandenburg and C. Bannwarth, Dispersion-corrected mean-field electronic structure methods, Chem. Rev., 2016, 116(9), 5105–5154 CrossRef CAS PubMed.
- K. R. Bryenton, A. A. Adeleke, S. G. Dale and E. R. Johnson, Delocalization error: The greatest outstanding challenge in density-functional theory, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2023, 13(2), e1631 CAS.
- G. J. Beran, I. J. Sugden, C. Greenwell, D. H. Bowskill, C. C. Pantelides and C. S. Adjiman, How many more polymorphs of ROY remain undiscovered, Chem. Sci., 2022, 13(5), 1288–1297 RSC.
- G. J. Beran, C. Greenwell, C. Cook and J. Rezac, Improved description of intra-and intermolecular interactions through dispersion-corrected second-order Møller–Plesset perturbation theory, Acc. Chem. Res., 2023, 56(23), 3525–3534 Search PubMed.
- G. B. Correa, S. Konstantinopoulos, B. I. Tan, Y. Zhang, F. W. Tavares, C. S. Adjiman and E. J. Maginn, Assessing Polymorph Stability and Phase Transitions at Finite Temperature: Integrating Crystal Structure Prediction, Lattice Dynamics, and Molecular Dynamics, J. Chem. Theory Comput., 2025, 21(23), 12197–12213 CrossRef CAS PubMed.
- A. M. Reilly and A. Tkatchenko, Role of dispersion interactions in the polymorphism and entropic stabilization of the aspirin crystal, Phys. Rev. Lett., 2014, 113(5), 055701 CrossRef CAS PubMed.
- C. Parks, A. Koswara, F. DeVilbiss, H.-H. Tung, N. K. Nere, S. Bordawekar, Z. K. Nagy and D. Ramkrishna, Solubility curves and nucleation rates from molecular dynamics for polymorph prediction–moving beyond lattice energy minimization, Phys. Chem. Chem. Phys., 2017, 19(7), 5285–5295 RSC.
- R. M. Bhardwaj, L. S. Price, S. L. Price, S. M. Reutzel-Edens, G. J. Miller, I. D. H. Oswald, B. F. Johnston and A. J. Florence, Exploring the Experimental and Computed Crystal Energy Landscape of Olanzapine, Cryst. Growth Des., 2013, 13(4), 1602–1617, DOI:10.1021/cg301826s.
- T. Beyer, T. Lewis and S. L. Price, Which organic crystal structures are predictable by lattice energy minimisation?, CrystEngComm, 2001, 3(44), 178–212, 10.1039/b108135g.
- S. L. Price, Why don't we find more polymorphs?, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69(4), 313–328, DOI:10.1107/s2052519213018861.
- S. Z. Ismail, C. L. Anderton, R. C. B. Copley, L. S. Price and S. L. Price, Evaluating a Crystal Energy Landscape in the Context of Industrial Polymorph Screening, Cryst. Growth Des., 2013, 13(6), 2396–2406, DOI:10.1021/cg400090r.
- P. G. Karamertzanis, A. V. Kazantsev, N. Issa, G. W. A. Welch, C. S. Adjiman, C. C. Pantelides and S. L. Price, Can the Formation of Pharmaceutical Cocrystals Be Computationally Predicted? 2. Crystal Structure Prediction, J. Chem. Theory Comput., 2009, 5(5), 1432–1448, DOI:10.1021/ct8004326.
- R. K. Hylton, G. J. Tizzard, T. L. Threlfall, A. L. Ellis, S. J. Coles, C. C. Seaton, E. Schulze, H. Lorenz, A. Seidel-Morgenstern, M. Stein and S. L. Price, Are the Crystal Structures of Enantiopure and Racemic Mandelic Acids Determined by Kinetics or Thermodynamics?, J. Am. Chem. Soc., 2015, 137(34), 11095–11104, DOI:10.1021/jacs.5b05938.
- P. W. Butler and G. M. Day, Reducing overprediction of molecular crystal structures via threshold clustering, Proc. Natl. Acad. Sci. U. S. A., 2023, 120(23), e2300516120 Search PubMed.
- G. M. Whitesides and B. Grzybowski, Self-assembly at all scales, Science, 2002, 295(5564), 2418–2421 Search PubMed.
- G. R. Desiraju, Supramolecular Synthons in Crystal Engineering—A New Organic Synthesis, Angew. Chem., Int. Ed. Engl., 1995, 34(21), 2311–2327, DOI:10.1002/anie.199523111.
- S. Parveen, R. J. Davey, G. Dent and R. G. Pritchard, Linking solution chemistry to crystal nucleation: the case of tetrolic acid, Chem. Commun., 2005,(12), 1531, 10.1039/b418603f.
