DOI:
10.1039/D5CE00898K
(Paper)
CrystEngComm, 2026,
28, 390-395
Systematic investigation of the effect of trialkylsilyl groups on the crystal structures of perfluorophenyl-substituted thiophene†
Received
15th September 2025
, Accepted 20th October 2025
First published on 21st October 2025
Abstract
Controlling the crystal packing of perfluorophenyl-substituted aryl molecules (Ar–ArF-type molecules) is crucial for their properties. Herein, we investigated the crystal structures of perfluorophenyl-substituted thiophene derivatives bearing three different trialkylsilyl groups: trimethylsilyl, tert-butyldimethylsilyl, and triisopropylsilyl. Single-crystal X-ray diffraction analysis revealed that the compounds exhibited distinct head-to-tail stacking patterns depending on the length and degree of branching of the alkyl chains on the trialkylsilyl groups. Notably, the photophysical properties of the crystals differed, despite being similar in solution. Specifically, the trimethylsilyl and tert-butyldimethylsilyl derivatives exhibited a larger red shift in their excitation and emission wavelengths, as well as higher photoluminescence quantum yields. These results indicate that trialkylsilyl groups can effectively modulate molecular packing to enhance the solid-state properties of these compounds.
Introduction
Controlling the crystal structures of molecules and their solid-state properties is a significant subject in crystal engineering. Combinations of noncovalent interactions, including attractive forces and steric hindrance, are essential for the design of crystal structures.1–8 Introducing trialkylsilyl groups into aryl molecules is a simple and effective strategy for modifying crystal structures and their solid-state properties.5,9–17 In this strategy, the π-stacking interactions of aryl molecules are combined with the repulsive steric hindrance and attractive dispersion forces of trialkylsilyl groups, which enables the modulation of π-stacking structures by varying the length or degree of branching of the alkyl groups on the silicon atom. For example, trialkylsilyl-substituted diethynylindenofluorenediones exhibit distinct π-stacking structures, allowing for the tuning of their electronic and optical properties.10 Although this strategy is primarily used for fused-ring compounds5,9–15 and polymers16 in organic electronic materials, there remain unexplored applications of this strategy. To enhance its practicality, we have recently reported several attempts of using trialkylsilyl groups for controlling crystal structures, employing triaryltriazine or halogen-substituted dipolar 2,3-difluorobenzene as the aryl molecule.17,18
Following those examples, we subsequently focused on the perfluorophenyl group.2,6,7,19–24 On the perfluorophenyl group, the electron-withdrawing fluorine atoms generate an electron-deficient region (π-hole), leading to aryl–perfluoroaryl interactions with other aromatic or heteroaromatic rings owing to the charge complementarity between the stacked rings. For example, hexafluorobenzene forms a molecular complex with benzene in the crystalline state.19,20 While both compounds are liquid at 295 K, the cocrystal has a higher melting point, indicating that aryl–perfluoroaryl interactions exhibit a strong binding energy (ca. −30 kJ mol−1, experimental value for C6F6/C6H6).20 Similarly, crystal structures of perfluorophenyl-substituted aryl molecules (Ar–ArF-type molecules) are strongly influenced by aryl–perfluoroaryl interactions.6,7,25,26 While phenyl groups typically exhibit edge-to-face or slipped face-to-face interactions (8–12 kJ mol−1), perfluorophenyl groups and other aryl groups tend to adopt a face-to-face stacking arrangement (23–29 kJ mol−1), producing well-ordered head-to-tail arrangements of Ar–ArF-type molecules (Fig. 1a).6,8 Furthermore, π-plane overlaps between the arylene and perfluoroaryl moieties can influence their solid-state photophysical properties.6,21–26 For example, the introduction of linear alkyl groups with different chain lengths induces distinct π-plane overlaps in Ar–ArF-type molecules in the crystalline state, accompanied by variations in photoluminescence quantum yields and slight shifts in emission wavelengths.25,26 However, the introduction of trialkylsilyl groups into Ar–ArF-type molecules has not been reported to date.
