
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMore than “just a drop”: the enigmatic role of liquid additives in mechanochemistry
Ilenia
D'Abbrunzo
 and
Dritan
Hasa
*
and
Dritan
Hasa
*
Department of Chemical and Pharmaceutical Sciences, University of Trieste, P.le Europa 1, 34127 Trieste, Italy. E-mail: dhasa@units.it
First published on 11th December 2025
Abstract
We debate the role of liquid additives in liquid-assisted mechanochemistry, highlighting how miniscule amounts of liquid profoundly and mostly inexplicably affect the mechanism and outcomes of mechanochemical reactions. A critical analysis of well-known mechanochemistry studies, along with those reporting emerging concepts (such as the competitive liquid effect and dual-function liquids) underpins the complexity and mystery surrounding mechanochemical processes and suggest where the most series gaps in our understanding of mechanochemistry lie.
1. Introduction
Mechanochemistry is an area of green chemistry that promotes and/or accelerates chemical reactions by applying mechanical energy through various techniques, including milling, shearing, extrusion, speed mixing, resonant acoustic mixing, etc.1,2Unlike conventional solution-based synthetic and crystallisation methods, which are traditionally applied using copious amounts of solvent, mechanochemical processes regularly enable chemical transformations in the solid state under solvent-free conditions, whereby minute amounts of solvents are added to facilitate these conversions. This approach dramatically reduces the need for hazardous and costly solvents (thereby aligning with the principles of green and sustainable chemistry3–6), opens new reaction pathways in synthetic chemistry, and enables supramolecular recognition processes that are otherwise inaccessible in the solutions,6–10 thus opening new avenues for controlling product formation and solid-state properties.11
Mechanochemistry has continuously attracted great interest across various fields,12–18 mostly in pharmaceutical sciences and pharmaceutical research and development,6,19,20 (Fig. 1) where such milling and other mechanochemical processes are now habitually used to optimise solid-state properties of active pharmaceutical ingredients (APIs); mostly to improve solubility, stability, and bioavailability through amorphisation, and salt and cocrystal formation.9,16,21–25
 | ||
| Fig. 1 Applications of mechanochemistry with a particular emphasis on the pharmaceutical science. | ||
Among the various mechanochemical techniques, liquid-assisted grinding (LAG) has emerged as a highly versatile and widely used method due to its simplicity and proven effectiveness across a broad range of systems, as extensively demonstrated in many mechanochemical studies and elaborated in comprehensive reviews over the past three decades.26–31
LAG involves the promotion of chemical reactivity and solid-phase transformations in the presence of low amounts (typically a few microliters per milligram of solid) of one (or more) liquid additive(s) to control reaction rates, product selectivity, and reaction pathways.32–34
Initially introduced for improving process reproducibility,28 the role of liquids in LAG has since evolved well beyond that of passive facilitators. Today, liquid additives are recognised as active and sometime essential components in the reaction environment, capable of modulating reactivity and directing supramolecular assembly, or even altering the chemical nature of the mechanochemical product.35–38 Depending on the quantity and physicochemical characteristics, the presence of liquid additives can promote or inhibit transformations, mediate polymorphic transitions or stabilise metastable intermediates.26,27,35,39–41
In this highlight, we discuss the role and applications of liquid additives in mechanochemical reactions, with a particular focus on their physicochemical properties – including polarity, hydrogen bonding capacity, viscosity, molecular mobility, etc. We also identify challenges and gaps in our understanding of LAG, and discuss emerging concepts such as the use of liquid mixtures and the competitive behaviour of multicomponent liquid additives (Fig. 3).36,37 Lastly, we highlight the use of molecular dynamics or other computational methods42–45 to enable rational design and control of mechanochemical reactions, as initial approaches for stepping away from current highly empirical trial-and-error approaches.
2. Historical perspective on the use of liquid additives in mechanochemical processes
The earliest documented use of a liquid additive in a mechanochemical reaction dates to the fourth century B.C., when cinnabar (HgS) was processed with vinegar to release elemental mercury. In this rudimentary process, the liquid (vinegar) presumably acted as a lubricant, facilitating particle contact and accelerating reactivity.46 Over the centuries, mechanochemistry has been sporadically explored in the context of inorganic materials, metals, and alloys, particularly for applications in metallurgy, ceramics, and catalysis.13 In these early studies, liquids were typically added to improve milling efficiency or control physical parameters of the mechanochemical environment. During these times, LAG was commonly referred to as “solvent-assisted grinding” or more generically “wet milling” (Fig. 2). These terms clearly highlight how liquid additives are used primarily to control mechanical processes, rather than controlling chemical reactivity and selectivity.26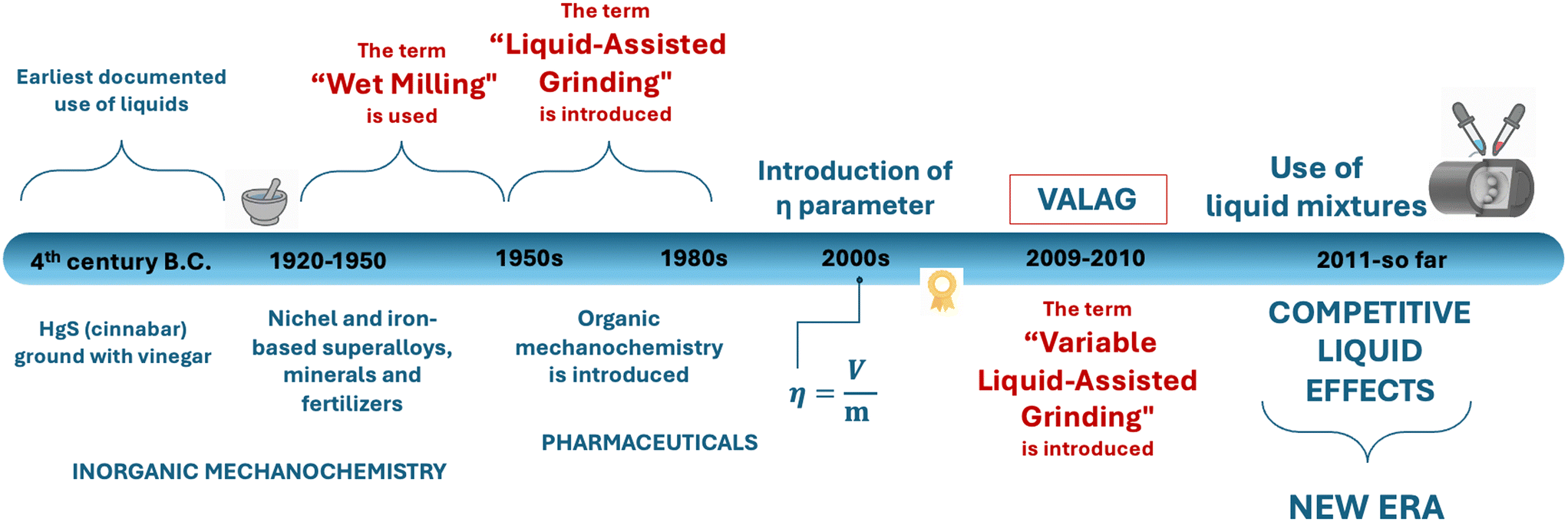 | ||
| Fig. 2 Schematic representation of some key stages of the development of liquid-assisted mechanochemistry. Adapted from ref. 32, with permission from Elsevier, Advanced Drug Delivery Reviews, Copyright 6164781277936. | ||
While the history of mechanochemistry spans over many centuries,7 it is only within the last three decades that the technique has been increasingly applied to soft matter, particularly in organic chemistry and pharmaceutical solid-form design.47,48 This shift has resulted in a surge of publications and patents, underlining the growing importance of mechanochemistry as a viable synthetic strategy. In this context, the term “kneading” was also introduced, particularly within pharmaceutical solid-state chemistry, to describe processes involving the incorporation of small amounts of liquid during milling. More refined terms such as “solvent-drop grinding” (SDG) and ultimately LAG (Fig. 2) have therefore emerged, mirroring a deeper understanding of the role of liquids in directing and enabling solid-state transformations in molecular systems.26
In line with these developments, the present highlight will focus on the use of liquid additives in organic mechanochemical reactions, and particularly on applications involving pharmaceuticals and related molecular systems.
At the core of early mechanochemical investigations of soft materials was the empirical observation that different quantities of a liquid can have different effects on the reaction outcome. In response to this, the η parameter39 (eqn (1)) was introduced to quantify the amount of liquid intentionally added to the mechanochemical reactor, and to standardise how LAG reactions are reported, thus enabling better reproducibility and comparison between systems.
 | (1) |
• η = 0; classed as neat grinding (NG), where no liquid is intentionally added.125
• 0 < η < 2; classed as LAG, where the addition of small amounts of liquid alters the process without dissolving the solid components.