- A. E. Van Driessche, N. Van Gerven, P. H. Bomans, R. R. Joosten, H. Friedrich, D. Gil-Carton, N. A. Sommerdijk and M. Sleutel, Molecular nucleation mechanisms and control strategies for crystal polymorph selection, Nature, 2018, 556(7699), 89–94 CrossRef CAS PubMed.
- R. C. Bernardi, M. C. R. Melo and K. Schulten, Enhanced sampling techniques in molecular dynamics simulations of biological systems, Biochim. Biophys. Acta, Gen. Subj., 2015, 1850(5), 872–877, DOI:10.1016/j.bbagen.2014.10.019.
- A. V. Dighe, P. Coliaie, P. K. R. Podupu and M. R. Singh, Selective desolvation in two-step nucleation mechanism steers crystal structure formation, Nanoscale, 2022, 14(5), 1723–1732, 10.1039/D1NR06346D.
- S. Hamad, C. Moon, C. R. A. Catlow, A. T. Hulme and S. L. Price, Kinetic Insights into the Role of the Solvent in the Polymorphism of 5-Fluorouracil from Molecular Dynamics Simulations, J. Phys. Chem. B, 2006, 110(7), 3323–3329, DOI:10.1021/jp055982e.
- J. Chen and B. L. Trout, Computational Study of Solvent Effects on the Molecular Self-Assembly of Tetrolic Acid in Solution and Implications for the Polymorph Formed from Crystallization, J. Phys. Chem. B, 2008, 112(26), 7794–7802, DOI:10.1021/jp7106582.
- K. Z. Takahashi, T. Aoyagi and J.-i. Fukuda, Multistep nucleation of anisotropic molecules, Nat. Commun., 2021, 12(1), 5278 CrossRef CAS PubMed.
- R. Wang, S. Mehdi, Z. Zou and P. Tiwary, Is the local ion density sufficient to drive NaCl nucleation from the melt and aqueous solution?, J. Phys. Chem. B, 2024, 128(4), 1012–1021 CrossRef CAS PubMed.
- D. Zahn, Thermodynamics and kinetics of prenucleation clusters, classical and non-classical nucleation, ChemPhysChem, 2015, 16(10), 2069–2075 CrossRef CAS PubMed.
- A. Rojnuckarin, S. Kim and S. Subramaniam, Brownian dynamics simulations of protein folding: Access to milliseconds time scale and beyond, Proc. Natl. Acad. Sci. U. S. A., 1998, 95(8), 4288–4292, DOI:10.1073/pnas.95.8.4288.
- D. E. Makarov and H. Metiu, A model for the kinetics of protein folding: Kinetic Monte Carlo simulations and analytical results, J. Chem. Phys., 2002, 116(12), 5205–5216, DOI:10.1063/1.1450123.
- P. Mereghetti, R. R. Gabdoulline and R. C. Wade, Brownian Dynamics Simulation of Protein Solutions: Structural and Dynamical Properties, Biophys. J., 2010, 99(11), 3782–3791, DOI:10.1016/j.bpj.2010.10.035.
- P. Carnevali, G. Tóth, G. Toubassi and S. N. Meshkat, Fast Protein Structure Prediction Using Monte Carlo Simulations with Modal Moves, J. Am. Chem. Soc., 2003, 125(47), 14244–14245, DOI:10.1021/ja036647b.
- S. C. Glotzer and W. Paul, Molecular and Mesoscale Simulation Methods for Polymer Materials, Annu. Rev. Mater. Res., 2002, 32(1), 401–436, DOI:10.1146/annurev.matsci.32.010802.112213.
- Z. M. Sherman and J. W. Swan, Dynamic, Directed Self-Assembly of Nanoparticles via Toggled Interactions, ACS Nano, 2016, 10(5), 5260–5271, DOI:10.1021/acsnano.6b01050.
- J. Van De Streek, H. Dietrich, D. Firaha, M. Ludwig, A. Ovchinnikov, K. Sasikumar, A. G. DiPasquale, L. Iuzzolino, A. W. Kelly, P. Lafarguette, A. Y. Lee, B. Robert, G. R. Woollam and M. A. Neumann, A Conceptual Framework for the Crystallizability of Organic Compounds, J. Am. Chem. Soc., 2025, 147(51), 46886–46896, DOI:10.1021/jacs.5c08933.
- J. Bauer, S. Spanton, R. Henry, J. Quick, W. Dziki, W. Porter and J. Morris, Ritonavir: an extraordinary example of conformational polymorphism, Pharm. Res., 2001, 18(6), 859–866 CrossRef CAS PubMed.
- D. K. Bučar, R. W. Lancaster and J. Bernstein, Disappearing polymorphs revisited, Angew. Chem., Int. Ed., 2015, 54(24), 6972–6993 CrossRef PubMed.
