DOI:
10.1039/D5CE00837A
(Paper)
CrystEngComm, 2026,
28, 2455-2467
Preparation of phosphine oxides with anthrylphenyl, pyrenylphenyl, anthrylethynyl, and pyrenylethynyl groups: luminescence properties and conformational polymorphs
Received
29th August 2025
, Accepted 11th March 2026
First published on 11th March 2026
Abstract
Precise control of crystal polymorphism provides a powerful strategy to tune luminescence, yet remains a major challenge in molecular material design. We show that introducing ethynylene linkers into phosphine oxide derivatives bearing anthryl and pyrenyl groups enables polymorphism through diverse intermolecular interactions, whereas phenylene-bridged analogues crystallize in a single form. Single-crystal X-ray diffraction confirmed distinct packing arrangements, and optical studies revealed extended π-conjugation with bathochromic shifts, as well as packing-dependent modulation of excited-state vibrational levels. These results establish a direct structure–property relationship between polymorphism and emission, offering a versatile molecular design principle for next-generation optoelectronic and photonic materials.
1. Introduction
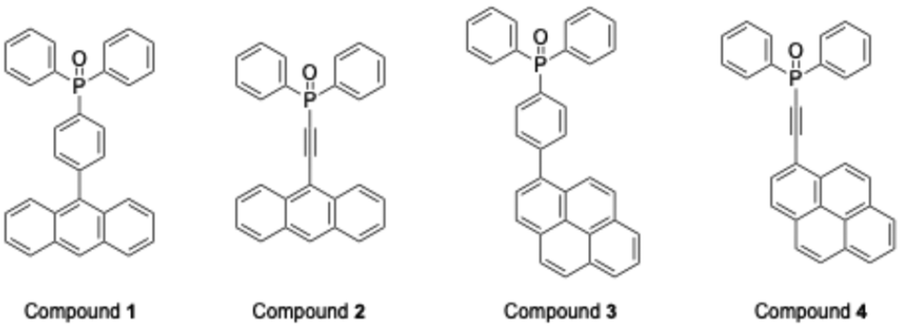
The design of luminescent organic materials with tunable emission color and high stability has attracted sustained attention owing to their potential applications in organic light-emitting diodes, optical sensors, and bioimaging.1,2 Among various candidates, lanthanide complexes coordinated with triphenylphosphine oxide (TPPO) are well-known examples,3 where TPPO acts as an antenna molecule to enhance emission efficiency.4 Previous studies have demonstrated that replacing the phenyl group of TPPO with extended π-conjugated substituents5 such as anthryl6 and pyrenyl7 units can further improve light-harvesting capability and luminescence intensity, highlighting the significance of molecular design in tailoring photophysical properties.8 Recently, increasing emphasis has been placed on controlling the luminescence of organic solids,9 where the molecular packing in the crystal lattice is a decisive factor. Subtle variations in weak intermolecular interactions, including π–π stacking, CH–π, and halogen bonding, can fine-tune the emission color and efficiency. However, precise control of such interactions remains difficult, and the relationship between crystal packing and luminescence is not yet fully understood. One promising concept to address this challenge is crystal polymorphism,10 in which a compound crystallizes in different packing motifs while maintaining the same chemical composition.11 Polymorphism is well recognized for inducing significant changes in physical properties, and in luminescent systems, it provides a pathway to regulate solid-state emission. While strong hydrogen-bonding interactions often fix molecular arrangements into predictable patterns,12 systems governed by multiple weak interactions are more likely to generate diverse crystal structures, thereby increasing the probability of polymorphism.13 In this context, phosphine oxide derivatives bearing anthryl and pyrenyl substituents present an attractive platform for studying structure–property correlations in luminescent materials. These aromatic groups are expected to promote π–π stacking interactions, leading to diverse molecular arrangements. Furthermore, the introduction of an ethynylene linker between the phosphine oxide backbone and the aromatic substituent not only extends the π-conjugation, but also enhances rotational flexibility, which could significantly influence the diversity of crystal packing. This structural feature is therefore anticipated to promote polymorphism and, consequently, provide new opportunities for tuning luminescence behavior in the solid state. Herein, we report the design and synthesis of four phosphine oxide derivatives—9-anthrylphenyl, 9-anthrylethynyl, 1-pyrenylphenyl, and 1-pyrenylethynyl compounds (Chart 1)—to systematically investigate the effect of ethynylene linkers on crystal packing and luminescence properties. Single-crystal X-ray diffraction analysis was performed under various recrystallization conditions to assess polymorph formation. In addition, the photophysical properties were examined in both solution and crystalline states, and the results were correlated with structural features. This study demonstrates how the incorporation of ethynylene groups into phosphine oxide frameworks can promote crystal polymorphism and provides a molecular design strategy for fine-tuning solid-state luminescence.
 |
| | Chart 1 Chemical structures of the aryldiphenylphosphine oxides. | |
2. Experimental
2.1. Materials and instruments
All commercially available reagents were used without further purification. 1H, 13C, and 31P nuclear magnetic resonance (NMR) spectra were recorded using an Agilent UNITY INOVA 500 spectrometer (500 MHz for 1H, 126 MHz for 13C, and 200 MHz for 31P). Mass spectral data were obtained using a JEOL JMS-T100LP mass spectrometer in the positive-ion detection mode. Single crystal X-ray diffraction (SCXRD) data for crystals 1, 2a, 2b, 3a, 3c, and 4 were collected on a RIGAKU Oxford Diffraction XtaLAB Pro equipped with a Dectris PILATUS 200K HPAD detector and microfocus sealed tube CuKα radiation with mirror optics (λ = 1.54184 Å). The diffraction data were integrated using CrysAlisPro14 and corrected for absorption effects using a combination of empirical (ABSPACK) and numerical corrections. The structures were solved using SHELXT and refined by full-matrix least-squares analysis (SHELXL) using the OLEX2 program package.15 Unless otherwise indicated, all non-hydrogen atoms were refined anisotropically. All the hydrogen atom positions were constrained to ideal geometries and refined using fixed isotropic displacement parameters (in terms of the riding model). X-ray diffraction images for crystals 1 and 2b were collected using an EIGER 4M detector with synchrotron radiation at a wavelength of 0.80000 Å at the BL26B1 station of SPring-8 (Hyogo, Japan). The distance between the crystal and detector was 50 mm. Images were processed using HKL2000 (HKL Research) or the XDS program package. The structural solution and refinement were performed using SHELXS-97 and SHELXL-2014/7 (Sheldrick).16,17 Powder X-ray diffraction (PXRD) patterns were recorded on a Bruker EMPYREAN diffractometer equipped with a CuKα radiation source (λ = 1.54184 Å) operating at 45 kV and 40 mA. The data were collected in the range of 2θ = 5–50° with a step size of 0.02° and a scanning speed of 4° min−1. Ultraviolet-visible (UV-vis) spectra were recorded using a JASCO model V650 spectrophotometer. Fluorescence spectra were recorded using a JASCO FP8050 spectrophotometer (in solution) and JASCO FP8600 (in solid). Photoluminescence quantum yields (PQLYs) were obtained on a HAMAMATSU Photonics K.K. Quantaurus-QY C11347-01 series with an integrating sphere. All theoretical calculations were performed using the Gaussian 09 package,18 details of which are given in the SI.
2.2. Synthesis procedure
2.2.1. Synthesis of (4-(9-anthryl)phenyl)diphenylphosphine oxide (compound 1).
Under a N2 atmosphere, 9-(4-bromophenyl)anthracene (0.359 g, 1.08 mmol) was dissolved in anhydrous tetrahydrofuran (12 mL). The solution was cooled to −78 °C, and n-BuLi was added (1.52 M in n-hexane, 1.5 mL, 2.3 mmol). After stirring for 30 min, chlorodiphenylphosphine (0.28 mL, 1.5 mmol) was added to the mixture and allowed to react at room temperature for 3 h. Subsequently, water was incorporated to deactivate any residual n-BuLi. Thereafter, hydrogen peroxide (30%, 1 mL) was added, and the mixture was stirred for 30 min. After the complete reaction, the mixture was quenched with water, extracted with chloroform, and washed with brine. The organic layer was then dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography using CHCl3 and CHCl3/MeOH (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v) as eluents. Compound 1 was obtained as a white solid (0.244 g, 50.0%). 1H NMR (500 MHz, CDCl3, 25 °C): δ 8.53 (s, 1H), 8.06 (d, J = 8.5 Hz, 2H), 7.89–7.81 (m, 6H), 7.63–7.54 (m, 10H), 7.48 (t, J = 7.0, 7.5 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3, 25 °C): δ 142.9, 135.4, 132.9, 132.3–132.1, 131.6, 131.5, 131.3, 129.9, 128.7, 128.6, 128.4, 127.2, 126.3, 125.7, 125.2 ppm; 31P NMR (200 MHz, CDCl3, 25 °C): δ 29.3 ppm. The MS (ESI-TOF) m/z calculated for C32H24OP was 455.15648. Found: 455.11788.
:1 v/v) as eluents. Compound 1 was obtained as a white solid (0.244 g, 50.0%). 1H NMR (500 MHz, CDCl3, 25 °C): δ 8.53 (s, 1H), 8.06 (d, J = 8.5 Hz, 2H), 7.89–7.81 (m, 6H), 7.63–7.54 (m, 10H), 7.48 (t, J = 7.0, 7.5 Hz, 2H), 7.38 (t, J = 7.5 Hz, 2H) ppm; 13C NMR (126 MHz, CDCl3, 25 °C): δ 142.9, 135.4, 132.9, 132.3–132.1, 131.6, 131.5, 131.3, 129.9, 128.7, 128.6, 128.4, 127.2, 126.3, 125.7, 125.2 ppm; 31P NMR (200 MHz, CDCl3, 25 °C): δ 29.3 ppm. The MS (ESI-TOF) m/z calculated for C32H24OP was 455.15648. Found: 455.11788.
2.2.2. Synthesis of (9-anthrylethynyl)diphenylphosphine oxide (compound 2).
Under a N2 atmosphere, 9-ethynylanthracene (0.145 g, 0.731 mmol) was dissolved in anhydrous tetrahydrofuran (10 mL). The solution was cooled to −78 °C, and sec-BuLi was added (1.2 M in n-hexane, 1 mL, 1.2 mmol). After being stirred for 30 min, chlorodiphenylphosphine (0.14 mL, 0.76 mmol) was added to the mixture and allowed to react at room temperature for 2 h. Subsequently, water was incorporated to deactivate any residual n-BuLi. Thereafter, hydrogen peroxide (30%, 1.5 mL) was added, and the mixture was stirred for 40 min. After the complete reaction, the mixture was quenched with water, extracted with chloroform, and washed with brine. The organic layer was then dried over anhydrous Na2SO4 and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography using hexane/EtOAc (1:1 v/v) as the eluent. Compound 2 was obtained as a yellow solid (0.215 g, 73.1%). 1H NMR (500 MHz, CDCl3, 25 °C): δ 8.55 (s, 1H), 8.44 (dd, J = 1.0, 8.5 Hz, 2H), 8.09–8.03 (m, 6H), 7.61–7.53 (m, 10H) ppm; 13C NMR (126 MHz, CDCl3, 25 °C): δ 133.9, 133.8, 132.8, 132.4, 131.1, 130.8, 129.0, 128.8, 128.0, 126.0, 113.1, 103.2, 94.4, 93.1 ppm; 31P NMR (200 MHz, CDCl3, 25 °C): δ 8.40 ppm. The MS (ESI-TOF) m/z value calculated for C28H20OP was 403.12518. Found: 403.14830.
2.2.3. Synthesis of (4-(1-pyrenyl)phenyl)diphenylphosphine oxide (compound 3).
A mixture of (4-bromophenyl)diphenylphosphine oxide (0.360 g, 1.01 mmol), 1-pyreneboronic acid (0.502 g, 2.04 mmol), Pd(PPh3)4 (0.123 g, 0.106 mmol), and K2CO3 aq. (0.687 g, 4.97 mmol) in toluene/MeOH (v/v = 8 mL/2 mL) was refluxed under N2 at 110 °C overnight. After cooling to room temperature, the mixture was filtered through Celite, quenched with water, extracted with chloroform, and washed with brine. The organic layer was then dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography using CHCl3/EtOAc (20:1 v/v) as the eluent. Compound 3 was obtained as a light-brown solid (0.303 g, 62.9%). 1H NMR (500 MHz, CDCl3, 25 °C): δ 8.26–8.20 (m, 3H), 8.15–8.11 (m, 3H), 8.10–8.03 (m, 2H), 7.98 (d, J = 8.0 Hz, 1H), 7.90–7.80 (m, 6H), 7.76 (dd, J = 2.5 Hz, 2H), 7.62 (td, J = 1.5, 0.2, 1.5 Hz, 2H), 7.55 (td, J = 3.0, 4.0 Hz, 4H) ppm; 13C NMR (126 MHz, CDCl3, 25 °C): δ 145.0, 136.2, 133.0, 132.2, 132.1, 131.4, 131.0, 130.9, 130.8, 130.7, 128.7, 128.6, 128.4, 127.9, 127.8, 127.4, 126.2, 125.4, 125.1, 124.9, 124.8, 124.7, 124.7 ppm; 31P NMR (200 MHz, CDCl3, 25 °C): δ 29.2 ppm. MS (ESI-TOF): m/z calcd for C34H24OP: 479.15648. Found: 479.13168.
2.2.4. Synthesis of (1-pyrenylethynyl)diphenylphosphine oxide (compound 4).
Under a N2 atmosphere, 1-ethynylpyrene (0.141 g, 0.623 mmol) was dissolved in anhydrous tetrahydrofuran (10 mL). The solution was cooled to −78 °C, and sec-BuLi was added (1.2 M in n-hexane, 0.7 mL, 0.8 mmol). After being stirred for 30 min, chlorodiphenylphosphine (0.14 mL, 0.76 mmol) was added to the mixture and allowed to react at room temperature for 2 h. Subsequently, water was incorporated to deactivate any residual n-BuLi. Thereafter, hydrogen peroxide (30%, 1.5 mL) was added, and the mixture was stirred for 30 min. After the complete reaction, the mixture was quenched with water, extracted with chloroform, and washed with brine. The organic layer was then dried over anhydrous Na2SO4, and evaporated under reduced pressure. The crude product was purified by silica gel column chromatography using hexane/EtOAc (20:1, 10:1, 1:1 v/v) as the eluent. Compound 4 was obtained as a yellow solid (0.244 g, 91.9%). 1H NMR (500 MHz, CDCl3, 25 °C): δ 8.48 (d, J = 9.5 Hz, 1H), 8.26–8.24 (m, 3H), 8.20–8.16 (m, 2H), 8.13 (d, J = 8.0 Hz, 1H), 8.10–8.03 (m, 6H), 7.62–7.59 (m, 2H), 7.58–7.54 (m, 4H) ppm; 13C NMR (126 MHz, CDCl3, 25 °C): δ 133.8, 133.2, 132.9, 132.8, 132.3, 131.1, 131.0, 130.7, 130.4, 128.8, 128.7, 127.1, 126.6, 126.4, 124.7, 124.4, 124.1, 123.9, 113.6, 105.2, 88.8, 87.5 ppm; 31P NMR (200 MHz, CDCl3, 25 °C): δ 8.46 ppm. The MS (ESI-TOF) m/z calculated for C30H20OP was 427.12518. Found: 427.11886.
3. Results and discussion
3.1. Synthesis
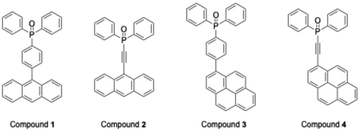
Phenylene-bridged triarylphosphine oxide derivatives 1 and 3 were synthesized by two distinct methodologies. In the first approach, 4-anthrylbromobenzene was obtained through a Suzuki cross-coupling reaction between 4-iodobromobenzene and anthracene boronic acid. Subsequent halogen–lithium exchange, followed by reaction with chlorodiphenylphosphine and oxidation with hydrogen peroxide, furnished compound 1 in 50% yield (Scheme S1). This procedure, however, failed to afford compound 3 in a pure form. In an alternative approach, 4-halophenyldiphenylphosphine oxide was initially prepared, and subsequent Suzuki cross-coupling with pyrenyl moieties afforded compound 3 in 63% yield (Scheme S2). Application of this methodology to the synthesis of compound 1 was hampered by difficulties associated with separation of by-products. Ethynylene-bridged analogues 2 and 4 were synthesized by three different routes. In the first route, ethynyldiphenylphosphine oxide was prepared and subjected to Sonogashira cross-coupling with 9-bromoanthracene and 1-bromopyrene (Scheme S3). In the second route, 9-haloanthracene and bromopyrene were coupled with trimethylsilylacetylene to afford ethynylanthracene and ethynylpyrene, respectively. The resulting intermediates were converted to the corresponding acetylide anions by treatment with butyllithium, which were subsequently reacted with chlorodiphenylphosphine and oxidized with hydrogen peroxide to yield the target compounds (Scheme S4). In the third route, ethynylanthracene and ethynylpyrene were prepared from the corresponding aldehydes via the Corey–Fuchs reaction.19 The resulting intermediates were subjected to the same sequence of transformations as in the second route to furnish the desired products (Scheme S5). While both Sonogashira-based methodologies failed to provide analytically pure products, the Corey–Fuchs route18 afforded compounds 2 and 4 in isolated yields of 54% and 92%, respectively, over four steps (Scheme 1).
 |
| | Scheme 1 Crystallization conditions. | |
3.2. Crystal structures
The four synthesized phosphine oxide compounds were recrystallized in various solvents, including chloroform, ethyl acetate, tetrahydrofuran (THF), N,N-dimethylformamide (DMF), methanol, and toluene. A single crystal was obtained by dissolving 5 mg of the compound in a solvent (0.5 mL); the mixture was heated and allowed to stand at room temperature. This crystal was then analyzed via SCXRD to identify its crystal structure (Fig. 1). The following scheme shows the correlation between the phosphine oxide crystallization conditions and the resulting crystal structures.
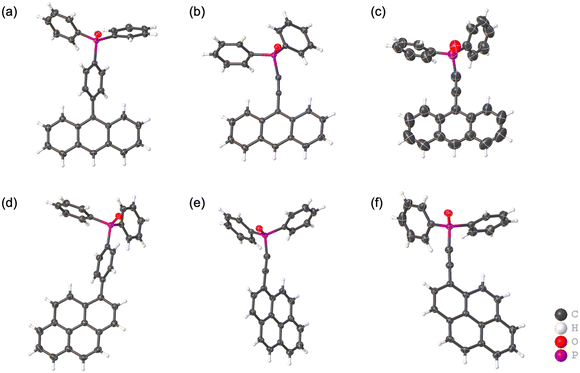 |
| | Fig. 1 Crystal structures of (a) (4-(9-anthryl)phenyl)diphenylphosphine oxide 1, (b) (9-anthrylethynyl)diphenylphosphine oxide 2a, (c) (9-anthrylethynyl)diphenylphosphine oxide 2b, (d) (4-(1-pyrenyl)phenyl)diphenylphosphine oxide 3, (e) (1-pyrenylethynyl)diphenylphosphine oxide 4a, and (f) (1-pyrenylethynyl)diphenylphosphine oxide 4b in the thermal ellipsoid model. The ellipsoids of all non-hydrogen atoms have been drawn at the 50% probability level. | |
3.2.1. (4-(9-Anthryl)phenyl)diphenylphosphine oxide 1.
Single crystals suitable for single-crystal X-ray diffraction (SCXRD) analysis were obtained from chloroform/methanol mixed solutions. The pale-yellow crystals crystallize in the monoclinic system with the space group P21/n. The anthryl substituent is attached at the 4-position of one of the three phenyl groups of the phosphine oxide core. In the solid state, the molecules are organized into a one-dimensional (1D) chain structure through C–H⋯π interactions between the anthryl unit and the phenyl rings of adjacent molecules (Fig. 2a). Additional C–H⋯π interactions involving the anthryl hydrogen atoms and neighboring phenyl groups extend this arrangement into a two-dimensional (2D) layer (Fig. 2b and c). The dihedral angle between the planes of the anthryl and phenylene units was determined to be 71.6°, clearly indicating that these moieties are not coplanar in the crystal structure (Fig. 2d). Hirshfeld surface analysis was performed to visualize intermolecular contacts (Fig. 2e). In the mapped surface, regions corresponding to hydrogen-bonding appear in red, reflecting relatively strong interactions. The contribution of C–H contacts to the overall intermolecular interactions was calculated to be 40.9% (Fig. 2f), suggesting a predominance of weak interactions among the multiple aromatic rings.
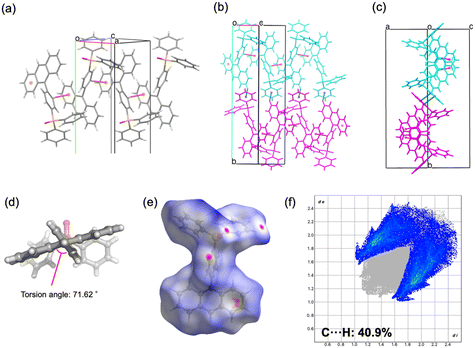 |
| | Fig. 2 Compound 1. (a) 1D linear chain of 1via CH/π interactions. The CH/π interactions are shown in red color. (b) 2D layer network via CH/π interactions (top view). The CH/π interactions are shown in green color. (c) 2D layer network via CH/π interactions (side view). The CH/π interactions are shown in green color. (d) Dihedral angle between the planes formed by an anthryl group and a phenylene group of 1. (e) Hirshfeld surface mapped with dnorm for 1. (f) Fingerprint plot for 1. | |
3.2.2. (9-Anthrylethynyl)diphenylphosphine oxide 2a.
Compound 2 was recrystallized from mixed solvents to afford single crystals (2a) suitable for SCXRD analysis. The pale-yellow crystals crystallize in the monoclinic system with the space group P21/c. In the molecular structure, the ethynylanthryl substituent is bound to the oxadiphenylphosphino moiety. Within the crystal packing, hydrogen bonds between the phenyl and oxophosphino groups organize the molecules into a zigzag one-dimensional (1D) chain along the c-axis (Fig. 3a). Additional C–H⋯π interactions between the anthryl unit of the ethynyl substituent and phenyl rings of adjacent molecules extend the packing into a two-dimensional (2D) layer along the b-axis (Fig. 3b and c). Furthermore, π–π interactions between anthryl units of neighboring molecules were also identified (Fig. 3b). The ethynyl moiety, typically expected to exhibit a linear geometry with a bond angle of 180°, was found to be significantly bent in the crystal structure (∠P–C–C = 164.5°, ∠C–C–C = 175.9°; Fig. 3d). Hirshfeld surface analysis was carried out to visualize intermolecular interactions (Fig. 3e). Regions corresponding to hydrogen-bonding and C–H⋯π contacts appear in red, reflecting relatively strong interactions. The contribution of C–H contacts to the overall intermolecular interactions was calculated to be 38.8% (Fig. 3f), indicating a predominance of weak aromatic interactions in the crystal lattice.
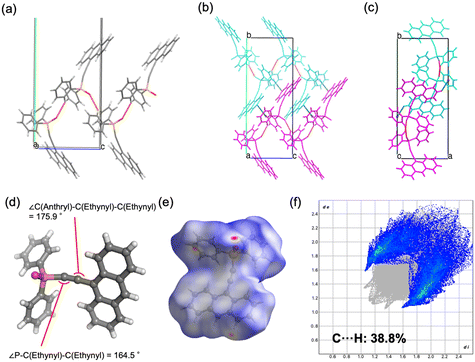 |
| | Fig. 3 Compound 2a. (a) 1D zigzag chain of 2avia P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯H interactions. The PO⋯H interactions are shown in red color. (b) 2D layer network via CH/π interactions (top view). The CH/π interactions are shown in green color. (c) 2D layer network via CH/π interactions (side view). The CH/π interactions are shown in green color. (d) Bond angles of the ethynyl group. (e) Hirshfeld surface mapped with dnorm for 2a. (f) Fingerprint plot for 2a. O⋯H interactions. The PO⋯H interactions are shown in red color. (b) 2D layer network via CH/π interactions (top view). The CH/π interactions are shown in green color. (c) 2D layer network via CH/π interactions (side view). The CH/π interactions are shown in green color. (d) Bond angles of the ethynyl group. (e) Hirshfeld surface mapped with dnorm for 2a. (f) Fingerprint plot for 2a. | |
3.2.3. (9-Anthrylethynyl)diphenylphosphine oxide 2b.
Single crystals of 2b suitable for SCXRD analysis were obtained from ethyl acetate solutions. The pale-yellow crystals represent a polymorphic form of 2a and crystallize in the triclinic system with the space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) . The molecular structure is identical to that of 2a, consisting of an ethynylanthryl substituent attached to the oxadiphenylphosphino moiety. In the crystal packing, two phosphine oxide molecules form a dimer through C–H⋯O hydrogen bonding between a phenyl hydrogen atom and the oxygen atom of the oxophosphino group (Fig. 4a). These dimers assemble into one-dimensional (1D) chains along the b-axis, mediated by additional C–H⋯π interactions between phenyl groups (Fig. 4b). Weak π–π interactions between the anthryl units of adjacent dimers align the 1D chains into a parallel arrangement (Fig. 4c). The centroid–centroid distance between the anthryl moieties was determined to be 3.880 Å. The ethynyl group displayed a geometry closer to linearity compared to polymorph 2a, with bond angles of ∠P–C–C = 165.9° and ∠C–C–C = 178.3° (Fig. 4d). Hirshfeld surface analysis was conducted to further examine intermolecular interactions (Fig. 4e). In the surface mapping, regions corresponding to hydrogen bonds are highlighted in red, whereas C–H⋯π and π–π interactions appear in white, suggesting interaction strengths comparable to van der Waals forces. The contribution of C–H⋯π contacts was calculated to be 36.5% (Fig. 4f), indicating a lower proportion of weak aromatic interactions relative to polymorph 2a.
. The molecular structure is identical to that of 2a, consisting of an ethynylanthryl substituent attached to the oxadiphenylphosphino moiety. In the crystal packing, two phosphine oxide molecules form a dimer through C–H⋯O hydrogen bonding between a phenyl hydrogen atom and the oxygen atom of the oxophosphino group (Fig. 4a). These dimers assemble into one-dimensional (1D) chains along the b-axis, mediated by additional C–H⋯π interactions between phenyl groups (Fig. 4b). Weak π–π interactions between the anthryl units of adjacent dimers align the 1D chains into a parallel arrangement (Fig. 4c). The centroid–centroid distance between the anthryl moieties was determined to be 3.880 Å. The ethynyl group displayed a geometry closer to linearity compared to polymorph 2a, with bond angles of ∠P–C–C = 165.9° and ∠C–C–C = 178.3° (Fig. 4d). Hirshfeld surface analysis was conducted to further examine intermolecular interactions (Fig. 4e). In the surface mapping, regions corresponding to hydrogen bonds are highlighted in red, whereas C–H⋯π and π–π interactions appear in white, suggesting interaction strengths comparable to van der Waals forces. The contribution of C–H⋯π contacts was calculated to be 36.5% (Fig. 4f), indicating a lower proportion of weak aromatic interactions relative to polymorph 2a.
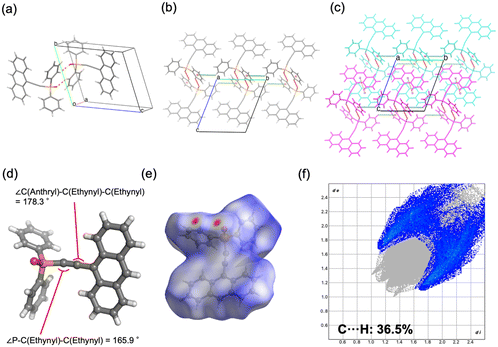 |
| | Fig. 4 Compound 2b. (a) Dimer of 2bvia PO⋯H interactions. The PO⋯H interactions are shown in red color. (b) 1D chain network via CH/π interactions. The CH/π interactions are shown in green color. (c) 2D layer network via π/π interactions. The π/π interactions are shown in orange color. (d) Bond angles of the ethynyl group. (e) Hirshfeld surface mapped with dnorm for 2b. (f) Fingerprint plot for 2b. | |
3.2.4. (4-(1-Pyrenyl)phenyl)diphenylphosphine oxide 3.
Single crystals of 3 suitable for SCXRD analysis were obtained from chloroform/methanol mixed solutions. The pale-yellow crystals crystallize in the monoclinic system with the space group P21/c. The molecular structure consists of a pyrenyl substituent attached to the 4-position of one of the three phenyl groups of the phosphine oxide core. In the crystal packing, molecules assemble into a one-dimensional (1D) chain along the b-axis through C–H⋯π interactions between pyrenyl moieties of adjacent molecules (Fig. 5a). Additional C–H⋯π interactions between phenyl groups extend this arrangement along the c-axis, resulting in a two-dimensional (2D) layered structure (Fig. 5b and c). The dihedral angle between the planes of the pyrenyl and phenylene groups was determined to be 54.3°, confirming their non-coplanarity in the solid state (Fig. 5d). Hirshfeld surface analysis was performed to visualize intermolecular interactions (Fig. 5e). Regions corresponding to C–H⋯π contacts are highlighted in white, indicative of relatively strong interactions. The contribution of C–H⋯π contacts to the overall intermolecular interactions was calculated to be 39.8% (Fig. 5f), suggesting that weak aromatic interactions play a predominant role in the packing arrangement.
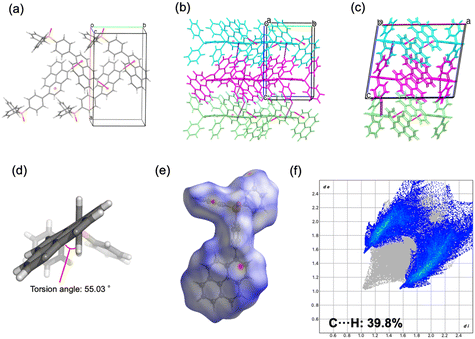 |
| | Fig. 5 Compound 3. (a) 1D linear chain of 3via CH/π interactions. The CH/π interactions are shown in red color. (b) 2D layer network via CH/π interactions (top view). The CH/π interactions are shown in purple color. (c) 2D layer network via CH/π interactions (side view). The CH/π interactions are shown in purple color. (d) Dihedral angle between the plane formed by an anthryl group and the plane formed by a phenylene group of 3. (e) Hirshfeld surface mapped with dnorm for 3. (f) Fingerprint plot for 3. | |
3.2.5. (1-Pyrenylethynyl)diphenylphosphine oxide 4a.
Recrystallization of 4 from mixed solvents afforded single crystals (4a) suitable for SCXRD analysis. The pale-yellow crystals crystallize in the monoclinic system with the space group P21/c. The molecular structure comprises an ethynylpyrenyl substituent bound to the oxadiphenylphosphino moiety. In the crystal packing, hydrogen bonds are formed between the phenyl and oxophosphino groups as well as between the pyrenyl and oxophosphino groups, generating a two-dimensional (2D) layered structure within the ab plane (Fig. 6a). Additional π–π interactions between pyrenyl moieties were also identified (Fig. 6b). The ethynyl unit adopts a geometry close to linearity, with bond angles of ∠P–C–C = 169.2° and ∠C–C–C = 179.1° (Fig. 6c), indicating a more linear configuration relative to compound 2. Hirshfeld surface analysis was carried out to examine intermolecular contacts (Fig. 6d). Regions corresponding to hydrogen-bonding interactions are highlighted in red, while those associated with C–H⋯π interactions appear in white, suggesting interaction strengths comparable to van der Waals contacts. The contribution of C–H⋯π contacts to the overall intermolecular interactions was calculated to be 43.8% (Fig. 6e), reflecting the prevalence of weak aromatic interactions in the crystal lattice.
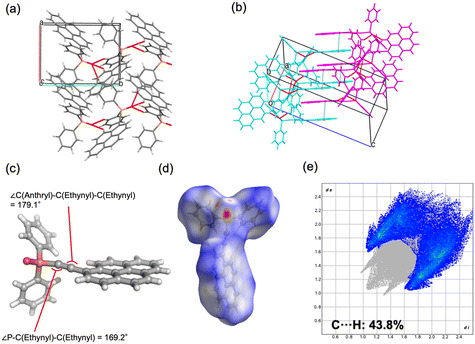 |
| | Fig. 6 Compound 4a. (a) 2D layer of 4avia PO⋯H interactions. The PO⋯H interactions are shown in red color. (b) 2D layer network via π/π interactions. The π/π interactions are shown in green color. (c) Bond angles of the ethynyl group. (d) Hirshfeld surface mapped with dnorm for 4a. (e) Fingerprint plot for 4a. | |
3.2.6. (1-Pyrenylethynyl)diphenylphosphine oxide 4b.
Single crystals of 4b suitable for SCXRD analysis were obtained from DMF solutions. The pale-yellow crystals represent a polymorphic form of 4a and crystallize in the monoclinic system with the space group P21/c. In the crystal packing, hydrogen bonds are formed between the oxophosphino group and multiple positions of the pyrenyl unit (C–H at the 5,6-positions and the 3-position) as well as with phenyl groups, thereby generating a two-dimensional (2D) layered structure within the ac plane (Fig. 7a). In addition, intermolecular π–π interactions between pyrenyl groups were observed (Fig. 7b). The ethynyl unit adopts a nearly linear geometry, with bond angles of ∠P–C–C = 174.5° and ∠C–C–C = 178.8° (Fig. 7c), indicating a more linear structure relative to polymorph 4a. Hirshfeld surface analysis was performed to examine intermolecular contacts (Fig. 7d). Regions corresponding to hydrogen-bonding interactions are highlighted in red, whereas those associated with π–π and C–H⋯π contacts appear in white, suggesting interaction strengths comparable to van der Waals forces. The contribution of C–H⋯π contacts to the overall interactions was calculated to be 36.9% (Fig. 7e), indicating reduced prevalence of weak aromatic interactions compared to polymorph 4a.
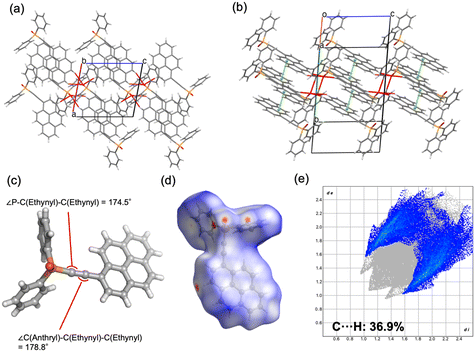 |
| | Fig. 7 Compound 4b. (a) 2D layer of 4bvia PO⋯H interactions. The PO⋯H interactions are shown in red color. (b) 2D layer network via π/π interactions. The π/π interactions are shown in green color. (c) Bond angles of the ethynyl group. (d) Hirshfeld surface mapped with dnorm for 4b. (e) Fingerprint plot for 4b. | |
3.2.7. Structural information and polymorphism.
Crystal polymorphism refers to the occurrence of multiple solid-state arrangements for a given molecule. The likelihood of polymorphism is generally correlated with the number of potential hydrogen-bonding sites, halogen-bonding sites, and aromatic ring–ring interaction sites. Aromatic substituents such as anthryl and pyrenyl units are therefore expected to promote intermolecular π–π interactions. In the phenylene-bridged compounds 1 and 3, the dihedral angles between the phenylene group and anthryl and pyrenyl groups are 71° and 55°, respectively (Table S6). This twisted conformation stabilizes the structures, indicating a loss of planarity between the phenylene moiety and the anthryl or pyrenyl groups. Accordingly, no polymorphism was observed for the phenylene-bridged compounds 1 and 3 synthesized in this study under various recrystallization conditions. This absence is most likely attributable to the restricted rotational freedom of the anthryl and pyrenyl substituents, which effectively fix the molecular geometry and reduce the diversity of weak interaction sites. By contrast, polymorphism was unambiguously confirmed for the ethynylene-bridged compounds 2 and 4, for which distinct crystalline forms were obtained depending on the recrystallization solvent (Fig. 8). This is thought to occur in the ethynylene-bridged type because the removal of torsion allows the anthryl and pyrenyl groups to rotate freely, thereby optimizing the interactions between the aromatic rings. The selected bond angles are summarized in Table S1. Furthermore, intermolecular interaction energies calculated using Crystal Explorer revealed that ethynylene-bridged molecules form stronger interactions with adjacent molecules than phenylene-bridged analogues. Specifically, the strongest interaction in 1 was −71.6 kJ mol−1, whereas those in 2a and 2b were −95.8 kJ mol−1 and −83.6 kJ mol−1, respectively. Similarly, the strongest interaction in 3 was −39.6 kJ mol−1, compared with −63.0 kJ mol−1 and −71.6 kJ mol−1 in 4a and 4b, respectively. The oxaphosphino group acts as a hydrogen bond acceptor in five of six crystals obtained. It exerts a strong influence on molecular arrangement and is considered to establish a structural framework that prevents excessive divergence of packing modes. The detailed structure of each polymorph is described below. In the case of compound 2, polymorph 2a (monoclinic, P21/c) exhibited zigzag one-dimensional chains stabilized by C–H⋯O and C–H⋯π interactions, whereas 2b (triclinic, P) formed dimeric units through C–H⋯O hydrogen bonding, which further assembled into chains. The geometry of the ethynyl moiety was more linear in 2b (∠P–C–C = 165.9°, ∠C–C–C = 178.3°) than in 2a (164.5° and 175.9°). Hirshfeld surface analysis further demonstrated a reduced contribution of C–H⋯π contacts in 2b (36.5%) compared to 2a (38.8%), reflecting differences in the balance of weak aromatic interactions. For compound 4, both polymorphs crystallized in the monoclinic system (P21/c), but with distinct packing motifs. Polymorph 4a formed layered structures within the ab plane through hydrogen bonding involving both phenyl and pyrenyl moieties, whereas 4b generated layers in the ac plane, stabilized by hydrogen bonding at multiple positions of the pyrenyl group. In both cases, π–π interactions between pyrenyl units contributed to the packing arrangement. The ethynyl moiety was again more linear in 4b (∠P–C–C = 174.5°, ∠C–C–C = 178.8°) relative to 4a (169.2° and 179.1°). Hirshfeld surface analysis revealed a lower proportion of C–H⋯π interactions in 4b (36.9%) than in 4a (43.8%), indicating reduced prevalence of weak aromatic contacts. Taken together, these results demonstrate that the conformational flexibility imparted by the ethynyl linkage, in contrast to the rigidity of the phenylene bridge, enables multiple packing modes in the solid state. The ability of the sp-hybridized ethynyl group to maintain a nominally linear geometry while tolerating slight distortions further augments the diversity of molecular arrangements, thereby increasing the probability of crystal polymorphism in compounds 2 and 4.
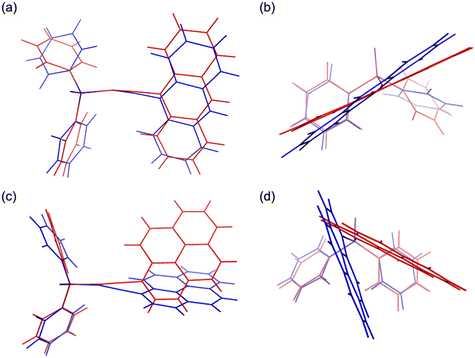 |
| | Fig. 8 Overlay of crystal polymorphs. (a) Top view of polymorphs 2a (red) and 2b (blue). (b) Side view of polymorphs 2a (red) and 2b (blue). (c) Top view of polymorphs 4a (red) and 4b (blue). (d) Side view of polymorphs 4a (red) and 4b (blue). | |
3.3. Luminescence properties in solution state
Fig. 9 presents the UV-vis absorption spectra of the synthesized phosphine oxides in acetonitrile solution. For the anthryl-substituted derivatives, the phenylene-bridged compound 1 exhibited absorption bands at 348, 365, and 385 nm, with a maximum absorption at 365 nm. In contrast, the ethynylene-bridged compound 2 displayed absorption at 374, 393, and 415 nm, with the maximum absorption red-shifted to 393 nm relative to 1. A similar trend was observed for the pyrenyl-substituted compounds. The phenylene-bridged derivative 3 exhibited absorptions at 279 and 343 nm, with the most intense band at 279 nm. Conversely, the ethynylene-bridged compound 4 showed relatively sharp absorption features at 290, 357, 375, and 389 nm, with a maximum absorption at 375 nm, red-shifted relative to 3. Thus, for both anthryl- and pyrenyl-containing derivatives, the ethynylene-bridged analogues consistently exhibited bathochromic shifts compared to their phenylene-bridged counterparts. This red-shift can be rationalized by the extended π-conjugation enabled by the ethynyl linkage, wherein the additional p-orbitals of the sp-hybridized carbon atoms contribute to delocalization. Notably, the absorption spectrum of 3 displayed significant broadening, indicative of enhanced electronic coupling between the pyrenyl, phenylene, and oxaphosphino moieties.
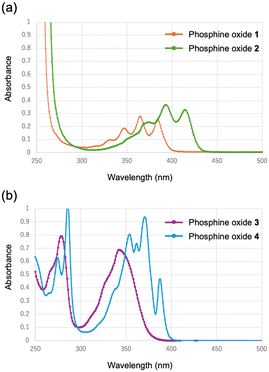 |
| | Fig. 9 UV-vis absorption spectra of (a) 1 (orange) and 2 (green), and (b) 3 (purple) and 4 (blue) in CH3CN. | |
3.4. Fluorescence spectra in solution state
Fig. 10 displays the fluorescence spectra of the phosphine oxides in acetonitrile solution. Emission bands characteristic of anthracene were observed, with maxima in the 400–500 nm region, consistent with monomeric anthracene fluorescence reported in the literature. For the anthryl-substituted derivatives, the phenylene-bridged compound 1 exhibited an emission maximum at 420 nm, whereas the ethynylene-bridged compound 2 showed an emission maximum at 455 nm. This bathochromic shift is consistent with the absorption behavior and can be attributed to the extended π-conjugation introduced by the ethynyl linkage. In contrast, the pyrenyl-substituted derivatives displayed emission bands in the 374–410 nm region, which are characteristic of monomeric pyrene fluorescence in solution. Specifically, the phenylene-bridged compound 3 displayed an emission at 410 nm, while the ethynylene-bridged compound 4 exhibited a maximum emission at 396 nm. These emission features are consistent with the locally excited state of pyrene monomers. Although the absorption spectra revealed red-shifts upon replacement of the phenylene unit with an ethynyl linker, the fluorescence wavelengths did not follow the same trend. Specifically, compound 3 displayed a longer emission wavelength than compound 4. The broadening of the fluorescence spectrum of 3 suggests pronounced electron delocalization and intramolecular polarization, which likely contribute to the observed bathochromic shift relative to 4.
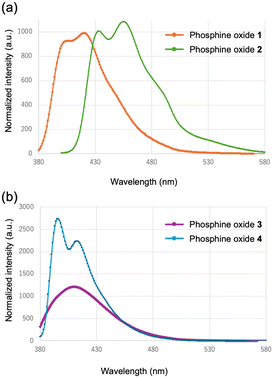 |
| | Fig. 10 Fluorescence spectra of (a) 1 (orange) and 2 (green), and (b) 3 (purple) and 4 (blue) in CH3CN. | |
3.5. Luminescence properties in solid state
Fig. 11 presents the UV-visible absorption spectra of the phosphine oxides in the solid state. Consistent with the solution-phase results, expansion of the conjugated system was confirmed by the conversion form the phenylene-bridged structure to the ethynylene-bridged structure. Specifically, the maximum absorption wavelengths of the ethynylene-bridged compounds 2 and 4 exhibited red shifts relative to the corresponding phenylene-bridged compounds 1 and 3. Since both compounds 2 and 4 exhibit crystalline polymorphism, the absorption spectra of their respective polymorphs were compared; however, no significant differences were observed.
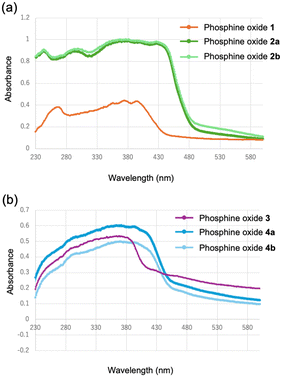 |
| | Fig. 11 UV-vis absorption spectra of (a) 1 (orange), 2a (green), and 2b (light green); (b) 3 (purple), 4a (blue), and 4b (light blue) in the solid state. | |
3.6. Fluorescence spectra in solid state
Fig. 12 depicts the solid-state fluorescence spectra of the synthesized phosphine oxides. For the anthryl-substituted derivatives, the maximum emission wavelengths were observed at 431 nm for the phenylene-bridged compound 1 and 433 nm for the ethynylene-bridged compound 2a. These emission bands are characteristic of monomeric anthracene fluorescence in the solid state. Despite the red shift in absorption associated with conjugation extension, the emission wavelengths remained essentially unchanged, indicating that extension of π-conjugation does not significantly alter the nature of the emissive excited state in these crystalline forms. By contrast, the ethynylene-bridged polymorph 2b exhibited a red-shifted maximum emission wavelength of 462 nm. This bathochromic shift and spectral broadening are indicative of excimer-like emission arising from enhanced π–π interactions between anthracene units in the crystal lattice. The observed dual-emission behavior in this system is therefore attributed to the coexistence of monomeric and excimer-like emissive states, which are stabilized differently depending on the molecular packing arrangement. These results suggest that variations in intermolecular interactions can modulate excited-state stabilization through packing-dependent electronic coupling. Although precise control of crystal polymorphism remains a considerable challenge, these findings imply that modulation of weak interactions—particularly those involving aromatic moieties—can subtly alter molecular packing and thereby enable fine-tuning of emission characteristics. In the case of pyrenyl-substituted derivatives, the maximum emission wavelengths were determined to be 446 nm for the phenylene-bridged compound 3, 484 nm for the ethynylene-bridged compound 4a, and 483 nm for crystalline polymorph 4b. The emission bands in compounds 4a and 4b, which appear in the 480 nm region, are consistent with excimer-type emission typically observed for pyrene aggregates in the solid state. In contrast, emission in compound 3 contains a larger contribution from monomer-like fluorescence. Consistent with the absorption spectra (Fig. 9 and 11), the replacement of the phenylene bridge with an ethynylene unit extends the π-conjugated system. Nevertheless, in the pyrenyl series, this structural modification exerts negligible influence on the fluorescence maxima, underscoring the limited role of extended conjugation in modulating the emissive properties of pyrenyl-based phosphine oxides.
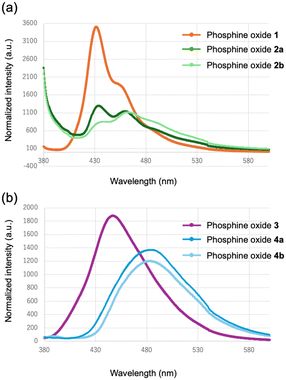 |
| | Fig. 12 Fluorescence spectra of (a) 1 (orange), 2a (green), and 2b (light green); (b) 3 (purple), 4a (blue), and 4b (light blue) in the solid state. | |
3.7. Photoluminescence quantum yields (PLQYs)
The PLQYs of the crystals, measured using an integrating sphere under deoxygenated conditions, were determined to be 51.4%, 6.7%, 0.8%, 45.6%, 47.7%, and 45.0% for crystals 1, 2a, 2b, 3, 4a, and 4b, respectively (Fig. S50–S55). Differences in crystal packing patterns modify intermolecular interactions and influence nonradiative decay pathways. In phenylene-bridged phosphine oxide 1, π–π stacking interactions between molecules are absent, resulting in relatively weak intermolecular packing; consequently, monomer-like emission is expected. The PLQY of approximately 50% supports this interpretation. In contrast, π–π stacking interactions are observed in the ethynylene-bridged polymorphs 2a and 2b. The extremely low quantum yields suggest the presence of excimer-like interactions that facilitate nonradiative decay. For phosphine oxides bearing pyrene moieties, nonradiative relaxation appears to be suppressed in both the phenylene-bridged and ethynylene-bridged derivatives, with only minimal reductions in PLQY observed. These results indicate that the presence of extended π–π interactions can significantly promote nonradiative pathways, particularly in the anthryl-containing systems.
3.8. Theoretical calculations
Quantum mechanical calculations were performed at the B3LYP/6-31g(d) level of theory to gain further insight into the structural and electronic properties of the crystals. Structural optimization was carried out using density functional theory (DFT) based on the geometries obtained from single-crystal X-ray diffraction data. Excited states were subsequently calculated with time-dependent DFT (TD-DFT) at the same level of theory, employing the optimized geometries. The optimized molecular geometries, HOMO and LUMO energy levels, and the corresponding frontier orbital distributions are shown in Fig. S56. Conversion from phenylene-bridged to ethynylene-bridged structures extends the π-conjugated system, leading to a red shift in the absorption wavelength. This interpretation is supported by the computational results, which show that the HOMO–LUMO gaps of the ethynylene-bridged species (2a, 2b, 4a and 4b) are smaller than those of the phenylene-bridged analogues (1 and 3). The reduction in the HOMO–LUMO gap quantitatively accounts for the observed bathochromic shifts in the absorption spectra of the ethynylene-bridged derivatives.
4. Conclusions
In this study, four phosphine oxides incorporating anthryl and pyrenyl luminescent moieties were successfully synthesized: (4-(9-anthryl)phenyl)diphenylphosphine oxide (1), 9-anthrylethynyldiphenylphosphine oxide (2), (4-(1-pyrenyl)phenyl)diphenylphosphine oxide (3), and 1-pyrenylethynyldiphenylphosphine oxide (4). Recrystallization from various solvents afforded high-quality single crystals, and polymorphism was confirmed in the ethynylene-bridged derivatives bearing anthryl and pyrenyl groups. Consequently, six distinct crystalline structures were characterized across the four compounds. Photophysical investigations in both solution and solid states revealed that the ethynylene-bridged derivatives generally exhibited red-shifted absorption and emission relative to their phenylene-bridged counterparts, consistent with the enhanced resonance effect and extended π-conjugation introduced by the ethynyl linkage. Importantly, polymorphism was found to modulate the emission behavior. In particular, although the anthryl-containing compound 2 did not exhibit a substantial shift in emission maxima between its polymorphs, differences in vibrational stabilization were observed, leading to subtle variations in fluorescence features. These findings indicate that while the overall luminescence color remains largely unaffected by polymorphism, the vibrational states contributing to emission can be tuned through subtle modifications in molecular packing. Overall, this work demonstrates that weak intermolecular interactions and crystal polymorphism provide an additional level of control over the excited-state dynamics of phosphine oxides. Furthermore, given that stimulus-induced structural transformations in the crystal lattice may also influence luminescence properties, the present results highlight the potential of crystal engineering as a strategy for the precise control of luminescence properties in organic functional materials.
Author contributions
All authors contributed to the preparation of the manuscript. All authors have read and approved the final version of the manuscript.
Conflicts of interest
The authors declare no conflicts of interest.
Data availability
All data supporting the findings of this study are provided in the supplementary information (SI).
Supplementary information is available. See DOI: https://doi.org/10.1039/d5ce00837a.
CCDC 2432548–2432553 (1, 2a, 2b, 3, 4a and 4b) contain the supplementary crystallographic data for this paper.20a–f
Acknowledgements
The authors thank Dr. Okumura and Dr. Kawamura (Japan Synchrotron Radiation Research Institute, JASRI) for their invaluable assistance with X-ray data collection. The synchrotron radiation experiments were performed at the BL26B1 beamline of SPring-8 with the approval of JASRI (Proposal No. 2023B1221).
References
-
(a) Y. Qin, G. Li, T. Qi and H. Huang, Mater. Chem. Front., 2020, 4, 1554–1568 RSC;
(b) Z. Wang, Q. Jingjing, X. Wang, Z. Zhang, Y. Chen, X. Huang and W. Huang, Chem. Soc. Rev., 2018, 47, 6128–6174 RSC;
(c) H. Wang and D. H. Kim, Chem. Soc. Rev., 2017, 46, 5204–5236 RSC;
(d) C.-T. Chen, Chem. Mater., 2004, 16, 4389–4400 CrossRef CAS.
-
(a) Y. Wang, H. Wu, W. Hu and J. F. Stoddar, Adv. Mater., 2022, 34, 2105405 CrossRef CAS PubMed;
(b) Y. Lei, W. Dai, J. Guan, S. Guo, F. Ren, Y. Zhou, J. Shi, B. Tong, Z. Cai, J. Zheng and Y. Dong, Angew. Chem., Int. Ed., 2020, 59, 16054–16060 CrossRef CAS PubMed;
(c) S. Kundu, B. Sk, P. Pallavi, A. Giri and A. Patra, Chem. – Eur. J., 2020, 26, 5557–5582 CrossRef CAS PubMed.
-
(a) X. Chen, T. Sun and F. Wang, Chem. – Asian J., 2020, 15, 21–33 CrossRef CAS PubMed;
(b) M. Tsurui, Y. Kitagawa, K. Fushimi, M. Gon, K. Tanaka and Y. Hasegawa, Dalton Trans., 2020, 49, 5352–5361 RSC;
(c) S. V. Eliseeva and J.-C. G. Bünzli, Chem. Soc. Rev., 2010, 39, 189–227 RSC;
(d) K. Binnemans, Chem. Rev., 2009, 109, 4283–4374 CrossRef CAS PubMed;
(e) A. de Bettencourt-Dias, Dalton Trans., 2007, 2229–2241 RSC.
-
(a) Y. Hirai, T. Nakanishi and Y. Hasegawa, J. Lumin., 2016, 170, 801–807 CrossRef CAS;
(b) K. Miyata, Y. Konno, T. Nakanishi, A. Kobayashi, M. Kato, K. Fushimi and Y. Hasegawa, Angew. Chem., Int. Ed., 2013, 52, 6413–6416 CrossRef CAS PubMed;
(c) M. L. P. Reddy, V. Divya and R. Pavithran, Dalton Trans., 2013, 42, 15249–15262 RSC;
(d) K. Miyata, T. Nakagawa, R. Kawakami, Y. Kita, K. Sugimoto, T. Nakashima, T. Harada, T. Kawai and Y. Hasegawa, Chem. – Eur. J., 2011, 17, 521–528 CrossRef CAS PubMed;
(e) K. Nakamura, Y. Hasegawa, H. Kawai, N. Yasuda, N. Kanehisa, Y. Kai, T. Nagamura, S. Yanagida and Y. Wada, J. Phys. Chem. A, 2007, 111, 3029–3037 CrossRef CAS PubMed;
(f) Y. Hasegawa, M. Yamamuro, Y. Wada, N. Kanehisa, Y. Kai and S. Yanagida, J. Phys. Chem. A, 2003, 107, 1697–1702 CrossRef CAS.
-
(a) Y. Kitagawa, M. Tsurui and Y. Hasegawa, RSC Adv., 2022, 12, 810–821 RSC;
(b) S. Miyazaki, K. Miyata, H. Sakamoto, F. Suzue, Y. Kitagawa, Y. Hasegawa and K. Onda, J. Phys. Chem. A, 2020, 124, 6601–6606 CrossRef CAS PubMed.
-
(a) C. Tang, X. Zhu, Y. Song, W. Liu, Q. Yang, Z. Lv and Y. Yang, J. Photochem. Photobiol., A, 2019, 376, 263–268 CrossRef CAS;
(b) H. Ye, G. Liu, S. Liu, D. Casanova, X. Ye, X. Tao, Q. Zhang and Q. Xiong, Angew. Chem., Int. Ed., 2018, 57, 1928–1932 CrossRef CAS PubMed.
-
(a) S. K. Rajagopal, P. S. Salini and M. Hariharan, Cryst. Growth Des., 2016, 16, 4567–4573 CrossRef CAS;
(b) L. Zou, X.-Y. Wang, K. Shi, J.-Y. Wang and J. Pei, Org. Lett., 2013, 15, 4378–4381 CrossRef CAS PubMed.
-
(a) P. Ludwig, J. Mayer, L. Ahrens, F. Rominger, G. Ligorio, F. Hermerschmidt, E. J. W. List-Kratochvil, J. Freudenberg and U. H. F. Bunz, Chem. – Eur. J., 2024, 30, e202303037 CrossRef CAS PubMed (1–8);
(b) G. S. Baviera and P. M. Donate, Beilstein J. Org. Chem., 2021, 17, 2028–2050 CrossRef CAS PubMed;
(c) K. Kumar, K. K. Kesavan, D. Thakur, S. Banik, J. Jayakumar, C.-H. Cheng, J.-H. Jou and S. Ghosh, ACS Omega, 2021, 6, 10515–10526 CrossRef CAS PubMed;
(d) M. Y. Lo, C. Zhen, M. Lauters, G. E. Jabbour and A. Sellinger, J. Am. Chem. Soc., 2007, 129, 5808–5809 CrossRef CAS PubMed.
-
(a) I. Ahmad, S. J. Prathapa and A. A. Dar, J. Mater. Chem. C, 2025, 13, 1161–11670 Search PubMed;
(b) A. A. Malik, Z. M. Saeed, I. Ahmad, T. Alkhidir, P. B. Managutti, S. Mohamed and A. A. Dar, ACS Appl. Opt. Mater., 2024, 2, 1709–1720 CrossRef CAS;
(c) A. A. Malik, A. A. Ganie, M. Wahid and A. A. Dar, ACS Appl. Opt. Mater., 2024, 2, 2229–2240 CrossRef CAS;
(d) A. A. Dar and A. A. Malik, J. Mater. Chem. C, 2024, 12, 9888–9913 RSC;
(e) A. A. Dar, A. A. Ahangar, C. Femina, A. A. Malik, J. V. Parambil and P. K. Sajith, J. Phys. Chem. C, 2024, 128, 18901–18912 Search PubMed;
(f) G. Chakraborty, J. N. Malegaonkar, S. V. Bhosale, P. K. Singh and H. Pal, J. Phys. Chem. B, 2021, 125, 11122–11133 CrossRef CAS PubMed;
(g) R. Liao, X. Wang, L. Peng, H. Sun and W. Huang, ACS Appl. Mater. Interfaces, 2021, 13, 27491–27499 CrossRef CAS PubMed;
(h) C. Botta, S. Benedini, L. Carlucci, A. Forni, D. Marinotto, A. Nitti, D. Pasini, S. Righetto and E. Cariati, J. Mater. Chem. C, 2016, 4, 2979–2989 RSC;
(i) R. Hagihara, N. Harada, S. Karasawa and N. Koga, CrystEngComm, 2015, 17, 8825–8834 RSC.
-
(a) J. Elguero, Cryst. Growth Des., 2011, 11, 4731–4738 CrossRef CAS;
(b) L. Yu, Acc. Chem. Res., 2010, 43, 1257–1266 CrossRef CAS PubMed;
(c) A. Nangia, Acc. Chem. Res., 2008, 41, 595–604 CrossRef CAS PubMed;
(d) W. I. F. David, K. Shankland, C. R. Pulham, N. Blagden, R. J. Davey and M. Song, Angew. Chem., Int. Ed., 2005, 44, 7032–7035 Search PubMed.
-
(a) D. S. Reddy, D. C. Craig and G. R. Desiraju, J. Am. Chem. Soc., 1996, 118, 4090–4093 CrossRef CAS;
(b) D. S. Reddy, Y. E. Ovchinnikov, O. V. Shishkin, Y. T. Strychkov and G. R. Desiraju, J. Am. Chem. Soc., 1996, 118, 4085–4089 CrossRef CAS.
-
(a) L. C. Gilday, S. W. Robinson, T. A. Barendt, M. J. Langton, B. R. Mullaney and P. D. Beer, Chem. Rev., 2015, 115, 7118–7195 CrossRef CAS PubMed;
(b) A. Mukherjee, Cryst. Growth Des., 2015, 15, 3076–3085 CrossRef CAS;
(c) T. M. Beale, M. G. Chdzinski, M. G. Sarwar and M. S. Taylor, Chem. Soc. Rev., 2013, 42, 1667–1680 RSC.
-
(a) S. Kikkawa, M. Takeno, T. Nakayama, D. Koike, Y. Saito, M. Tashiro, Y. Aoyama, H. Hikawa and I. Azumaya, Cryst. Growth Des., 2024, 24, 9564–9570 CrossRef CAS;
(b) S. Kikkawa, I. Maeno, K. Katagiri, Y. Murayama, M. Nozawa, H. Hikawa and I. Azumaya, Cryst. Growth Des., 2021, 21, 4380–4389 CrossRef CAS;
(c) S. Kikkawa, M. Okayasu, H. Hikawa and I. Azumaya, Cryst. Growth Des., 2021, 21, 1148–1158 CrossRef CAS;
(d) S. Kikkawa, H. Masu, K. Katagiri, M. Okayasu, K. Yamaguchi, H. Danjo, M. Kawahata, M. Tominaga, Y. Sei, H. Hikawa and I. Azumaya, Cryst. Growth Des., 2019, 19, 2936–2946 CrossRef CAS.
-
CrysAlis Pro version 171.39.20a, Rigaku Oxford Diffraction, Tokyo, Japan, 2015 Search PubMed.
- O. V. Dolomanov, L. J. Bourthis, R. J. Gildea, J. A. K. Howard and H. Puschmann, OLEX2: a complete structure solution, refinement and analysis program, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- G. M. Scheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- G. M. Scheldrick, Acta Crystallogr., Sect. C: Struct. Chem., 2015, 71, 3–8 Search PubMed.
-
M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheesman, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2013 Search PubMed.
-
(a) O. Tosic and J. Mattay, Eur. J. Org. Chem., 2011, 371–376 CrossRef CAS;
(b) E. J. Corey and P. L. Fuchs, Tetrahedron Lett., 1972, 3769–3772 CrossRef CAS.
-
(a)
CCDC 2432548: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mn89w;
(b)
CCDC 2432549: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mn8bx;
(c)
CCDC 2432550: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mn8cy;
(d)
CCDC 2432551: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mn8dz;
(e)
CCDC 2432552: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mn8f0;
(f)
CCDC 2432553: Experimental Crystal Structure Determination, 2026, DOI:10.5517/ccdc.csd.cc2mn8g1.
|
| This journal is © The Royal Society of Chemistry 2026 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *ab
*ab