• 2 < η < 12; described as slurrying, where more liquid is added to create a thick slurry that facilitates mixing and grinding.
• η > 12; described as solution-based reaction, in which the components are fully dissolved in a solvent to achieve complete mixing and reaction.
Within the η values for LAG, the liquid does not necessarily dissolve the solid components but can enhance the mobility of the reactants at the interfaces and improve the frequency and efficiency of the collisions among different solid particles.6 Such picture, however, was proved too simplistic as a growing number of reports demonstrated that not all liquids were equally effective under identical milling conditions. Indeed, it was suggested that the chemical nature of the liquid – such as polarity, hydrogen bonding ability, and dielectric constant – are likely to affect product selectivity, crystallinity, and reaction rates.34,37,49–59
Subsequently, the concept of variable amount liquid-assisted grinding (VALAG) was proposed to reflect the more nuanced continuum of solid–liquid interactions. Rather than drawing strict boundaries between NG, LAG, and slurry methods, VALAG emphasises that the outcome of a mechanochemical reaction is dynamically modulated by the precise liquid loading and its interaction with the solid components.32 Several notable examples showed that even slight variations of the volume could steer the system toward different solid forms – whether by stabilising metastable polymorphs or enabling the formation of multicomponent solids that were inaccessible by solution methods.32,35,39,41,59 In this context, in one of the most recent examples, Lombard et al. observed that by simply varying the amount of liquid used during the mechanochemical formation of multicomponent crystals was sufficient to alter the chemical nature of the dominant intermolecular interactions within the system. Specifically, higher liquid amounts (η ≈ 6 μL mg−1) favoured the formation of halogen-bonded cocrystals, whereas lower liquid levels (η ≈ 0.25 μL mg−1) increasingly promoted hydrogen-bonding.60 This finding neatly illustrates how quantitative control over the liquid can go beyond structural modulation and determine the predominant type of non-covalent interaction in a solid, thereby directing the supramolecular character of the resulting solid form. A series of striking examples published earlier are discussed below.
3. Multifunctional roles of liquid additives in LAG
The impact of liquid additives on a given mechanochemical process is governed by both intrinsic physicochemical properties and extrinsic parameters including the stoichiometric ratio, nature of the solid components, and some other mechanochemical experimental conditions. Depending on these factors, liquids can serve a wide range of functions within the same system.35,40,41,59,61,62 In what follows, we categorise these functions into four major categories (Fig. 3): | ||
| Fig. 3 Classifying the role of liquid additives in a mechanochemical reaction into four major categories. | ||
• Promoters, where the presence of a specific amount of a liquid additive (or a liquid mixture) enables product formation.
• Inhibitors and prohibitors, where the addition of the liquid slow down or entirely suppress specific reactions.
• Solvate-forming and structure-directing liquid agents, where the additive leads to the formation of solvated crystals or drives the mechanochemical product to a specific polymorph including metastable phases.
• Dual-role liquids, where the presence of a single liquid acts both as a solvate-forming agent and a reaction promoter when used in excess.
3.1 Promoters of reactions
In a typical role, the liquid additive acts as reaction promoter by enhancing molecular diffusion and improving contact between solid particles. This is particularly effective when the added liquid improves surface wetting or reduces interparticle friction, thereby facilitating energy transfer during milling.29 In pharmaceutical mechanochemistry, such effect is particularly evident during the synthesis of crystalline multicomponent solids. Indeed, several studies have shown that NG often leads only to physical mixtures or amorphous materials, whereas the addition of a small amount of a liquid enables the formation of new and highly crystalline phases. One of the first reported examples is the carbamazepine–nicotinamide cocrystal, where LAG with ethanol or acetonitrile drastically accelerates the reaction compared to NG.62 The same study also suggested that liquids enhance molecular mobility without dissolving the reactants, leading to rapid and complete cocrystal formation. Similarly, caffeine-based cocrystals (for example: caffeine–glutaric acid and caffeine–citric acid) show significant rate enhancements in the presence of polar protic solvents like methanol or water.63,64 This promoting effect has also been observed in the cocrystallisation of sulfadimidine and saccharin, where LAG with isopropanol yields a highly crystalline product within minutes, while NG is inefficient, or alternatively, leads to amorphous phases.65 A further compelling example is the mechanochemical formation of salts between ethacridine and salicylic or acetylsalicylic acid. In this case, only LAG with small amounts of aqueous solvents afforded highly crystalline, anhydrous salts in high yields, while both solution-based approaches and NG led to hydrated or undefined phases.103.2 Inhibitors and prohibitors: when liquids slow or suppress reactions
Unexpectedly, certain liquids can slow down or even completely suppress mechanochemical transformations. The inhibitive and prohibitive nature of liquid additives,40,41 was first systematically investigated using a vinpocetine–oxalic acid salt as model compound.41 The mechanochemical screens were conducted under LAG conditions using a range of different liquids (Fig. 4), and revealed that: | ||
| Fig. 4 (A) Molecular structures of vinpocetine and oxalic acid and the crystal packing of (top) the polymorphic forms (top) I and (bottom) II of the vinpocetine–oxalic acid salt, (B) summary of LAG experiments performed with liquid additives that vary in their chemical nature and quantities used, (C), diagram summarising all interconversion LAG experiments performed and highlighting the catalytic, inhibitive and prohibitive actions of the studied liquid additives. Adapted from ref. 41, published under a CC-BY license. | ||
• Acetonitrile and acetone slowed down or delayed product formation. The desired multicomponent solid was nonetheless accessible through extended milling times. These liquid additives were therefore described as inhibitive.
• Nitromethane and 2-pyrrolidone prevented solid-state reaction. A product could not be obtained even after prolonged milling of reactants. These additives were hence described as prohibitive.
• Dimethyl sulfoxide acted as reversing agent, redirecting the reaction pathway and favouring the stabilisation of starting materials instead of the salt.
A possible explanation of such different behaviour could be attributed to the competitive solvation and strong interactions between the liquid and one or more of the reactants that can prevent the necessary intermolecular contacts leading to product formation. These findings challenge the traditional assumption that all polar protic or hydrogen-bonding solvents are necessarily beneficial in LAG. For example, while water often promotes reactivity, it can also act as an inhibitor or template, depending on the system.66 The described studies also suggest that the chemical affinity of the added liquid and the solid reactants must be carefully considered together with the physical parameters such as viscosity or volatility. What appears as a simple additive can modulate the energy landscape of the reaction, shifting equilibria, stabilising metastable forms, or even diverting entirely the reaction pathway. It is therefore clear that the role of liquids in LAG goes beyond the promotion of solid-state reactions and transformations, and that liquids can be powerful gatekeepers in mechanochemical systems. Recognising and classifying such behaviours is an initial necessary step toward the rational design of LAG protocols.
3.3 Solvate-forming and structure-directing liquid agents
Liquids can also play a central role in product selectivity and supramolecular assembly. This includes both solvation effects, where a solvent guides the formation of solvated crystal structures, and phase mediation, where the choice of the liquid determines the formation of specific metastable polymorphs, or even new solid forms.27,35,49,55,59,61,67–71A particularly illustrative case was recently reported by Brekalo and co-authors,72 who employed LAG to systematically map the rich polymorphic landscape of zinc imidazolate (ZnIm2), one of the most polymorphic members of the zeolitic imidazolate framework (ZIF) family. Central to this work is the demonstration that the liquid additive is not merely a reaction accelerator but a decisive structure-directing agent capable of stabilising, shaping, and even templating distinct ZnIm2 topologies. The roles of the polarity, size, shape, and chemical functionality of a liquid additive were probed in a mechanochemical screen using 45 liquids. The screen highlighted how geometric and chemical features of the additive control the outcome of a mechanochemical reaction, and in this case, determine which ZnIm2 framework emerges. Specifically, the use of the 45 liquid additives enabled the formation of eight framework topologies, fourteen distinct crystals forms, two new crb-type structures, and several amorphous phases. The liquid therefore likely acted as a template, occupying and stabilising transient or metastable cavities during nucleation. For low-density frameworks (e.g., cag, neb, crb), this stabilisation is essential, as these materials are thermodynamically disfavoured in their ‘empty’ forms, but the encapsulation of appropriately shaped guests (in form of the liquid additive) enabled their mechanochemical synthesis. Interestingly, several liquids induced time-dependent or condition-dependent transformations: short milling times yielded one framework, whereas prolonged milling led to another, reflecting gradual shifts in guest–framework stability or rearrangement barriers. In specific cases liquid additives also stabilised amorphous ZnIm2 phases that later crystallise when the guest is removed, highlighting the inhibitive role of specific liquids. The authors also pursued periodic DFT calculations of empty frameworks to corroborate experimentally observed trends. It was suggested that dense structures lie lowest in energy, while open metal–organic frameworks (MOFs) cluster at 20–25 kJ mol−1 higher in energy. Liquid additives therefore operate primarily by lowering the effective free energy of specific loaded frameworks, thus enabling access to topologies that would otherwise be difficult to access. The strong parallel between solvothermal templation strategies and the mechanochemical results further demonstrates that many templating principles can be directly transferred to solid-state synthesis, necessitating an expansion of the variety of liquid additives that are habitually used in mechanochemical reactions. Overall, this study establishes that the liquid additive is a dominant steering factor in mechanochemical MOF form discovery, not only accelerating reactions but actively directing framework topology via pore filling, geometric matching, kinetic modulation, and stabilisation of metastable forms, providing an excellent foundation for the additive-directed MOF synthesis.