- D. Erdemir, A. Y. Lee and A. S. Myerson, Nucleation of crystals from solution: classical and two-step models, Acc. Chem. Res., 2009, 42(5), 621–629 CrossRef CAS PubMed.
- R. McGraw and D. T. Wu, Kinetic extensions of the nucleation theorem, J. Chem. Phys., 2003, 118(20), 9337–9347 CrossRef CAS.
- A. V. Dighe, R. R. Bhawnani, P. K. R. Podupu, N. K. Dandu, A. T. Ngo, S. Chaudhuri and M. R. Singh, Microkinetic insights into the role of catalyst and water activity on the nucleation, growth, and dissolution during COF-5 synthesis, Nanoscale, 2023, 15(21), 9329–9338, 10.1039/D2NR06685H.
- R. R. Bhawnani, O. M. Barreto, P. K. Podupu, Y. Colón, G. Giri and M. R. Singh, Next-Generation Computational and Experimental Tools for Understanding Nucleation and Growth of Metal–Organic Frameworks, ACS Mater. Lett., 2025, 7, 906–927 CrossRef CAS.
- D. Kashchiev and G. M. van Rosmalen, Review: Nucleation in solutions revisited, Cryst. Res. Technol., 2003, 38(7–8), 555–574, DOI:10.1002/crat.200310070.
- D. Kashchiev, Solution of the non-steady state problem in nucleation kinetics, Surf. Sci., 1969, 14(1), 209–220, DOI:10.1016/0039-6028(69)90055-7.
- E. Ruckenstein and B. Nowakowski, A kinetic theory of nucleation in liquids, J. Colloid Interface Sci., 1990, 137(2), 583–592, DOI:10.1016/0021-9797(90)90432-n.
- E. Ruckenstein and Y. S. Djikaev, Recent developments in the kinetic theory of nucleation, Adv. Colloid Interface Sci., 2005, 118(1–3), 51–72, DOI:10.1016/j.cis.2005.06.001.
- P. G. Vekilov, The two-step mechanism of nucleation of crystals in solution, Nanoscale, 2010, 2(11), 2346–2357 RSC.
- T. Kovács and H. K. Christenson, A two-step mechanism for crystal nucleation without supersaturation, Faraday Discuss., 2012, 159, 123, 10.1039/c2fd20053h.
- J. F. Lutsko, How crystals form: A theory of nucleation pathways, Sci. Adv., 2019, 5(4), eaav7399 CrossRef CAS PubMed.
- C. D. Fu, L. F. L. Oliveira and J. Pfaendtner, Determining energy barriers and selectivities of a multi-pathway system with infrequent metadynamics, J. Chem. Phys., 2017, 146(1), 014108 CrossRef PubMed.
- D. Reguera, J. M. Rubí and A. Pérez-Madrid, Fokker–Planck equations for nucleation processes revisited, Phys. A, 1998, 259(1–2), 10–23, DOI:10.1016/s0378-4371(98)00259-3.
- T. Matsoukas and Y. Lin, Fokker-Planck equation for particle growth by monomer attachment, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2006, 74(3), 031122 CrossRef PubMed.
- J. Elgin, The Fokker-Planck equation: methods of solution and applications, Opt. Acta, 1984, 31(11), 1206–1207 CrossRef.
- A. N. Firoozsalari, A. A. Aghaei and K. Parand, A machine learning framework for efficiently solving Fokker–Planck equations, J. Comput. Appl. Math., 2024, 43(6), 389 Search PubMed.
- G. M. Maggioni and M. Mazzotti, Modelling the stochastic behaviour of primary nucleation, Faraday Discuss., 2015, 179, 359–382, 10.1039/c4fd00255e.
- C. Y. Ma, J. J. Liu and X. Z. Wang, Measurement, modelling, and closed-loop control of crystal shape distribution: Literature review and future perspectives, Particuology, 2016, 26, 1–18, DOI:10.1016/j.partic.2015.09.014.
- P. K. R. Podupu, V. V. Gande, R. Hari, A. Korde, M. S. Kelkar, N. K. Nere and M. R. Singh, Analytical solution to the discretized population balance equation for pure breakage with application to kernel identification, Chem. Eng. Res. Des., 2025, 217, 295–303, DOI:10.1016/j.cherd.2025.03.019.
- J. Anwar and D. Zahn, Uncovering Molecular Processes in Crystal Nucleation and Growth by Using Molecular Simulation, Angew. Chem., Int. Ed., 2011, 50(9), 1996–2013, DOI:10.1002/anie.201000463.
- R. J. Davey, S. L. M. Schroeder and J. H. ter Horst, Nucleation of Organic Crystals—A Molecular Perspective, Angew. Chem., Int. Ed., 2013, 52(8), 2166–2179, DOI:10.1002/anie.201204824.