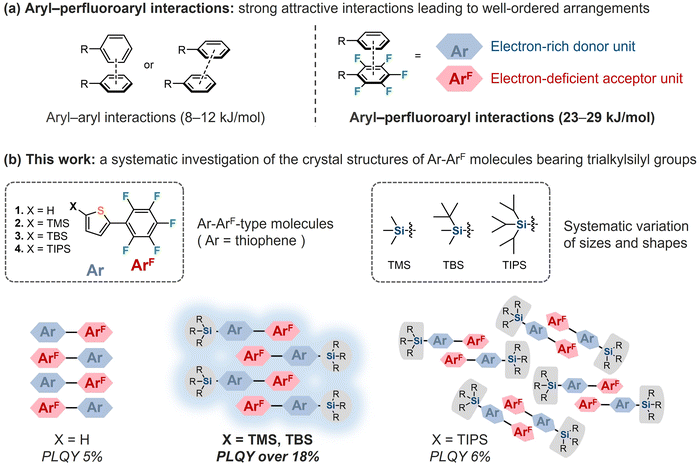 |
| | Fig. 1 (a) Aryl–perfluoroaryl interactions. (b) This work: investigation of the crystal structures of Ar–ArF molecules bearing trialkylsilyl groups with systematic variation in size and shape. | |
Herein, as a first systematic study of the silyl substituent effect on Ar–ArF-type molecules, we synthesized 2-(perfluorophenyl)thiophene (1) and introduced three different trialkylsilyl groups, namely, trimethylsilyl (TMS; 2), tert-butyldimethylsilyl (TBS; 3), and triisopropylsilyl (TIPS; 4) at the 5-position of thiophene.27,28 X-ray diffraction (XRD) analysis demonstrated that although the crystals of 1–4 have head-to-tail arrangements in common, their face-to-face stacking arrangements varied depending on the sizes and shapes of the trialkylsilyl groups. In addition, the excitation and emission spectra of the four crystals differed, as well as the photoluminescence quantum yields, as a result of the differences in the aryl–perfluoroaryl interactions caused by the various steric effects of the trialkylsilyl groups (Fig. 1b).
Results and discussion
Synthesis and crystal preparation
Ar–ArF-type molecules 2, 3, and 4 were synthesized in two steps, as shown in Scheme 1. The trialkylsilyl (TMS, TBS, and TIPS) groups were introduced to the thiophene moiety in good yields using the corresponding trialkylsilyl trifluoromethane sulfonates (R-OTf) after the lithiation of the C–Br bond by n-BuLi.29 In the second step, the palladium-catalyzed Suzuki–Miyaura cross-coupling reaction of pentafluorophenyl boronic acid pinacol ester with either 2-bromothiophene or 2a–4a was conducted. Because the reaction of pentafluorophenyl boronic acid pinacol ester often gives low yields, we adopted a mechanochemical approach.30 Using a Retsch MM400 mill [stainless-steel milling jar (5 mL); 30 Hz; stainless-steel ball (10 mm)], the palladium-catalyzed solid-state perfluoroarylation of aryl halides afforded the targeted compounds 1–4 in 28%, 14%, 57%, and 52% yields, respectively. 1–4 were dissolved in hexane, and the crystals of 1–4 were obtained by slow evaporation at room temperature in air.
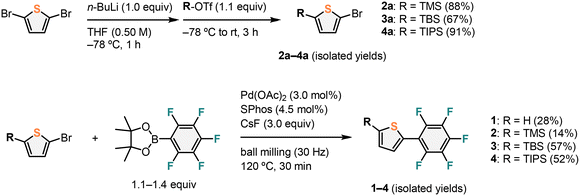 |
| | Scheme 1 Synthesis of Ar–ArF-type molecules 1–4. | |
Crystal structure analysis of 1–4
1–4 furnished single crystals that were suitable for single-crystal XRD analysis. The stacking structures in the crystal structures, along with the observed short contact distances, are shown in Fig. 2. In the crystal structure of 1 (Fig. 2a), the perfluorophenyl group and thiophene adopted a coplanar conformation with angle Φ of 6.78–7.05°, and molecules of 1 stacked vertically with the distance dπ in the range of 3.509–3.536 Å and θ = 73.84–76.67° in an antiparallel manner, driven by aryl–perfluoroaryl interactions. As shown in Fig. 2b, the crystal structure of 2 was similar to that of 1, with Φ = 0.13–2.04°, dπ in the range of 3.400–3.450 Å, and θ in the range of 71.44–72.44°. However, the neighboring molecules were oriented in a crossed head-to-head fashion because steric hindrance between the TMS and perfluorophenyl groups prevented the antiparallel orientation of the two molecules. In addition, the crossed head-to-head stacking structure brought the TMS group and the perfluorophenyl group into close proximity, resulting in hydrogen bonds of 2.587–2.857 Å between a hydrogen atom of TMS and a fluorine atom of the perfluorophenyl group. In the crystal of 3 (Fig. 2c), Φ was 12.58°, and dimer structures with dπ = 3.545–3.573 Å and θ = 76.21° stacked diagonally with inter-dimer dπ and θ values of 3.534–3.562 Å and 49.07°, respectively. This is attributed to the fact that the two methyl groups in the TBS group are small enough for the dimer structure to be formed, while the bulky tert-butyl group separates the dimers from each other. Fig. 2d shows the crystal structure of 4 with angle Φ = 4.51° and composed of slipped stacking dimer structures with dπ = 3.474 Å and θ = 51.77°. The bulky TIPS groups completely separated dimers from other neighboring dimers, suggesting that the TIPS group is too bulky to construct antiparallel molecular stacks. Similar phenomena have also been observed in the crystal structures of pentacene-based semiconductors, in which intermolecular CH–π interactions were hindered by bulky TIPS groups, thereby leading to intermolecular π–π interactions.9 In all compounds 1–4, steric hindrance from the silyl substituents influences the aryl–perfluoroaryl interactions, causing the orientation of adjacent aromatic rings to deviate from the optimal π–π stacking arrangement. This phenomenon, where silyl groups introduce steric hindrance that affects other dominant intermolecular interactions, is commonly observed in other silyl-substituted aromatic systems.9–17 In addition to the stacking structures of the molecules, there were apparent differences in the interface structure of the stacking columns or dimer structures. The intercolumnar structure of 1 showed a layered structure of π-stacked domains (Fig. 3a). In the intercolumnar interface of 2, half of the TMS groups formed layered domains, resulting in the separation of the π-stacked columns (Fig. 3b). The intercolumnar structure of 3 was alternating layered domains consisting of π-stacked domains and TBS domains (Fig. 3c). In the interdimer interface of 4, the bulky TIPS groups dominated the packing structure, shielding the π-stacked dimers (Fig. 3d).
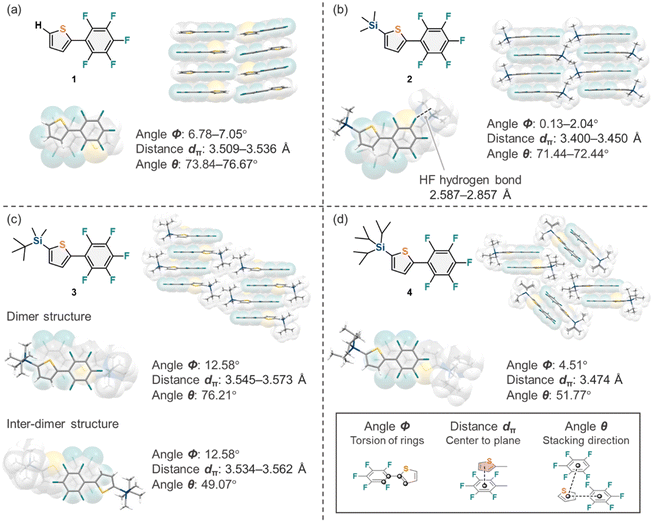 |
| | Fig. 2 (a–d) Crystal structures of 1–4, respectively. Definition of the parameters: Φ refers to the torsion angle of the perfluorophenyl group and thiophene calculated from the four points shown in the figure, dπ refers to the distance between the center of thiophene ring and the plane of the perfluorophenyl group, and θ refers to the angle of the three centers which represents the stacking directions. | |
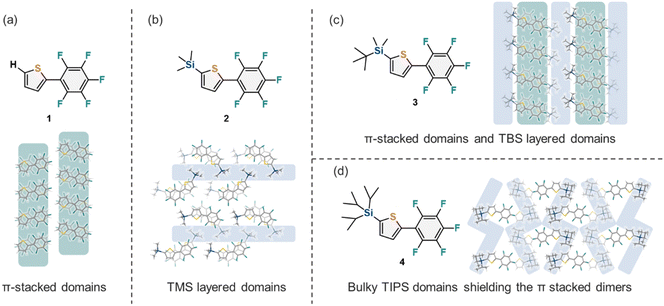 |
| | Fig. 3 (a–d) The intercolumnar or interdimer structure in the crystal structures of 1–4, respectively. | |
Photophysical properties in solution
DFT calculations suggested that the HOMOs and LUMOs of 1–4 have similar values (Fig. S1). Dilute solutions of 1–4 were prepared in chloroform, and their UV-vis excitation and emission spectra recorded. Fig. 4a shows the obtained excitation and emission spectra and Table 1 summarizes the corresponding photophysical data. Solutions of 1–4 exhibited similar excitation and emission wavelengths, with λex = 312–322 nm and λem = 385–395 nm. Solutions of 1–4 also exhibit a low ΦPL of 0.01–0.02 and a short τ of 0.7–4.4 ns.
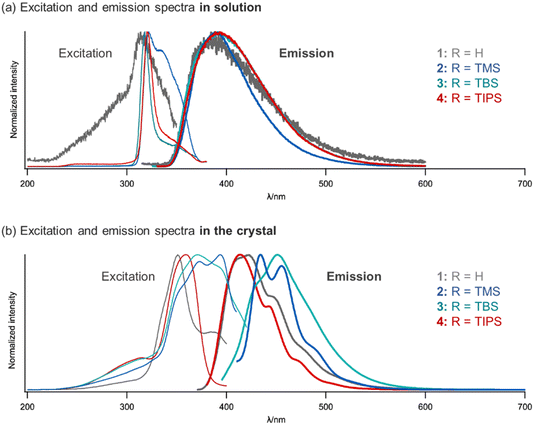 |
| | Fig. 4 Excitation and emission spectra of 1–4 in (a) dilute chloroform solutions (concentration: 10−3 mol L−1 for both spectra) and (b) the crystalline state. | |
Table 1 Photophysical data for dilute solutions and crystals of 1–4
|
|
In dilute solution |
In the crystal form |
|
|
λ
ex [nm] |
λ
em [nm] |
Φ
PL
|
τ [ns] |
λ
ex [nm] |
λ
em [nm] |
Φ
PL
|
τ
av [ns] |
|
λ
ex: maximum excitation wavelength, λem: maximum emission wavelength, ΦPL: photoluminescence quantum yield, τ: emission lifetime, τav: average of emission lifetime. |
| 1 |
312 |
385 |
0.01 |
1.84 |
351 |
421 |
0.05 |
0.67 |
| 2 |
318 |
395 |
0.01 |
1.77 |
394 |
434 |
0.21 |
1.06 |
| 3 |
322 |
393 |
0.02 |
0.73 |
371 |
451 |
0.18 |
1.19 |
| 4 |
320 |
393 |
0.02 |
4.40 |
359 |
414 |
0.06 |
0.90 |
Photophysical properties in the crystal form
Although the photophysical properties of 1–4 were similar in solution, their photophysical properties in the crystal form were expected to vary because their crystal structures differed considerably. Therefore, we measured the excitation and emission spectra (Fig. 4b), and the photophysical data are summarized in Table 1. Compared to those in solution, the excitation and emission wavelengths were red-shifted, especially for 2 (λex = 394 nm and λem = 434 nm,) and 3 (λex = 371 nm and λem = 451 nm). Moreover, the value of ΦPL improved in all the crystals, especially for 2 (ΦPL = 0.21) and 3 (ΦPL = 0.18). In Fig. 5, the red-shifts (in eV) and ΦPL values for 1–4 are summarized along with the π-plane overlaps in their crystal structures. Larger red-shifts in λex for 2 (0.75 eV) and λem for 3 (0.41 eV), as well as higher ΦPL values for 2 (0.21) and 3 (0.18), are highlighted in red. It is suggested that the disparities in π-plane overlaps, as shown by the red areas in Fig. 5, led to the variation in the photophysical properties in the crystal form. In aryl–perfluoroaryl interactions, photophysical properties are associated with charge transfer transitions between the electron-rich aryl ring and the electron-deficient perfluorophenyl group; therefore, the distances between the thiophene and perfluorophenyl groups (dπ) and the stacking directions (θ) directly influenced the values of λex, λem, and ΦPL. Furthermore, 2 showed a pronounced vibrational structure in its emission spectrum compared with 1, 3, and 4 (Fig. 4). The molecular arrangement in the crystal of 2 might favor emission from the locally excited state, whereas the charge-transfer state would be more dominant in the crystals of 1, 3, and 4.
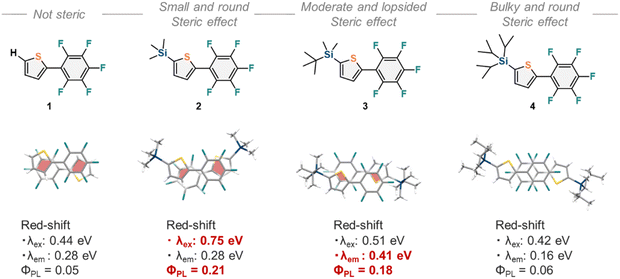 |
| | Fig. 5 A summary of the molecular structures and their arrangement within the crystal structure. π-plane overlaps are shown by red areas. | |
Conclusions
We synthesized 2-(perfluorophenyl)thiophene (1) and introduced TMS (2), TBS (3), and TIPS (4) groups at the 5-position of the thiophene to systematically investigate the effect of the bulkiness of the silyl substituents on their crystal structures. Single-crystal XRD analysis revealed that the crystal structures of 1–4 exhibited distinct head-to-tail stacking structures according to the difference in the length or degree of branching of the alkyl groups on the trialkylsilyl groups. Notably, the photophysical properties of the crystals differed, despite being similar in solution. Specifically, 2 and 3 showed large red shifts in the excitation and emission wavelengths as well as a large increase in photoluminescence quantum yields. Therefore, it is suggested that trialkylsilyl groups can effectively modify molecular arrangements to improve the solid-state properties of materials.
Author contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Data availability
Supplementary information is available. See DOI: https://doi.org/10.1039/d5ce00898k.
CCDC 2487542–2487545 (1–4) contain the supplementary crystallographic data for this paper.31a–d
Acknowledgements
This work was financially supported by the Japan Society for the Promotion of Science (JSPS) via KAKENHI grants JP22K18333, and JP22H00318; by the JST via the CREST grant JPMJCR19R1 and the FOREST grant JPMJFR232C; and by the Institute for Chemical Reaction Design and Discovery (ICReDD), established by the World Premier International Research Initiative (WPI), MEXT, Japan.
References
- R. Berger, G. Resnati, P. Metrangolo, E. Weberd and J. Hulliger, Chem. Soc. Rev., 2011, 40, 3496–3508 RSC.
- M. T. Messina, P. Metrangolo, S. Pappalardo, M. F. Parisi, T. Pilati and G. Resnati, Chem. – Eur. J., 2000, 6, 3495–3500 CrossRef CAS.
- D. Braga and F. Grepioni, Coord. Chem. Rev., 1999, 183, 19–41 CrossRef CAS.
- C. B. Aakeröy, Acta Crystallogr., Sect. B:Struct. Sci., 1997, 53, 569–586 CrossRef.
- P. Yu, Y. Zhen, H. Dong and W. Hu, Chem, 2019, 5, 2814–2853 CAS.
- W. Wang, W. X. Wu, Y. Zhang and W. J. Jin, Chem. Phys. Rev., 2024, 5, 031303 CrossRef CAS.
- K. Reichenbächer, H. I. Süss and J. Hulliger, Chem. Soc. Rev., 2005, 34, 22–30 RSC.
- M. O. Sinnokrot, E. F. Valeev and C. D. Sherrill, J. Am. Chem. Soc., 2002, 124, 10887–10893 CrossRef CAS PubMed.
- J. E. Anthony, J. S. Brooks, D. L. Eaton and S. R. Parkin, J. Am. Chem. Soc., 2001, 123, 9482–9483 CrossRef CAS.
- B. D. Rose, D. T. Chase, C. D. Weber, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Org. Lett., 2011, 13, 2106–2109 CrossRef CAS.
- K. J. Thorley, O. D. Jurchescu, D. Dremann, H. F. Iqbal and J. E. Anthony, Mol. Syst. Des. Eng., 2022, 7, 374–380 RSC.
- M. L. Tang, A. D. Reichardt, T. Siegrist, S. C. B. Mannsfeld and Z. Bao, Chem. Mater., 2008, 20, 4669–4676 CrossRef CAS.
- V. S. Barlier, C. W. Schlenker, S. W. Chinb and M. E. Thompson, Chem. Commun., 2011, 47, 3754–3756 RSC.
- L. Zhang, A. Fonari, Y. Liu, A.-L. M. Hoyt, H. Lee, D. Granger, S. Parkin, T. P. Russell, J. E. Anthony, J.-L. Bredas, V. Coropceanu and A. L. Briseno, J. Am. Chem. Soc., 2014, 136, 9248–9251 CrossRef CAS.
- Z. W. Schroeder, P. R. Rami, M. Adam, M. J. Ferguson, F. Hampel and R. R. Tykwinski, Chem. – Eur. J., 2024, 30, e202402651 CrossRef CAS PubMed.
- A. Iida, K. Nagura and S. Yamaguchi, Chem. – Asian J., 2008, 3, 1456–1464 CrossRef CAS PubMed.
- M. Jin, R. Kitsu, N. Hammyo, A. Sato-Tomita, M. Mizuno, A. S. Mikherdov, M. Tsitsvero, A. Lyalin, T. Taketsugu and H. Ito, J. Am. Chem. Soc., 2023, 145, 27512–27520 CrossRef CAS PubMed.
- N. Hammyo, M. Jin and H. Ito, Cryst. Growth Des., 2023, 23, 4514–4521 CrossRef CAS.
- C. R. Patrick and G. S. Prosser, Nature, 1960, 187, 1021 CrossRef CAS.
- O. R. Lozman, R. J. Bushby and J. G. Vinter, J. Chem. Soc., Perkin Trans. 2, 2001, 1446–1452 RSC.
- S. A. Sharber, W. J. Mullin and S. W. Thomas III, Chem. Mater., 2021, 33, 6640–6661 CrossRef CAS.
- Q. Liao, Q. Li and Z. Li, Adv. Mater., 2023, 2306617 Search PubMed.
- L. Wang, J. Deng, M. Jiang, C. Zhen, F. Li, S. Li, S. Bai, X. Zhang and W. Zhu, J. Mater. Chem. A, 2023, 11, 11235 RSC.
- A. Mandal, A. Choudhury, P. K. Iyer and P. Mal, J. Phys. Chem. C, 2019, 123, 18198–18206 CrossRef CAS.
- S. Yamada and T. Konno, Chem. Rec., 2023, 23, e202300094 CrossRef CAS.
- S. Yamada, A. Mitsuda, K. Miyano, T. Tanaka, M. Morita, T. Agou, T. Kubota and T. Konno, ACS Omega, 2018, 3, 9105–9113 CrossRef CAS.
- E. M. Breitung, C.-F. Shu and R. J. McMahon, J. Am. Chem. Soc., 2000, 122, 1154–1160 CrossRef CAS.
- R. Yoshida, T. Tachikawa and S. Ito, Cryst. Growth Des., 2022, 22, 547–558 CrossRef CAS.
- W. Zhang, G. M. Ng, H. L. Tam, M. S. Wong and F. Zhu, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 1865–1873 CrossRef CAS.
- R. Takahashi, T. Seo, K. Kubota and H. Ito, ACS Catal., 2021, 11, 14803–14810 CrossRef CAS.
-
(a) CCDC 2487542: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2phh90;
(b) CCDC 2487543: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2phhb1;
(c) CCDC 2487544: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2phhc2;
(d) CCDC 2487545: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2phhd3.
Footnote |
| † “Dedicated to Professor Resnati, celebrating a career in fluorine and noncovalent chemistry on the occasion of his 70th birthday.” |
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *ab and
Mingoo
Jin
*ab and
Mingoo
Jin