 | ||
| Fig. 5 (A) Molecular structures of caffeine and anthranilic acid, and the crystal packing of the three known polymorphic forms of the cocrystal, (B) summary of LAG experiments performed with different amounts of alcohols having different molecular weight, (C) summary of LAG experiments with ethanol performed with different volumes of liquids previously saturated with either pure caffeine or anthranilic acid. Green, blue, and yellow balls represent solid products containing X-ray pure cocrystal form I, form II, and form III, respectively, while red balls indicate a solid product containing a mixture of either polymorphic form I and II or form I and III. Adapted from ref. 35, published under a CC-BY license. | ||
Other studies have also shown that subtle variations in liquid properties such as polarity, hydrogen bonding ability, surface solvation effects or dielectric constant can bias the system toward one polymorph over another.27,49,51,56,57,71,73–76 Numerous studies have substantiated this behaviour, demonstrating that LAG can reproducibly mediate polymorphic interconversion as a function of the liquid used. For example, polymorphic salts of trimethoprim with L- and D-lactic acids were shown to undergo reversible interconversion between two forms, α and β, depending on the liquid environment: methanol favoured the formation of the α-polymorph, while a 5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 acetonitrile–water mixture directed the system toward the β-form.77 Also, in the mechanochemical synthesis of nicotinamide-ascorbic acid cocrystals, ethanol LAG led to form I, with form II being an intermediate, while a reaction involving methanol as additive yielded form I, which subsequently transformed to form II – a phenomenon not correlated with bulk solubility, suggesting a templating role of the liquid through transient solvation or stabilisation of specific intermediates.78
:1 acetonitrile–water mixture directed the system toward the β-form.77 Also, in the mechanochemical synthesis of nicotinamide-ascorbic acid cocrystals, ethanol LAG led to form I, with form II being an intermediate, while a reaction involving methanol as additive yielded form I, which subsequently transformed to form II – a phenomenon not correlated with bulk solubility, suggesting a templating role of the liquid through transient solvation or stabilisation of specific intermediates.78
In a more recent study79 Germann et al. monitored the mechanochemical formation of caffeine–glutaric acid cocrystal system by using in situ synchrotron X-ray powder diffraction. Specifically, LAG experiments were performed using a highly polar liquid such as acetonitrile and a low-polar additive such as 1-dodecanol, and the kinetics of cocrystal formation were compared to those obtained through NG (Fig. 6). It was observed that LAG with acetonitrile at η = 0.05 μL mg−1 yielded cocrystal form I almost instantaneously. This was soon after followed by a transformation into form II as the final product, reaching a steady state within 15 minutes. Interestingly, additional LAG experiments at η = 0.12 and 0.17 μL mg−1 suggested that, while the overall rate of cocrystallisation is higher, the kinetics of conversion of cocrystal form I to form II were not significantly affected by a higher amount liquid additive (Fig. 6). The kinetics of polymorph conversion (form I → form II), however, strongly depended on the amount of the liquid additive in the case of LAG reactions with a less polar liquid such as 1-dodecanol. Additional LAG experiments with such liquid revealed that, form I was obtained as the final product within 10 minutes of milling during LAG at η = 0.05 μL mg−1, while form II started appearing after ca. 30 min at η values of 0.12 and 0.17 μL mg−1, respectively. Generally, the development of in situ techniques has provided significant contributions to the understanding of LAG.80
 | ||
| Fig. 6 Molecular structures of caffeine and glutaric acid, the crystal packing of the two known polymorphic forms of the cocrystal, and quantitative analyses of the reactants and products from sequential Rietveld refinement of the time-resolved monitoring reaction under different experimental conditions. Adapted from ref. 79. | ||
Another intricate example of liquid-mediated polymorphic control was demonstrated by Belenguer et al.,81 who reported the reversible and quantitative interconversion of three distinct polymorphs of a disulfide compound via ball milling. In that study, LAG enabled selective and reversible interconversion between three crystalline polymorphs of a disulfide compound, including a newly discovered form. LAG with specific solvents such as water or acetonitrile directed polymorph interconversion by stabilising crystal surfaces at the nanoscale, shifting the apparent thermodynamic order and allowing access to metastable forms unattainable by recrystallisation. The authors showed that each polymorph is obtained with a characteristic and reproducible crystal size, confirming that LAG governs both nucleation and growth under milling equilibrium. The work highlights the complex interplay between liquid properties, surface effects, and nucleation pathways, emphasising the important and diverse role of liquids in directing polymorphic outcomes.56,81
In the early work by Losev and Boldyreva,82 it was reported that the mechanochemical reaction between β-alanine and DL-tartaric acid gave distinctly different outcomes depending on the crystallisation protocol. Slow evaporation or LAG reliably produced the stable cocrystal, whereas NG and fast anti-solvent precipitation promoted the formation of a metastable salt due to rapid nucleation. The authors show that even trace amounts of water in the form of atmospheric moisture can shift the mechanochemical pathway, revealing that nominally ‘dry’ milling in some cases tends to be an inadvertent LAG. Once the cocrystal has nucleated, it dominates all subsequent crystallisation attempts, rendering the salt a disappearing solid form. Interestingly, they also observed that in a VALAG experiment the amount of the added liquid could further influence the nature and stability of the resulting solid forms: ‘dry’ milling conditions (facilitated by the moisture form air) favoured the metastable salt, intermediate amounts (η ≈ 0.08 μL mg−1) promoted the formation of the thermodynamically stable cocrystal, while great amounts of water (η ≈ 1.43 μL mg−1) occasionally led to the formation of physical mixtures, or a reversion to metastable phases (Fig. 7).
 | ||
| Fig. 7 Schematic representation of the mechanochemical reaction outcome between β -alanine and DL-tartaric acid under NG and VALAG conditions. Adapted from ref. 82. | ||
A more recent study by Daolio et al.38 provided additional insight into this phenomenon by investigating the mechanochemical reactivity between quinoline and pamoic acid. Under NG conditions, the authors obtained a neutral cocrystal, whereas LAG in the presence of tetrahydrofuran or dimethyl sulfoxide led to the formation of two distinct solvated salts. This transformation not only altered the stoichiometry of the final products but also caused a profound change in the interaction networks, shifting from neutral hydrogen bonding to ionic interactions being stabilised by solvent coordination. The authors proposed that the coordinating ability of the polar aprotic solvents, combined with the enhanced mobility, and the solvation environment provided during LAG, favoured proton transfer and stabilised the resultant salt forms.
Together, these examples underscore how subtle changes in the reaction environment – particularly the presence of liquid additives, as well as their nature and quantity – can trigger a proton transfer that ultimately leads to salt formation.
 | ||
| Fig. 8 Chiral reorganisation mediated through liquids in mechanochemistry. Adapted from ref. 83. | ||
This process highlights the dynamic nature of solid-state interactions under mechanochemical conditions, where the presence of a small amount of liquid enables molecular reorganisation without requiring full solubilisation. The liquid additive thus acts as a molecular mobility enhancer, facilitating the reassembly of components into a new crystalline architecture with altered stereochemical characteristics. The authors attributed this transformation to the increased local diffusion and interfacial rearrangement enabled by the liquid, which allowed for equilibration between homochiral and heterochiral assemblies. This example illustrates how LAG can be used to manipulate chiral recognition and assembly processes in the solid-state.
:1 monosolvate and a 2:3 sesquisolvate (Fig. 9).59 The use of lower liquid loadings (η ≈ 0.4 μL mg−1) favoured the rapid formation of a monosolvate, whereas higher values (η > 0.4 μL mg−1) led to a slower transformation into the thermodynamically more stable sesquisolvate. This transformation did not occur directly but proceeded through a stepwise mechanism: the monosolvate formed first and, under conditions of excess liquid, the monosolvate gradually transformed into sesquisolvate. To better understand such behaviour, the authors conducted ex situ kinetic experiments, interrupting the milling process at different time intervals and applying quantitative powder X-ray diffraction and Rietveld refinement (Fig. 9). These analyses revealed the sequential nature of the transformation and provided a direct link between the observed crystallisation kinetics and the physical properties of the solvates. Specifically, attachment energy calculations showed that the cleavage planes of the monosolvate are significantly weaker – almost four times less cohesive – than those of the more stable sesquisolvate. This suggests that crystal breakage and surface generation, which are necessary for solvent contact and molecular rearrangement, act as the rate-limiting steps in the transformation. The stronger interfacial cohesion in the sesquisolvate thus explains its slower formation kinetics. This example underscores the importance of finely tuning the amount of liquid additive, and not just its identity, to steer the crystallisation pathway.
 | ||
| Fig. 9 Molecular structures of theophylline and 2-pyrrolidone, the hydrogen bonding of the two theophylline-2-pyrrolidone solvates, and ex situ experiments performed using different stoichiometric ratios between theophylline and 2-pyrrolidone. Adapted from ref. 59. | ||
3.4 Dual-role liquids: simultaneous solvate-forming agents and crystallisation promoters
In some mechanochemical reactions, the added liquid can simultaneously act both as a structural component and as a reaction promoter. A recent example is provided by the mechanochemical synthesis of a cocrystal solvate of praziquantel, niclosamide and acetic acid, where the latter was added in excess to promote the reaction and improve crystallinity.61 The role of acetic acid was found to be highly dependent on its quantity. Specifically, when milling was performed using a stoichiometric amount of acetic acid necessary for the formation of a 1:1:1 cocrystal solvate, the reaction did not yield a pure ternary phase. Instead, a physical mixture of the ternary cocrystal solvate and the known binary and anhydrous cocrystal was consistently observed.25 When an excess of acetic acid was added, the mechanochemical reaction moved toward the formation of the ternary cocrystal as a pure phase (Fig. 10). Such example emphasises the need of carefully considering the quantity and properties of the liquid additive, as it can have multiple roles in a reaction. Such multi-role behaviour can offer strategies to overcoming synthetic limitations or accessing difficult-to-obtain multicomponent forms with desirable crystallinity and stability.
 | ||
| Fig. 10 Some experimental results showing the use of acetic acid as liquid additive in stoichiometric amount (η = 0.13 μL mg−1) and in excess (η = 0.40 μL mg−1) for the synthesis of the ternary cocrystal as a pure phase. Adapted from ref. 61, published under a CC-BY license. | ||
4. Multicomponent liquid additives: a new dimension
While much of the LAG literature focuses on pure liquid additives, recent studies began to explore the intricate field of mixtures of liquid additives, where two (or more) liquids interact to influence solid-state reactions in unexpected ways. This emerging version of LAG can provide further possibilities for directing product formation during LAG. For example, liquid components in the mixture can also compete, producing outcomes that do not simply sum their individual effects.One of the earliest demonstrations of this concept was reported by Friščić et al., who investigated the hydration behaviour of magnesium naproxen using LAG with mixtures of water and ethanol.87 In that study, the authors observed that the modulation of the water activity through water–ethanol mixtures enabled the rational selection of distinct hydrated phases. Specifically, the authors observed the formation of a monohydrate coordination polymer, a discrete tetrahydrate complex, and a highly hydrated phase, depending on the composition of the solvent mixture. This work established a critical precedent: co-solvent composition can serve as an important variable in mechanochemical synthesis, enabling selective crystallisation outcomes in pharmaceutical systems.
In a more recent study involving theophylline,36 LAG experiments with water yielded the well-known monohydrate form of this molecule, while LAG with 2-pyrrolidone produced a monosolvate or sesquisolvate (depending on the amount of 2-pyrrolidone). Interestingly, during LAG experiments with water:2-pyrrolidone mixtures (tested in various molar and volumetric ratios), neither water nor 2-pyrrolidone fully incorporated into the solid unless used in significant excess (Fig. 11A). Interconversion experiments revealed that 2-pyrrolidone solvate preferentially formed compared to the monohydrate form, demonstrating a clear competitive mechanism (Fig. 11B). Several mechanism were proposed to rationalise the outcome of the described reactions: (i) different liquids can adsorb onto different solid surfaces, favouring one interaction over another and controlling the reaction outcome; (ii) co-solvents may compete for solvation of reactants or intermediates; the one with higher affinity will possibly dominate, blocking alternate pathways; (iii) interactions between co-solvents can modify the microenvironment (e.g., viscosity, wetting), either through synergistic or antagonistic effects, impacting nucleation and growth dynamics.
 | ||
| Fig. 11 A) Solid phase recovered at the end of the milling and slurry experiments using individual solvents or H2O:2-pyr mixtures, based on Rietveld refinement and quantitative phase analysis of the PXRD patterns. Column “0.81:0.19” corresponds to isovolumetric H2O:2-pyr mixtures. Each solid phase corresponds to a different colour in the pie chart: yellow represents the amount of anhydrous theophylline, blue, green and dark green are the amounts of the theophylline monohydrate (theo:H2O-mh), theophylline monosolvate (theo:2-pyr-ms) and theophylline sesquisolvate (theo:2-pyr-ss), respectively. B) Solid phase composition (weight fractions) recovered at the end of the interconversion studies starting from preformed theo:H2O-mh, theo:2-pyr-ms, theo:2-pyr-ss phases in the presence of individual liquids or H2O:2-pyr mixtures. Each solid phase corresponds to a different colour in the pie chart: yellow represents the amount of anhydrous theophylline, blue, green and dark green colours indicate the amounts of the theo:H2O-mh, theo:2-pyr-ms and theo:2-pyr-ss, respectively. C) Solid phases recovered from each molar fraction of H2O:AA/2-pyr/EtOH/EA/HXN mixtures. Green circles stand for solid phase present and red circles for solid phase absent. D) Solid phases recovered from the interconversion milling experiments. Adapted from ref. 36 and 37, published under a CC-BY license. | ||
Building on this, another recent study,37 explored the formation of praziquantel hemihydrate through LAG using binary liquid systems composed of water and one of five co-solvents with varying water miscibility: acetic acid (AA), 2-pyrrolidone (2-pyr), ethanol (EtOH), ethyl acetate (EA), and hexane (HXN). It was noticed that: (i) water-miscible solvents (AA and 2-pyr) significantly reduced or completely inhibited hemihydrate formation compared to pure water; (ii) hexane, immiscible with water, allowed hemihydrate formation (Fig. 11C). Furthermore, in co-solvent systems, AA and 2-pyr preferentially replaced water in the crystal structure – underpinning the competitive inclusion seen in crystalline theophylline.
The recognition of synergistic or competitive behaviour among liquids unlocks new possibilities in mechanochemical syntheses:
• Competitive systems can allow selective access to multicomponent forms, including mixed solvates or metastable polymorphs.
• By strategically combining liquids with different physicochemical properties – such as polarity, hydrogen-bonding capacity, or miscibility – one can fine-tune the local solvent environment to modulate crystallisation pathways. For instance, adjusting the water activity by introducing water-miscible co-solvents can be used to selectively stabilise or destabilise hydrated phases.87
• Rational design of competitive liquid environments can not only improve reproducibility and control over reaction outcomes but also enable the tuning of reaction kinetics. This can be achieved by integrating systematic experimentation through LAG with mixtures of liquids on model drug systems, and computational studies, such as crystal structure prediction, attachment energy modelling, and emerging machine learning frameworks.42–45 In mechanochemical systems, nucleation and growth processes remain inherently non-linear and can proceed through both primary and secondary pathways, like traditional crystallisation phenomena. However, under LAG conditions, these processes are continuously perturbed by mechanical impacts and shear forces that can dynamically modify local supersaturation, defect generation, and surface energy distribution. As a result, the mechanical input does not replace nucleation and growth but acts as an additional variable that couples with these non-linear pathways to influence reaction kinetics and phase evolution. Integrating systematic LAG experimentation with computational and modelling approaches can therefore provide deeper insights into how these coupled effects can be steered toward predictive control in mechanochemical synthesis.
5. Is it currently possible a rational design of the liquid additive?
As mentioned above, the exact mechanisms by which the liquid additives impact LAG reactions are so far not understood. It is, however, generally accepted that the added liquid primarily facilitates molecular diffusion and enhances contact between solid particles, thus promoting reactivity.29 To better rationalise and predict such effects, several physicochemical parameters have been qualitatively correlated with LAG outcomes. These features can help rationalising differences in reaction kinetics and product selectivity:• Polarity and polarizability: liquids with different dielectric constants or polarizabilities can promote molecular diffusion and reactant wetting to different extents. These factors can determine the direction of a mechanochemical reaction or the stabilisation of certain phases (e.g., polymorphs).88,89
• Viscosity and surface tension: these two parameters can possibly affect the distribution efficiency of a specific liquid over solid particles during milling, and the efficiency of the energy transfer among the solid particles. Viscosity plays a direct role in molecular mobility within the liquid layer. The Einstein–Stokes relationship is often invoked to qualitatively describe how higher viscosity can reduce diffusivity and molecular rearrangement.90 However, it must be noted that this equation, derived for spherical Brownian particles in a continuous viscous fluid, does not strictly apply to the heterogeneous solid–liquid systems typical of LAG. Another reference can be the Stokes drag framework, which similarly relates mobility to viscous resistance but allows for discussion in terms of mechanical energy dissipation and frictional forces at particle interfaces. In mechanochemical reactions, mechanical impacts and shear forces continuously disturb local equilibria, transiently enhancing molecular mobility even within highly viscous or confined environments. In LAG, these non-equilibrium effects explain why liquids with different viscosities can lead to markedly different reaction kinetics and product selectivity despite deviations from ideal diffusive behaviour.91,92
• Molecular geometry and conformational flexibility: the spatial arrangement and rigidity of the liquid molecules can possibly affect the outcome of a mechanochemical reaction. It is indeed possible that rigid molecules such as acetonitrile would interact differently at particle surfaces compared to more flexible, multidentate species such as ethylene glycol. This difference in molecular geometry and flexibility can possibly explain the distinct polymorph selectivity observed for these two liquids in the study of Hasa et al.35
• Volatility and evaporation dynamics: highly volatile liquids can partially or totally evaporate during milling, altering η over time and introducing variability in reaction outcomes. This can affect not only the kinetics but also the crystallinity and reproducibility of the final product.93,94
• Hydrogen-bond donor and acceptor abilities: quantified through parameters such as the Kamlet–Taft α (donor) and β (acceptor) scales.95,96 These properties can be very useful for a better understanding of possible interactions of a specific liquid with functional groups of molecules present on the surface of reactants. For example, during cocrystal formation, the ability of a solvent to engage in hydrogen bonding has been linked to polymorph selectivity and templating behaviour.43,49,55,56
In addition to these descriptors, it must be considered that during mechanochemical processes such as ball-milling, the mechanical energy is delivered through repetitive collisions and shear forces. The presence of a liquid introduces a medium for mechanical energy dissipation. According to the fluctuation–dissipation principle, the microscopic fluctuations that occur at thermal equilibrium are conceptually related to the response of a system to an external mechanical perturbation. In this context, the liquid additive can act as a medium that not only facilitates molecular motion but also mediates how the mechanical energy is distributed within the reacting mixture.97 Furthermore, the way a liquid can wet a solid surface depends on parameters such as surface energy, interfacial tension, and contact angle; a lower contact angle reflects better wetting and increased surface coverage, which in turn promotes more effective molecular interactions between reactants. These interfacial phenomena can possibly affect the way crystal nucleation and growth of a specific system proceed. It must be mentioned, however, that in a typical LAG reaction the confined liquid layers are just a few molecules thick, thus the behaviour of the solvent can significantly deviate from the bulk properties.98,99
Despite significant progress, the lack of universal predictive models – that is, comprehensive theoretical or computational frameworks capable of quantitatively relating liquid properties, reactant surface chemistry, and milling parameters to reaction outcomes – still constrains rational LAG design. Liquid effects are inherently system-specific and depend not only on the liquid properties but also on the reactant surface chemistry and the milling conditions (e.g., frequency, jar material etc.),100–103 as well as on the balance between kinetic and thermodynamic control regimes.30,32,44 A deeper integration of physical models from tribology, materials science, and molecular simulations can help uncover the underlying mechanisms and enable more predictive control over mechanochemical reactions.104
6. The (long) road ahead
Over the past three decades, the importance of liquid additives in mechanochemistry has significantly increased. What was once dismissed as non grata in mechanochemical reactions has ultimately established itself as a key component, a shift made possible by studies showing that mechanochemical transformations, reaction kinetics and product selectivity can be finely tuned through the choice of liquid additives. Although LAG is transforming mechanochemistry, we are still far from fully grasping how and why it works so well. Several aspects require exploration to advance our understanding of the fascinating processes involved. For example, although at a first glance the scenario within a milling jar can appear unsophisticated, there are numerous scenarios that require consideration: is the liquid additive remaining “liquid” or it is affecting the milling atmosphere (or both)? What are the consequences of creating a liquid layer around the particles of the reactants? Does the thickness of such layer changes within time considering that the particle size and size distribution of the solid reactants change as the mechanochemical process proceeds? Why are certain liquids preventing specific reactions, while others are dismantling preformed products?41 A more multidisciplinary approach, involving computational and experimental work, must become the norm in mechanochemical research to offer answers to these questions. Such approaches have recently addressed how the molecule of the additive affects reactions at the surface of the solid reactants in LAG. For example, Ferguson and co-workers have pioneered the use of molecular dynamics to probe the early stages of mechanochemical reactions.42,43 Their work has demonstrated how solvent molecules engage in transient, directional interactions with the surfaces of solid reactants, guiding the assembly of supramolecular structures and intermediates. These simulations have not only predicted what liquids can drive the mechanochemical reaction towards specific products but have also helped rationalising why certain solvates or polymorphs emerge under defined LAG conditions. In a more recent study,43 Ferguson and co-workers showed how different solvents with different polarity and hydrogen-bonding ability exhibit distinct interaction patterns with solid surfaces. The simulations captured transient clustering of liquid molecules at reactive sites, which in turn correlated with experimentally observed polymorph selection and reaction rates, offering one of the first atomistically grounded insights into solvent effects in LAG (Fig. 12). | ||
| Fig. 12 (top) Schematic representation of the simulation strategy employed in the work. (bottom) Snapshots of molecular dynamics depicting the local environments of the liquid additive molecules in a LAG reaction. Adapted from ref. 43, published under a CC-BY license. | ||
Yet, despite these promising advances, significant challenges persist. High computational time required renders extremely difficult understanding the effect of liquid additives combined with other experimental parameters. An interesting example is reported from Germann and co-authors103 who reported that the mechanochemical formation of a nicotinamide–adipic acid cocrystal and the interconversion of polymorphs is strongly affected, and can be controlled, by the choice of milling assembly. Specifically, the numbers and the size of the milling media and the material of the jar (steel or polymeric) can be used to direct polymorphism of mechanochemical cocrystallisation, enabling the selective synthesis, and even reversible and repeatable interconversion of cocrystal polymorphs. Importantly, by using in situ diffraction techniques Germann and co-authors observed that the modification of energy input in the mechanochemical system permitted the selective synthesis of either the more stable room temperature cocrystal polymorph or the new metastable high-temperature form. Indeed, in situ techniques – ranging from X-ray diffraction to Raman spectroscopy – allow researchers to peek inside the mechanochemical reaction vessel, thus allowing us to monitor structural and chemical changes as a dynamic process, rather than forcing us to piece together an understanding using several ex situ snapshots of the process. However, the difficulties on the experimental setup and the challenges related to the limits of specific analytical techniques render in situ real-time experiments still sporadic. Indeed, future efforts need to go beyond the modified mill at the beamline and reduce possible analytic limitations through combining different characterisation techniques. In this context, some very interesting efforts have been reported, both for the in situ monitoring mechanochemical reactions without using synchrotron radiation and the combination of different analytical techniques.105,106 Efforts could be spent on the expansion of the analytical tools. For example, tomographic techniques can offer a powerful means to visualise the internal structure of mechanochemical systems under realistic conditions. By providing three-dimensional information on particle size, distribution, and mixing efficiency, tomography can possibly reveal how the addition of liquids influences the evolution of microstructures during a mechanochemical reaction. These spatially resolved insights help bridge the gap between macroscopic reaction behaviour and microscopic material transformations, possibly offering a more holistic understanding of LAG processes. Another interesting integration would be on the use of isotopically or chemically labelled liquids for tracing their fate during LAG. Labels allow researchers to follow how the liquid distributes across particle surfaces, participates in intermediate formation, or becomes incorporated into reaction products. Such measurements reveal whether the liquid acts merely as a dispersant or plays a direct chemical role in bond formation and transformation.107 By illuminating these subtle pathways, labelled liquids can provide some of the most direct evidence available for deciphering some of the enigmatic mechanisms that govern LAG processes. Complementing techniques such as real-time and Raman spectroscopy or NMR provide direct access with the use isotopically or chemically labelled liquids can possibly allow researchers to trace the precise course of the additive within the mechanochemical setting.
The complexity of a multicomponent environment – where liquid–solid affinity, co-solvent competition and kinetic versus thermodynamic control continues to limit our ability to reliably predict the outcome of a LAG process. While descriptors such as polarity, dielectric constant, or hydrogen-bonding parameters have proven useful, they fall short of fully capturing the nuanced roles that liquids can play. Moreover, liquid mixtures remain a largely underexplored and unsystematised aspect of LAG. Only a handful of studies have investigated how co-solvent systems modulate reaction outcomes, and comprehensive frameworks to interpret such behaviour are still lacking. An especially underexplored aspect is also the use of chiral liquid additives. Although chirality plays a critical role in crystal engineering and pharmaceutical development, the influence of chiral solvents on mechanochemical outcomes has not been systematically studied. Understanding how enantiopure or racemic liquids affect reaction kinetics, stereoselectivity, or phase formation could open entirely new avenues for asymmetric or enantiospecific mechanochemistry.
Looking forward, several promising directions are emerging. The integration of machine learning and data-driven methodologies holds great promise for uncovering relationships between liquid properties and mechanochemical reactions, enabling more rational selection of optimal solvent systems – be they single components, well-defined co-solvent mixtures, or dynamically variable compositions such as those used in VALAG protocols. Another approach that may become increasingly valuable is resonant acoustic mixing (RAM), which eliminates the need for milling media. In particular, liquid-assisted RAM could provide new opportunities to improve selectivity and efficiency in mechanochemical transformations, complementing conventional LAG strategies.108–115 The exploration of increasingly complex liquid environments – including ionic liquids,96,116 deep eutectic solvents,117 and porous liquids118 – offers exciting opportunities to create microenvironments that stabilise specific intermediates or promote unique reactivity pathways inaccessible under more conventional experimental conditions. Such “designer liquids” could become key tools for programming selectivity, improving efficiency, or enabling transformations under milder conditions. Furthermore, the development of continuous-flow platforms for liquid-assisted mechanochemistry can bridge the gap between laboratory discovery and industrial scalability.119,120 Unlike traditional batch milling, continuous-flow systems allow solid reactants and liquid additives to be fed steadily into a reactor, where shear and grinding forces are applied in a controlled manner. This configuration provides unique advantages: (i) precise and reproducible dosing of liquids, (ii) tuneable residence time and shear rate, (iii) efficient mixing under constant operating conditions, and (iv) facile coupling with several real-time analytical tools such as Raman or PXRD for in situ monitoring. As a result, continuous-flow mechanochemistry enables scale-up while simultaneously improving reproducibility, reducing solvent waste, and allowing direct translation of optimised laboratory conditions into industrially relevant processes.121–123
Although the road ahead is still windy and steep, we envision and anticipate a future in which the selection of liquid additive to be used in LAG is not driven by empirically derived intuition, but through predictive models based in experimental validation and computational simulations. Such concepts are already well-established in solution-based synthesis and there is no reason to believe that their systematic application cannot be extended to LAG. The achievement of such knowledge will possibly unlock unprecedented levels of control and functionality in solid-state synthesis.
Conflicts of interest
There are no conflicts to declare.Data availability
No new research results, software or code have been included and no new data were generated or analysed as part of this highlight article. Some of the figures are adapted images from specific research papers, for which the respective reference manuscripts have been cited in each figure caption.Acknowledgements
The authors are grateful to Dejan-Krešimir Bučar (Department of Chemistry, UCL, UK) for insightful discussions.References
- Research Journal of Chemical Sciences: Mechanochemistry: A green chemistry for green technology - ISCA, https://www.isca.me/rjcs/Archives/v14/i1/8.ISCA-RJCS-2023-022.php, (accessed 7 July 2025).
- S. Arfelis, A. I. Martín-Perales, R. Nguyen, A. Pérez, I. Cherubin, C. Len, I. Malpartida, A. Bala and P. Fullana-i-Palmer, Heliyon, 2024, 10, e34655 CrossRef.
- V. Štrukil, L. Fábián, D. G. Reid, M. J. Duer, G. J. Jackson, M. Eckert-Maksić and T. Friščić, Chem. Commun., 2010, 46, 9191 RSC.
- K. J. Ardila-Fierro and J. G. Hernández, ChemSusChem, 2021, 14, 2145–2162 CrossRef.
- V. André, M. T. Duarte, C. S. B. Gomes and M. C. Sarraguça, Molecules, 2021, 27, 241 CrossRef.
- M. Solares-Briones, G. Coyote-Dotor, J. C. Páez-Franco, M. R. Zermeño-Ortega, C. M. de la O Contreras, D. Canseco-González, A. Avila-Sorrosa, D. Morales-Morales and J. M. Germán-Acacio, Pharmaceutics, 2021, 13, 790 CrossRef PubMed.
- S. L. James, C. J. Adams, C. Bolm, D. Braga, P. Collier, T. Friščić, F. Grepioni, K. D. M. Harris, G. Hyett, W. Jones, A. Krebs, J. Mack, L. Maini, A. G. Orpen, I. P. Parkin, W. C. Shearouse, J. W. Steed and D. C. Waddell, Chem. Soc. Rev., 2012, 41, 413–447 RSC.
- S. Pagola, Crystals, 2023, 13, 124 CrossRef.
- L. Dong, L. Li, H. Chen, Y. Cao and H. Lei, Adv. Sci., 2025, 12(24), e2403949 CrossRef.
- A. Mirocki, M. Lopresti, L. Palin, E. Conterosito, E. Sikorska, A. Sikorski and M. Milanesio, Sci. Rep., 2024, 14, 1834 CrossRef.
- J. Lombard, H. Laker, F. Prins, H. Wahl, T. le Roex and D. A. Haynes, CrystEngComm, 2021, 23, 7380–7384 RSC.
- M. E. Skala, S. M. Zeitler and M. R. Golder, Chem. Sci., 2024, 15, 10900–10907 RSC.
- R. Janot and D. Guerard, Prog. Mater. Sci., 2005, 50, 1–92 CrossRef.
- P. J. Beldon, L. Fábián, R. S. Stein, A. Thirumurugan, A. K. Cheetham and T. Friščić, Angew. Chem., Int. Ed., 2010, 49, 9640–9643 CrossRef.
- B. Karadeniz, D. Žilić, I. Huskić, L. S. Germann, A. M. Fidelli, S. Muratović, I. Lončarić, M. Etter, R. E. Dinnebier, D. Barišić, N. Cindro, T. Islamoglu, O. K. Farha, T. Friščić and K. Užarević, J. Am. Chem. Soc., 2019, 141, 19214–19220 CrossRef PubMed.
- T. Friščić, C. Mottillo and H. M. Titi, Angew. Chem., Int. Ed., 2020, 59, 1018–1029 CrossRef.
- C. Bolm and J. G. Hernández, Angew. Chem., Int. Ed., 2019, 58, 3285–3299 CrossRef CAS.
- P. Baláž, M. Achimovičová, M. Baláž, P. Billik, Z. Cherkezova-Zheleva, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and K. Wieczorek-Ciurowa, Chem. Soc. Rev., 2013, 42, 7571 RSC.
- D. Tan, L. Loots and T. Friščić, Chem. Commun., 2016, 52, 7760–7781 RSC.
- A. Delori, T. Friščić and W. Jones, CrystEngComm, 2012, 14, 2350 RSC.
- W. Jones and M. D. Eddleston, Faraday Discuss., 2014, 170, 9–34 RSC.
- J.-L. Do and T. Friščić, ACS Cent. Sci., 2017, 3, 13–19 CrossRef CAS.
- L. Takacs, Chem. Soc. Rev., 2013, 42, 7649 RSC.
- A. V. Trask, D. A. Haynes, W. D. S. Motherwell and W. Jones, Chem. Commun., 2006, 51–53 RSC.
- I. D'Abbrunzo, E. Bianco, L. Gigli, N. Demitri, R. Birolo, M. R. Chierotti, I. Škoríc, J. Keiser, C. Häberli, D. Voinovich, D. Hasa and B. Perissutti, Int. J. Pharm., 2023, 644, 123315 CrossRef PubMed.
- G. A. Bowmaker, Chem. Commun., 2013, 49, 334–348 RSC.
- K. Trzeciak, M. K. Dudek and M. J. Potrzebowski, Chem. – Eur. J., 2024, 30(71), e202402683 CrossRef CAS.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- T. Friščić and W. Jones, Cryst. Growth Des., 2009, 9, 1621–1637 CrossRef.
- D. Hasa, G. Schneider Rauber, D. Voinovich and W. Jones, Angew. Chem., Int. Ed., 2015, 54, 7371–7375 CrossRef CAS.
- I. A. Tumanov, A. A. L. Michalchuk, A. A. Politov, E. V. Boldyreva and V. V. Boldyrev, CrystEngComm, 2017, 19, 2830–2835 RSC.
- D. Hasa and W. Jones, Adv. Drug Delivery Rev., 2017, 117, 147–161 CrossRef CAS.
- D. Douroumis, S. A. Ross and A. Nokhodchi, Adv. Drug Delivery Rev., 2017, 117, 178–195 CrossRef CAS.
- N. Shan, F. Toda and W. Jones, Chem. Commun., 2002, 2372–2373 RSC.
- D. Hasa, E. Miniussi and W. Jones, Cryst. Growth Des., 2016, 16, 4582–4588 CrossRef CAS.
- I. D'Abbrunzo, M. Spadaro, M. Arhangelskis, G. Zingone, D. Hasa and B. Perissutti, Cryst. Growth Des., 2023, 23, 8094–8102 CrossRef.
- I. D'Abbrunzo, D. Voinovich and B. Perissutti, Crystals, 2024, 14, 374 CrossRef.
- A. Daolio, M. Prencipe, T. Abodunrin, P. Pelagatti, P. P. Mazzeo and A. Bacchi, Chem. – Eur. J., 2025, 31(33), e202500956 CrossRef CAS PubMed.
- T. Friščić, S. L. Childs, S. A. A. Rizvi and W. Jones, CrystEngComm, 2009, 11, 418–426 RSC.
- L. Gonnet, T. H. Borchers, C. B. Lennox, J. Vainauskas, Y. Teoh, H. M. Titi, C. J. Barrett, S. G. Koenig, K. Nagapudi and T. Friščić, Faraday Discuss., 2023, 241, 128–149 RSC.
- M. Arhangelskis, D.-K. Bučar, S. Bordignon, M. R. Chierotti, S. A. Stratford, D. Voinovich, W. Jones and D. Hasa, Chem. Sci., 2021, 12, 3264–3269 RSC.
- M. Ferguson, M. S. Moyano, G. A. Tribello, D. E. Crawford, E. M. Bringa, S. L. James, J. Kohanoff and M. G. Del Pópolo, Chem. Sci., 2019, 10, 2924–2929 RSC.
- M. Ferguson, Y. Xie, A. Moores and T. Friščić, ChemRxiv, 2025, preprint, DOI:10.26434/chemrxiv-2025-mtvdw.
- L. Gui, A. Armstrong, A. Galindo, F. B. Sayyed, S. P. Kolis and C. S. Adjiman, Mol. Syst. Des. Eng., 2024, 9, 1254–1274 RSC.
- R. Birolo, F. Bravetti, E. Alladio, E. Priola, G. Bianchini, R. Novelli, A. Aramini, R. Gobetto and M. R. Chierotti, Cryst. Growth Des., 2023, 23, 7898–7911 CrossRef CAS.
- L. Takacs, JOM, 2000, 52, 12–13 CrossRef CAS.
- S. Nakamatsu, S. Toyota, W. Jones and F. Toda, Chem. Commun., 2005, 3808 RSC.
- E. Boldyreva, Chem. Soc. Rev., 2013, 42, 7719 RSC.
- T. Friščić and W. Jones, Development of liquid-assisted grinding (LAG) for the synthesis of hydrogen-bonded and coordination frameworks, 31-40. Paper presented at 6th International Conference on Mechanochemistry and Mechanical Alloying, INCOME 2008, Jamshedpur, India.
- J. L. Howard, Y. Sagatov, L. Repusseau, C. Schotten and D. L. Browne, Green Chem., 2017, 19, 2798–2802 RSC.
- F. Fischer, A. Heidrich, S. Greiser, S. Benemann, K. Rademann and F. Emmerling, Cryst. Growth Des., 2016, 16, 1701–1707 CrossRef.
- W. Yuan, J. O'Connor and S. L. James, CrystEngComm, 2010, 12, 3515 RSC.
- L. Konnert, M. Dimassi, L. Gonnet, F. Lamaty, J. Martinez and E. Colacino, RSC Adv., 2016, 6, 36978–36986 RSC.
- J.-L. Do, C. Mottillo, D. Tan, V. Štrukil and T. Friščić, J. Am. Chem. Soc., 2015, 137, 2476–2479 CrossRef PubMed.
- A. M. Belenguer, G. I. Lampronti, A. J. Cruz-Cabeza, C. A. Hunter and J. K. M. Sanders, Chem. Sci., 2016, 7, 6617–6627 RSC.
- A. M. Belenguer, G. I. Lampronti, N. De Mitri, M. Driver, C. A. Hunter and J. K. M. Sanders, J. Am. Chem. Soc., 2018, 140, 17051–17059 CrossRef PubMed.
- C.-H. Gu, H. Li, R. B. Gandhi and K. Raghavan, Int. J. Pharm., 2004, 283, 117–125 CrossRef PubMed.
- K. K. Sarmah, T. Rajbongshi, A. Bhuyan and R. Thakuria, Chem. Commun., 2019, 55, 10900–10903 RSC.
- D. Hasa, M. Pastore, M. Arhangelskis, B. Gabriele, A. J. Cruz-Cabeza, G. S. Rauber, A. D. Bond and W. Jones, CrystEngComm, 2019, 21, 2097–2104 RSC.
- J. Lombard, T. le Roex and D. A. Haynes, Cryst. Growth Des., 2020, 20, 7384–7391 CrossRef.
- I. D'Abbrunzo, L. Gigli, N. Demitri, C. Sabena, C. Nervi, M. R. Chierotti, S. Bertoni, I. Škorić, C. Häberli, J. Keiser, D. Hasa and B. Perissutti, J. Drug Delivery Sci. Technol., 2025, 109, 106974 CrossRef.
- Z. Rahman, C. Agarabi, A. S. Zidan, S. R. Khan and M. A. Khan, AAPS PharmSciTech, 2011, 12, 693–704 CrossRef.
- A. Mukherjee, R. D. Rogers and A. S. Myerson, CrystEngComm, 2018, 20, 3817–3821 RSC.
- S. Karki, T. Friščić, W. Jones and W. D. S. Motherwell, Mol. Pharmaceutics, 2007, 4, 347–354 CrossRef.
- M. Bashimam and H. El-Zein, Heliyon, 2022, 8, e11872 CrossRef PubMed.
- I. A. Tumanov, A. A. L. Michalchuk, A. A. Politov, E. V. Boldyreva and V. V. Boldyrev, Dokl. Chem., 2017, 472, 17–19 CrossRef.
- D. Zanolla, L. Gigli, D. Hasa, M. R. Chierotti, M. Arhangelskis, N. Demitri, W. Jones, D. Voinovich and B. Perissutti, Pharmaceutics, 2021, 13, 1606 CrossRef PubMed.
- R. Thakuria, M. Arhangelskis, M. D. Eddleston, E. H. H. Chow, K. K. Sarmah, B. J. Aldous, J. F. Krzyzaniak and W. Jones, Org. Process Res. Dev., 2019, 23, 845–851 CrossRef.
- L. M. Martínez, J. Cruz-Angeles, M. Vázquez-Dávila, E. Martínez, P. Cabada, C. Navarrete-Bernal and F. Cortez, Pharmaceutics, 2022, 14, 2003 CrossRef PubMed.
- L. Qi, C. Li, X. Cheng, H. Hao and C. Xie, Cryst. Growth Des., 2024, 24, 6196–6203 CrossRef.
- A. Chatziadi, E. Skořepová, J. Rohlíček, M. Dušek, L. Ridvan and M. Šoóš, Cryst. Growth Des., 2020, 20, 139–147 CrossRef.
- I. Brekalo, K. Lisac, J. R. Ramirez, P. Pongrac, A. Puškarić, S. Valić, Y. Xu, M. Ferguson, J. M. Marrett, M. Arhangelskis, T. Friščić and K. T. Holman, J. Am. Chem. Soc., 2025, 147, 27413–27430 CrossRef.
- N. Bouvart, R.-M. Palix, S. G. Arkhipov, I. A. Tumanov, A. A. L. Michalchuk and E. V. Boldyreva, CrystEngComm, 2018, 20, 1797–1803 RSC.
- A. V. Trask, N. Shan, W. D. S. Motherwell, W. Jones, S. Feng, R. B. H. Tan and K. J. Carpenter, Chem. Commun., 2005, 880 RSC.
- H. Kulla, C. Becker, A. A. L. Michalchuk, K. Linberg, B. Paulus and F. Emmerling, Cryst. Growth Des., 2019, 19, 7271–7279 CrossRef.
- N. Tumanova, N. Tumanov, F. Fischer, F. Morelle, V. Ban, K. Robeyns, Y. Filinchuk, J. Wouters, F. Emmerling and T. Leyssens, CrystEngComm, 2018, 20, 7308–7321 RSC.
- L. Ma, Q. Zheng, D. K. Unruh and K. M. Hutchins, Chem. Commun., 2023, 59, 7779–7782 RSC.
- T. Stolar, S. Lukin, M. Tireli, I. Sović, B. Karadeniz, I. Kereković, G. Matijašić, M. Gretić, Z. Katančić, I. Dejanović, M. di Michiel, I. Halasz and K. Užarević, ACS Sustainable Chem. Eng., 2019, 7, 7102–7110 CrossRef.
- L. S. Germann, S. T. Emmerling, M. Wilke, R. E. Dinnebier, M. Moneghini and D. Hasa, Chem. Commun., 2020, 56, 8743–8746 RSC.
- A. A. L. Michalchuk and F. Emmerling, Angew. Chem., Int. Ed., 2022, 61(21), e202117270 CrossRef PubMed.
- A. M. Belenguer, G. I. Lampronti, A. A. L. Michalchuk, F. Emmerling and J. K. M. Sanders, CrystEngComm, 2022, 24, 4256–4261 RSC.
- E. A. Losev and E. V. Boldyreva, CrystEngComm, 2018, 20, 2299–2305 RSC.
- T. Friščić, L. Fábián, J. C. Burley, W. Jones and W. D. S. Motherwell, Chem. Commun., 2006, 5009–5011 RSC.
- Y. Zhoujin, M. Zhang, S. Parkin, T. Li, F. Yu and S. Long, RSC Adv., 2021, 11, 24836 RSC.
- A. B
![[e with combining overline]](https://www.rsc.org/images/entities/char_0065_0305.gif) rziņš, E. Skarbulis, T. Rekis and A. Actiņš, Cryst. Growth Des., 2014, 14, 2654–2664 CrossRef.
rziņš, E. Skarbulis, T. Rekis and A. Actiņš, Cryst. Growth Des., 2014, 14, 2654–2664 CrossRef. - T. H. H. Sohi, F. Maass, C. Czekelius and V. Vasylyeva, Crystals, 2023, 13, 1512 CrossRef.
- T. Friščić, I. Halasz, F. C. Strobridge, R. E. Dinnebier, R. S. Stein, L. Fábián and C. Curfs, CrystEngComm, 2011, 13, 3125 RSC.
- S. Okumu, J. Chem., 2024, 3, 24–35 CrossRef.
- M. Jorge, J. R. B. Gomes and M. C. Barrera, J. Mol. Liq., 2022, 356, 119033 CrossRef CAS.
- A. Einstein, Ann. Phys., 1905, 322, 549–560 CrossRef.
- N. Chideme and P. de Vaal, JAFM, 2024, 17, 2652–2657 Search PubMed.
- Q. Yin, C. Li, L. Dong, X. Bai, Y. Zhang, M. Yang, D. Jia, R. Li and Z. Liu, Int. J. Precis. Eng. Manuf. - Green Technol., 2021, 8, 1629–1647 CrossRef.
- A. G. L. Williams, G. Karapetsas, D. Mamalis, K. Sefiane, O. K. Matar and P. Valluri, J. Fluid Mech., 2021, 907, A22 CrossRef CAS.
- W. Foudhil, P. Chen, K. Fahem, S. Harmand and S. Ben Jabrallah, Heat Mass Transfer, 2021, 57, 1773–1790 CrossRef CAS.
- T. Islam, Md. Z. Islam Sarker, A. H. Uddin, K. Bin Yunus, R. Prasad, Md. A. R. Mia and S. Ferdosh, Anal. Chem. Lett., 2020, 10, 550–561 CrossRef CAS.
- N. Weiß, C. H. Schmidt, G. Thielemann, E. Heid, C. Schröder and S. Spange, Phys. Chem. Chem. Phys., 2021, 23, 1616–1626 RSC.
- R. Kubo, Rep. Prog. Phys., 1966, 29, 255 CrossRef CAS.
- R. G. dos Santos, in Fundamentals of Surface Thermodynamics, Springer International Publishing, Cham, 2024, pp. 161–174 Search PubMed.
- J. Ma, I. Zarin and N. Miljkovic, Phys. Rev. Lett., 2022, 129, 246802 CrossRef CAS.
- L. E. Wenger and T. P. Hanusa, RSC Mechanochem., 2024, 1, 235–243 RSC.
- M. F. Rappen, L. Beissel, J. Geisler, S. T. Tietmeyer, S. Grätz and L. Borchardt, RSC Mechanochem., 2024, 1, 386–392 RSC.
- K. Linberg, F. Emmerling and A. A. L. Michalchuk, Cryst. Growth Des., 2023, 23, 19–23 CrossRef CAS.
- L. S. Germann, M. Arhangelskis, M. Etter, R. E. Dinnebier and T. Friščić, Chem. Sci., 2020, 11, 10092–10100 RSC.
- A. A. L. Michalchuk, Chem. Commun., 2024, 60, 14750–14761 RSC.
- F. Emmerling and A. A. L. Michalchuk, in Encyclopedia of Green Chemistry, 2025, pp. 437–447 Search PubMed.
- P. A. Julien, M. Arhangelskis, L. S. Germann, M. Etter, R. E. Dinnebier, A. J. Morris and T. Friščić, Chem. Sci., 2023, 14, 12121–12132 RSC.
- S. Lukin, M. Tireli, T. Stolar, D. Barišić, M. V. Blanco, M. di Michiel, K. Užarević and I. Halasz, J. Am. Chem. Soc., 2019, 141, 1212–1216 CrossRef CAS.
- D. J. am Ende, S. R. Anderson and J. S. Salan, Org. Process Res. Dev., 2014, 18, 331–341 CrossRef CAS.
- C. B. Lennox, T. H. Borchers, L. Gonnet, C. J. Barrett, S. G. Koenig, K. Nagapudi and T. Friščić, Chem. Sci., 2023, 14, 7475–7481 RSC.
- J. G. Osorio and F. J. Muzzio, Powder Technol., 2015, 278, 46–56 CrossRef CAS.
- C. J. Wright, P. J. Wilkinson, S. E. Gaulter, D. Fossey, A. O. Burn and P. P. Gill, Propellants, Explos., Pyrotech., 2021, 46, 1–15 CrossRef.
- R. Tanaka, S. Osotprasit, J. Peerapattana, K. Ashizawa, Y. Hattori and M. Otsuka, Pharmaceutics, 2021, 13, 56 CrossRef CAS PubMed.
- F. Effaty, L. Gonnet, S. G. Koenig, K. Nagapudi, X. Ottenwaelder and T. Friščić, Chem. Commun., 2023, 59, 1010–1013 RSC.
- K. Nagapudi, E. Y. Umanzor and C. Masui, Int. J. Pharm., 2017, 521, 337–345 CrossRef CAS.
- A. A. L. Michalchuk, K. S. Hope, S. R. Kennedy, M. V. Blanco, E. V. Boldyreva and C. R. Pulham, Chem. Commun., 2018, 54, 4033–4036 RSC.
- W. M. Reichert, J. D. Holbrey, K. B. Vigour, T. D. Morgan, G. A. Broker and R. D. Rogers, Chem. Commun., 2006, 4767–4779 RSC.
- P. P. Mazzeo, M. Prencipe, T. Feiler, F. Emmerling and A. Bacchi, Cryst. Growth Des., 2022, 22, 4260–4267 CrossRef CAS PubMed.
- T. D. Bennett, F.-X. Coudert, S. L. James and A. I. Cooper, Nat. Mater., 2021, 20, 1179–1187 CrossRef CAS.
- R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 RSC.
- D. E. Crawford, Beilstein J. Org. Chem., 2017, 13, 65–75 CrossRef PubMed.
- H. L. D. Hayes and C. J. Mallia, Org. Process Res. Dev., 2024, 28, 1327–1354 CrossRef.
- R. S. Atapalkar and A. A. Kulkarni, React. Chem. Eng., 2024, 9, 10–25 RSC.
- R. R. A. Bolt, J. A. Leitch, A. C. Jones, W. I. Nicholson and D. L. Browne, Chem. Soc. Rev., 2022, 51, 4243–4260 RSC.
- E. A. Losev and E. V. Boldyreva, CrystEngComm, 2014, 16, 3857–3866 Search PubMed.
- Please note that NG (η = 0) does not necessarily mean that the reaction is performed under strict dry conditions. Indeed, water in the form of environmental humidity124 or as a liquid generated as side products during the mechanochemical process (e.g., dehydration/desolvation of a hydrate/solvate64) are not contemplated in the η value.
| This journal is © The Royal Society of Chemistry 2026 |