- L. Filion, M. Hermes, R. Ni and M. Dijkstra, Crystal nucleation of hard spheres using molecular dynamics, umbrella sampling, and forward flux sampling: A comparison of simulation techniques, J. Chem. Phys., 2010, 133(24), 244115, DOI:10.1063/1.3506838.
- G. T. Beckham, B. Peters and B. L. Trout, Evidence for a Size Dependent Nucleation Mechanism in Solid State Polymorph Transformations, J. Phys. Chem. B, 2008, 112(25), 7460–7466, DOI:10.1021/jp710192u.
- H. Tan, M. Goldschmidt, R. Boerefijn, M. Hounslow, D. Salman and J. Kuipers, Population balance modelling of fluidized bed melt granulation: an overview, Chem. Eng. Res. Des., 2005, 83(7), 871–880 CrossRef CAS.
- A. Bravais, Etudes cristallographiques, Gauthier-Villars, 1866 Search PubMed.
- M. G. Friedel, Études Sur la loi de Bravais, Bull. Soc. Fr. Mineral., 1907, 9, 326–455 Search PubMed.
- J. Donnay and D. Harker, A new law of crystal morphology extending the law of Bravais, Am. Mineral., 1937, 22(5), 446–467 CAS.
- A. V. Dighe, P. K. Podupu, V. V. Gande, U. Diwekar and M. R. Singh, Group contribution method for rapid estimation of crystal growth rates, Chem. Eng. Res. Des., 2024, 203, 140–148 CrossRef CAS.
- P. Hartman and P. Bennema, The attachment energy as a habit controlling factor, J. Cryst. Growth, 1980, 49(1), 145–156, DOI:10.1016/0022-0248(80)90075-5.
- P. Hartman, The attachment energy as a habit controlling factor, J. Cryst. Growth, 1980, 49(1), 166–170, DOI:10.1016/0022-0248(80)90077-9.
- P. Hartman, The attachment energy as a habit controlling factor II. Application to anthracene, tin tetraiodide and orthorhombic sulphur, J. Cryst. Growth, 1980, 49(1), 157–165, DOI:10.1016/0022-0248(80)90076-7.
- W. Burton, N. Cabrera and F. Frank, The growth of crystals and the equilibrium structure of their surfaces, Philos. Trans. R. Soc., A, 1951, 299–358 CrossRef CAS.
- A. Chernov, The spiral growth of crystals, Phys.-Usp., 1961, 4(1), 116–148 Search PubMed.
- D. Winn and M. F. Doherty, A new technique for predicting the shape of solution-grown organic crystals, AIChE J., 1998, 44(11), 2501–2514, DOI:10.1002/aic.690441117.
- S. Boerrigter, G. Josten, J. Van De Streek, F. Hollander, J. Los, H. Cuppen, P. Bennema and H. Meekes, MONTY: Monte Carlo crystal growth on any crystal structure in any crystallographic orientation; application to fats, J. Phys. Chem. A, 2004, 108(27), 5894–5902 CrossRef CAS.
- D. Winn and M. F. Doherty, Modeling crystal shapes of organic materials grown from solution, AIChE J., 2000, 46(7), 1348–1367 CrossRef CAS.
- C. J. Tilbury and M. F. Doherty, Modeling layered crystal growth at increasing supersaturation by connecting growth regimes, AIChE J., 2017, 63(4), 1338–1352 CrossRef CAS.
- D. Ramkrishna and M. R. Singh, Population balance modeling: Current status and future prospects, in Annual Review of Chemical and Biomolecular Engineering, Annual Reviews Inc., 2014, vol. 5, pp. 123–146 Search PubMed.
- E. L. Haseltine, D. B. Patience and J. B. Rawlings, On the stochastic simulation of particulate systems, Chem. Eng. Sci., 2005, 60(10), 2627–2641 Search PubMed.
- W. Heineken, D. Flockerzi, A. Voigt and K. Sundmacher, Dimension reduction of bivariate population balances using the quadrature method of moments, Comput. Chem. Eng., 2011, 35(1), 50–62 CrossRef CAS.
- X. Z. Wang and C. Y. Ma, Morphological population balance model in principal component space, AIChE J., 2009, 55(9), 2370–2381 CrossRef CAS.
- A. V. Dighe, P. K. Podupu and M. R. Singh, Emulsification of Supersaturated Solutions Amplifies Induction Time Variation in Crystallization, Cryst. Growth Des., 2023, 23(9), 6290–6297 CrossRef CAS.
- J. Kuipers and G. Barkema, Limitations of a Fokker-Planck description of nucleation, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2010, 82(1), 011128 CrossRef CAS PubMed.
- E. Komp, N. Janulaitis and S. Valleau, Progress towards machine learning reaction rate constants, Phys. Chem. Chem. Phys., 2022, 24(5), 2692–2705 RSC.
Footnote |
| † Authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |