
A deep dive into mechanochemical organic reactions by accurate crystallographic analysis via TAAM refinement
Ana M.
Constantin
 a,
Francesco
Mele
a,
Vinayak
Botla
a,
Nicola Della
Ca'
a,
Raimondo
Maggi
a,
Giovanni
Maestri
a,
Alessandro
Cerveri
a,
Remie M.
Sundermann
a,
Daniele
Cauzzi
a,
Francesco
Pancrazzi
*a and
Paolo P.
Mazzeo
*ab
a,
Francesco
Mele
a,
Vinayak
Botla
a,
Nicola Della
Ca'
a,
Raimondo
Maggi
a,
Giovanni
Maestri
a,
Alessandro
Cerveri
a,
Remie M.
Sundermann
a,
Daniele
Cauzzi
a,
Francesco
Pancrazzi
*a and
Paolo P.
Mazzeo
*ab
aDepartment of Chemistry, Life Sciences and Environmental Sustainability, Università di Parma, Parco Area delle Scienze 17/A, 43124 Parma, Italy. E-mail: francesco.pancrazzi@unipr.it; paolopio.mazzeo@unipr.it
bBiopharmanet-tec, Parco Area delle Scienze 95, 43124 Parma, Italy
First published on 17th October 2025
Abstract
An original oxidative functionalization of 8-aminoquinolines in the presence of an organic peroxide under batch conditions has been recently reported. It was proposed that the reaction involved a highly unusual C8-to-C7 amino group shift, coupled with the concomitant oxygenation at the C8 position of the heterocycle. Although single crystal X-ray diffraction was performed with acceptable statistical parameters, we propose a substantial reinterpretation of the actual structure by an accurate refinement of data via the transferable aspherical atom model (TAAM). Indeed, we unambiguously clarify that no amino-group-shift took place and that direct oxygenation at C7 occurred instead. Moreover, we report herein that this reactivity could be also performed under mechanochemical conditions, suggesting the feasibility to perform similar oxidation processes minimizing chemical waste.
Introduction
The quinoline motif is a versatile heterocyclic core, being present in many natural products, drugs and functional materials (Scheme 1a).1–5 For instance, 8-aminoquinolines have been broadly used as auxiliary directing groups for C–H activation methods since the original discovery of their chelating properties in 2005.4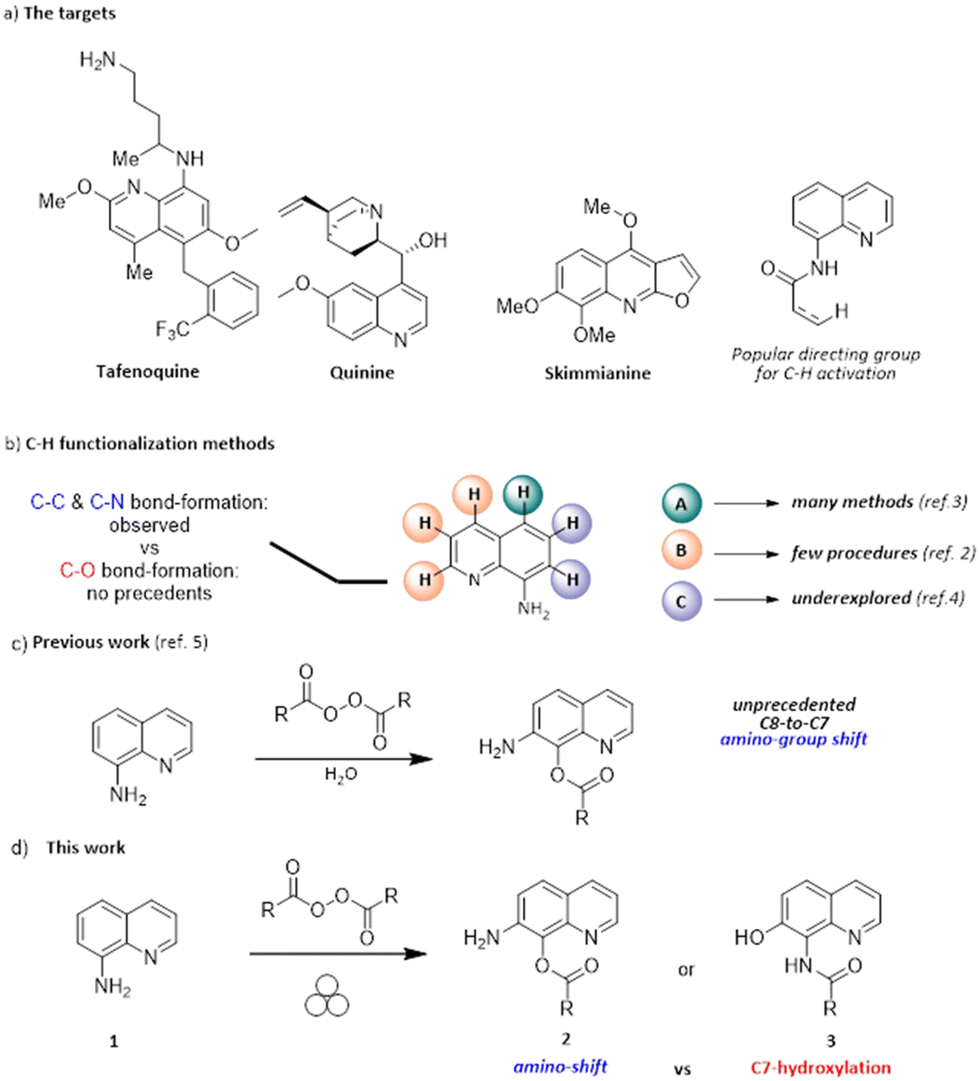 | ||
| Scheme 1 a) Selected examples of functional molecules containing the 8-aminoquinoline core; b) reported strategies for the C–H functionalization of 8-aminoquinolines; c) the originally proposed tandem amino-group shift/C-benzoylation in the presence of peroxides and water providing 2; d) the present mechanochemical approach and the actual structure of the oxidation product, namely 3. | ||
Significant efforts have been made by scientists to design and develop new strategies for the functionalization of the quinoline scaffold because of its importance.6–9 The presence of the amino moiety on the C8 position imparts unique properties and reactivity to the heterocycle, making it a valuable platform for various derivatization processes (Scheme 1b).1–4,6–9 The possibility of activating quinoline C–H bonds in a regioselective manner is likely the most appealing derivatization strategy. The functionalization at the C5 position is by far the most accomplished transformation reported in the literature,6–16 although only a few studies reported the functionalization at the C7 position.17–21 They almost invariably involve the use of metal catalysts, except for a nitration method using tert-butyl nitrite.19 No methods forming a new C–O bond at the C7 position have been reported yet.
In 2019, it was reported that aminoquinoline 1 undergoes the unprecedented C8-to-C7 amino-group shift and benzoxylation at C7, upon treatment with benzoyl peroxide in water at room temperature (2a, 69% yield, Scheme 1c).22
A similar reactivity was not previously reported, nor proposed in alternative methods since then, regardless of the employed reactants or catalytic systems. The ability to drive chemical transformation by means of mechanical force has been recently rediscovered as an attractive and cost-effective tool23–26 for the synthesis of organic,27–29 metal–organic,30–34 and inorganic compounds and materials.35–37 Many traditional solution-based chemical reactions can be performed via mechanochemistry with no (or minimal) use of solvent,25,38–41 providing a more sustainable and cheaper strategy for synthesis compared to solution methods.42,43 We report herein that the same apparent reactivity observed in solution22 by treating 1 with an organic peroxide can be obtained under mechanochemical conditions, minimizing chemical waste (Scheme 1d). Moreover, during this study, we gained clear-cut evidence for the actual structure of the product of this unusual oxidation, which led us to reassign the initially proposed structure 2. Indeed, the reaction involves the original C-hydroxylation on the C7 position of the heterocycle, together with the concomitant N-acylation of the primary amino group, eventually delivering 3.
Results and discussion
By aiming at developing a metal- and solvent-free method for the synthesis of 7-aminoquinolin-8-ols, the model reaction of 1 with benzoyl peroxide (BPO) was initially tested in a 15 ml stainless steel jar loaded with one 15 mm stainless steel (SS) ball and accelerated by means of a vertical ball mill at 15 Hz for 1 hour (Table 1). A single product formed by milling 8-amino quinoline together with a small excess of benzoyl peroxide, which was added in a single portion. Although this result showed the feasibility of the mechanochemical approach (Table 1, entry 1), the reaction was optimized in terms of mechanochemical variables as well as different addition schemes of BPO into the milling jar (Table 1). The best result was reached when BPO was added in three portions (55%, entry 7). We further investigated on the effect of a small amount of solvent water in a liquid assisted grinding (LAG) protocol. However, it is worth reminding that traces of water were intrinsically present in the reaction environment because commercial BPO contains ca. 25% of water. We then explored the effect of further addition of water (η = 1 and η = 3, entries 10 and 11, respectively) and observed a significant drop in terms of the reaction yield. Neither the use of acetonitrile nor ethyl acetate led to a better outcome (entries 12 and 13). Finally, the use of a solid auxiliary (SAA) was explored even though no benefit was proved (entries 14–16). To strengthen these findings and in order to thoroughly compare the data reported in the literature,22 we also prepared derivative 3b in which BPO was replaced with 4-methylbenzoylperoxide (see the SI page S4); the sample was obtained in 40% yield, and its spectroscopic data were both identical to those already described and consistent with the trend presented herein for 3a.|
|
|||||
|---|---|---|---|---|---|
| Entrya | BPO (equiv.) | Time | Freq. | LAG/SAA | Yield of 3a (%) |
| a Reaction conditions: 2 mmol of 1, BPO, milled at 15 Hz for 2 hours in the presence of 1 SS 15 mm ball, isolated yields. b Milling for 1 hour. c Milling for 4 hours. d Milling at 30 Hz. e Using two SS 5 mm balls. | |||||
| 1b | 1.1 | 1 h | 15 Hz | — | 10 |
| 2 | 1.1 | 2 h | 15 Hz | — | 28 |
| 3c | 1.1 | 4 h | 15 Hz | — | 10 |
| 4 | 1.5 | 2 h | 15 Hz | — | 18 |
| 5d | 1.1 | 2 h | 30 Hz | — | 21 |
| 6 | 1.1 (0.18 × 6) | 2 h | 15 Hz | — | 30 |
| 7 | 1.1 (0.37 × 3) | 2 h | 15 Hz | — | 55 |
| 8 | 1.5 (0.50 × 3) | 2 h | 15 Hz | — | 50 |
| 9e | 1.1 (0.37 × 3) | 2 h | 15 Hz | — | 35 |
| 10 | 1.1 (0.37 × 3) | 2 h | 15 Hz | H2O; η = 1 | 18 |
| 11 | 1.1 (0.37 × 3) | 2 h | 15 Hz | H2O; η = 3 | 17 |
| 12 | 1.1 (0.37 × 3) | 2 h | 15 Hz | EtOAc; η = 1 | 25 |
| 13 | 1.1 (0.37 × 3) | 2 h | 15 Hz | MeCN; η = 1 | 27 |
| 14 | 1.1 (0.37 × 3) | 2 h | 15 Hz | Na2SO4 | 17 |
| 15 | — | 2 h | 15 Hz | NaCl | 17 |
| 16 | — | 2 h | 15 Hz | SiO2 | 20 |
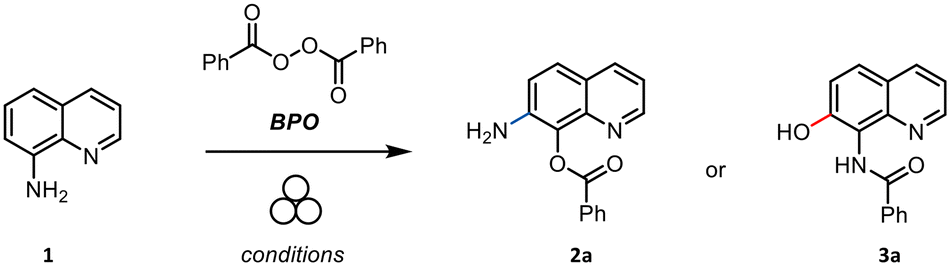
Upon the milling step, the crude was recovered with ethyl acetate and purified by column chromatography to separate the unreacted starting material from the functionalized quinoline. After purification, the product was characterized via NMR spectroscopy (1H and 13C NMR) and mass spectrometry (see the SI). Although spectroscopic data of the obtained compound were identical to those reported in the literature,22 it is worth noting that neither NMR nor MS spectra allowed for an unequivocally discrimination of the actual structure of the heterocycle. Therefore, it is not possible to conclude in a clear-cut fashion from these analyses whether a tandem 8,7-amino shift/C8 benzoylation occurred, providing 2, or direct C7 hydroxylation/N-acylation took place, affording 3 instead.
Furthermore, we recorded the 1H–15N HSQC spectra of both the starting material (8-aminoquinoline) and the product 3a. The starting material shows characteristic correlations of the amino group at δ 5.0 (1H)/46.6 (15N) ppm, while, for the product 3a, the corresponding signal shifts to δ 11.0 (1H)/117.9 (15N) ppm, which is consistent with the presence of an amide NH group (see SI Fig. S4 and S7).
Single crystal X-ray diffraction (SCXRD) was then used to unambiguously rationalize the reaction outcome. The compound was recrystallized by slow evaporation of DCM. The process led to the formation of clear prismatic crystals.
The fine interpretation of the electron density of the sample allowed us to discriminate between the structure of 2a and that of 3a. Indeed, two intramolecular H-bond rings are clearly observed in the structure, which clearly reveals the presence of an amidic group in the product. This, in turn, indicates that compound 3a was actually obtained. In particular, an S(5) intermolecular H-bond ring between the NH in position 8 and the quinolinic nitrogen, and an S(7) intermolecular H-bond ring between the OH decoration in position 7 and the carboxyl group of the amidic moiety were observed (Fig. 1). Diffraction data were modelled with the aspherical atom refinement algorithm to achieve a more accurate structure model. In particular, transferable aspherical atom model (TAAM)44,45 refinement was used by means of the DiscambMATTS2tsc software implemented in the non-spherical atoms tool in Olex2 (ref. 46) (NoSpherA2).47 The program built upon the new version of the DiSCaMB48 library computes non-spherical atomic scattering factors from a multipolar model parametrized using the MATTS databank.49 The analysis of the differential electron density (DED) map (i.e. the difference between experimental and modelled electron density maps) is presented in Fig. 2. 2D slices of the DED map are shown along with their related superimposed crystal structures. Blue and red areas represent positive and negative values of the DED map, respectively. Blue areas represent unmodelled electron density or underestimation of the modelled electron density, while red areas represent overestimation of the modelled electron density. The DED map of product 3a is fully consistent with the hydroxylation at the C7 position (Fig. 2a). Indeed, this hypothesis provides a map whose results are almost flat (min −0.3 e− Å−3, max 0.2 e− Å−3), and no significant positive or negative deviation peaks emerged. Additionally, thermal ellipsoids are all consistent in terms of dimensions and orientation, which is further symptomatic of the correct assignment of the heteroatoms in the molecule.
 | ||
| Fig. 1 Crystallographic outcome of the analysis on 3a, emphasizing the formation of the intramolecular H-bond rings. Non-hydrogen atoms are reported in ellipsoids at the 50% probability level according to the color code C = grey, N = blue, and O = red. Bonds involving hydrogen atoms are reported in a stick style for the sake of clarity. | ||
 | ||
| Fig. 2 2D slices of the differential electron density (DED) map (coloured contour style) observed for the structural model consistent with a. the C7 hydroxylation (left) vs. b. the C8-to-C7 amino shift (right). Blue and red areas represent under- vs. over-estimation of the modelled electron density, respectively. The color scale is automatically adapted according to the maximum and minimum values of the map (a. min = −0.2, max = 0.05 e− Å−3; b. min = −0.5, max = 0.3 e− Å−3). Superimposed structural models are reported with non-hydrogen atoms in an ellipsoid style at the 50% level of probability according to the colour code C = grey, N = blue, and O = red. Hydrogen atoms are reported in a ball-and-stick style for the sake of clarity. All out-of-plane atoms are plotted in transparency (i.e. part of the aryl ring of the benzoyl arm). | ||
On the other hand, the originally proposed hypothesis of a C8 to C7 amino shift, which would have provided 2a, gives rise to a DED map (Fig. 2b) that shows important mismatches between the modelled and the experimental electron density. Indeed, positive (blue) DED peaks are observed around the nitrogen atom of the amino group and close to the ethereal oxygen atom of the ester functionality. These deviations indicate that an amount of experimentally measured electron density is not accounted for by the proposed structural assignment. Moreover, negative (red) DED peaks are observed on the ethereal oxygen atom of the ester and around one of the hydrogen atoms of the –NH2 group. These deviations indicate that the proposed structural assignment involves the presence of an amount of electronic density that was not experimentally measured. Furthermore, R1 values finally obtained for the two refined structures are respectively 4.38% (3a) and 7.56% (2a).
All these data are symptomatic of the misinterpretation of the functional group allocation in the original structural assignment.22
In particular, i) the nitrogen atom assigned on the C7 position underestimates the electron density, while the balance would be correct if an oxygen atom is present on this position, ii) only a single hydrogen atom should be linked to the heteroatom bound to C7, rather than two, iii) the oxygen atom bound to C8 overestimates the measured electron density, while the balance would be correct if a nitrogen atom is present on this site, and iv) a missing hydrogen atom should be bonded thereof.
The outcome of the crystallographic analysis described herein is consistent with the data found in the crystal structure that was previously reported in the literature. Indeed, by further refining the data deposited in the CCDC by Botla et al. in 2019,22 we have noticed a significant mismatch between the calculated and experimental electron densities suggesting that direct C7 hydroxylation is more likely to occur rather than an 8,7-amino shift (see the SI). The presence of a secondary amide and of a phenol group, which are consistent with the structure of 3a, and the concomitant absence of an ester and a primary aniline fragment, which would be hallmarks of the structure of 2a, were similarly confirmed by IR analyses (Fig. 3, details in the SI).
 | ||
| Fig. 3 FTIR spectrum of compound 3a. | ||
Indeed, the HATR-FTIR absorption spectrum of the compound shows the expected amide I and amide II bands at 1633 and 1528 cm−1, respectively. In contrast, the region above 1700 cm−1, in which the diagnostic band of Ar–CO2–Ar′ esters is found, did not present any extra peak.
Experimental
Reagents and consumables
All reagents were used as received from commercial sources without further purification. All solvents were dried over activated 4 Å molecular sieves for 24 h. All reactions were analysed by TLC and by GC using a 30 m SE-30 capillary column. Flash column chromatography was performed on silica gel 60 (70–230 mesh).Mechanochemical apparatus
The ball mill grinding experiments were all performed using a Fritsch P23 vertical movement vibrational mill. This mill has an amplitude of 9 mm and an adjustable frequency from 15 Hz to 50 Hz with an adjustable timer. Mechanochemical reactions were performed either in stainless steel or zirconia jars (see the SI).General procedure for the ball milling reaction
To a ball milling vessel (stainless steel, 15 mL) loaded with one grinding ball (stainless steel, diameter 15 mm, 13.5 g) was added sequentially 8-aminoquinoline (1 eq., 0.2 mmol, 28.8 mg) and benzoyl peroxide portion wise (1.1 eq., 0.22 mmol, 3 times). After all the reagents were added, the specified solvent was put above if requested for LAG protocols. The vessel was closed in air and the jar was placed in a vibratory ball mill (Fritsch Pulverisette P23) at 15 Hz. At the end of the reaction, the reaction mixture was recovered with EtOAc or DCM. The resulting reaction mixture was concentrated under reduced pressure and finally purified by flash column chromatography (eluent hexane/EtOAc 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Additionally, to prove that any metal contamination due to iron leaching from the jar could have influenced the reaction proposed, the same set up with a zirconia jar and milling balls was used instead of the commonly used stainless steel ones returning no significant difference in the product outcome (see the SI for details).
:1). Additionally, to prove that any metal contamination due to iron leaching from the jar could have influenced the reaction proposed, the same set up with a zirconia jar and milling balls was used instead of the commonly used stainless steel ones returning no significant difference in the product outcome (see the SI for details).
X-ray diffraction
The diffraction intensities of selected single crystals of 3a were collected under nitrogen flux at 200 K on a Bruker D8 Venture diffractometer using micro-focused Mo Kα radiation (λ = 0.71073 Å), equipped with a kappa goniometer and an Oxford Cryostream. Lorentz polarization and absorption corrections were applied for all of the experiments. Data reduction was carried out using APEX v5 software. The structure was solved by direct methods using SHELXT50 and refined with the aspherical atom refinement algorithm to achieve a more accurate structure model. In particular, transferable aspherical atom model (TAAM)44,45 refinement was used by means of the DiscambMATTS2tsc software implemented in the non-spherical atoms tool in Olex2 (ref. 46) (NoSpherA2).47 The program built upon the new version of the DiSCaMB48 library computes non-spherical atomic scattering factors from a multipolar model parametrized using the MATTS databank.49 All atoms were refined with anisotropic thermal displacement parameters (see Fig. S10 for the ORTEP representation). Table 2 in the main text (Table S5 here reported for the sake of reading) reports crystal data collection parameters and refinement results. The structure was solved in the P21/c space group. Two symmetry related molecules are present in the structures of 3a. CCDC 2388814 contains the supplementary crystallographic data for this paper. In addition, CCDC 2388841 contains the re-refined crystallographic data originally deposited by Botla et al.22 with the assigned ref code VIXMAT, CCDC 1559199.| Identification code | 3a | CCDC 1559199 | |
|---|---|---|---|
| Original | Re-refined | ||
| Empirical formula | C16H12N2O2 | C17H14N2O2 | C17H14N2O2 |
| Formula weight | 264.286 | 278.30 | 278.30 |
| Temperature/K | 200.00 | 294.15 | 294.15 |
| Crystal system | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P21/c | P21/c |
| a/Å | 14.5096(9) | 11.954(2) | 11.954(2) |
| b/Å | 17.8805(11) | 7.454(2) | 7.454(2) |
| c/Å | 10.3906(6) | 15.468(3) | 15.468(3) |
| α/° | 90 | 90 | 90 |
| β/° | 110.379(2) | 95.33(3) | 95.33(3) |
| γ/° | 90 | 90 | 90 |
| Volume/Å3 | 2527.0(3) | 1372.4(5) | 1372.4(5) |
| Z | 8 | 4 | 4 |
| ρ calc g cm−3 | 1.389 | 1.347 | 1.347 |
| μ mm−1 | 0.094 | 0.090 | 0.090 |
| F(000) | 1104.7 | 584.0 | 584.0 |
| Crystal size/mm3 | 0.8 × 0.1 × 0.08 | 0.42 × 0.38 × 0.24 | 0.42 × 0.38 × 0.24 |
| Radiation | Mo Kα | MoKα | MoKα |
| (λ = 0.71073) | (λ = 0.71073) | (λ = 0.71073) | |
| 2Θ range for data collection/° | 4.76 to 52.9 | 5.29 to 49.958 | 5.29 to 49.958 |
| Index ranges | −18 ≤ h ≤ 18 | −14 ≤ h ≤ 14 | −14 ≤ h ≤ 14 |
| −22 ≤ k ≤ 22 | −8 ≤ k ≤ 8 | −8 ≤ k ≤ 8 | |
| −13 ≤ l ≤ 13 | −18 ≤ l ≤ 18 | −18 ≤ l ≤ 18 | |
| Reflections collected | 50783 |
24733 |
24733 |
| Independent reflections | 4343 | 2413 | 2413 |
| R int = 0.0849 | R int = 0.0235 | R int = 0.0235 | |
| R sigma = 0.0381 | R sigma = 0.0143 | R sigma = 0.0143 | |
| Data/restraints/parameters | 4343/9/578 | 2413/0/192 | 2413/1/209 |
| Goodness-of-fit on F2 | 1.151 | 1.060 | 1.055 |
| Final R indices [I ≧ 2σ(I)] | R 1 = 0.0438 | R 1 = 0.0696 | R 1 = 0.0390 |
| wR = 0.0861 | wR2 = 0.2049 | wR2 = 0.1119 | |
| Final R indices [all data] | R 1 = 0.0560 | R 1 = 0.0749 | R 1 = 0.0429 |
| wR2 = 0.0907 | wR2 = 0.2126 | wR2 = 0.1173 | |
| Largest diff. peak/hole/e Å−3 | 0.23/−0.21 | 0.50/−0.54 | 0.19/−0.12 |
IR spectrometry
IR spectra were collected on a PerkinElmer Spectrum Two spectrometer paired with a Diamond Smart Orbit accessory. The spectra were recorded in the range of 3800 cm−1 to 550 cm−1.Conclusions
A metal free mechanochemical reaction has been carried out for the direct C7-hydroxylation of an 8-aminoquinoline using a simple vertical vibrational ball milling setup with a stainless-steel jar. The outcome of the reaction has been unambiguously clarified by means of single crystal X-ray diffraction analysis via transferable aspherical atom model (TAAM) refinement, by means of the DiscambMATTS2tsc software implemented in the non-spherical atoms tool in Olex2 (NoSpherA2).This study unambiguously identified the actual positions of the heteroatoms on the product, thus solving potential doubts derived from the interpretation of the spectroscopic data. The data indicated that the originally proposed amino shift did not occur, leading to reassigning the structure that was initially proposed in the literature and disclosing that C–O bond forming events can take place on the C7 position of these heterocycles.
Author contributions
AMC and FM contributed equally to this work. Conceptualization: NDC and RM. Investigation and formal analysis: AMC, FM, AC and VB performed the mechanochemical experiments; DC, RS, and PPM performed the spectroscopic and X-ray analyses. Writing – original draft: PPM and FP. Writing – review & editing: PPM, FP, GM, NDC, and RM. Fundings: NDC, RM, and PPM.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, NMR Spectra, IR spectra and crystallographic analyses. See DOI: https://doi.org/10.1039/d5ce00677e.CCDC 2388814 and 2388841 contain the supplementary crystallographic data for this paper.51a,b
Acknowledgements
This work has benefited from the equipment and framework of the COMP-HUB and COMP-R Initiatives, funded by the ‘Departments of Excellence’ programs of the Italian Ministry for University and Research (MUR, 2018–2027). We also warmly thank support from MUR (Grant Xylonite, No. P2022HSF3R). This work also benefitted from the call – Missione 4 “Istruzione e Ricerca” – Componente 2 – Investimento 1.1 “Fondo per il Programma Nazionale di Ricerca e Progetti di Rilevante Interesse Nazionale (PRIN)” with projects ‘Crystals4Bees’ X4BEES PRIN2022-D53D23010140001. This work was supported by MSCA-IF “METACYL” (grant no. 894026). Chiesi Farmaceutici S.p.A. is acknowledged for the use of the Bruker D8 Venture single-crystal diffractometer.Notes and references
- J. P. Michael, Nat. Prod. Rep., 2005, 22, 627–646 RSC.
- J. G. Díaz, J. G. Sazatornil, M. L. Rodríguez, L. R. Mesía and G. V. Arana, J. Nat. Prod., 2004, 67, 1667–1671 CrossRef.
- H. M. R. Hoffmann and J. Frackenpohl, Eur. J. Org. Chem., 2004, 2004, 4293–4312 CrossRef.
- M. Corbet and F. De Campo, Angew. Chem., Int. Ed., 2013, 52, 9896–9898 CrossRef CAS.
- A. Daolio, M. Prencipe, T. Abodunrin, P. Pelagatti, P. P. Mazzeo and A. Bacchi, Chem. – Eur. J., 2025, e202500956 CrossRef CAS.
- H. Qiao, K. Zhao, Y. Li, L. Yang and F. Yang, Green Chem., 2023, 25, 8385–8410 RSC.
- Z. Xu, X. Yang, S. F. Yin and R. Qiu, Top. Curr. Chem., 2020, 378, 1–99 CrossRef.
- B. Khan, H. S. Dutta and D. Koley, Asian J. Org. Chem., 2018, 7, 1270–1297 CrossRef CAS.
- S. M. Prajapati, K. D. Patel, R. H. Vekariya, S. N. Panchal and H. D. Patel, RSC Adv., 2014, 4, 24463–24476 RSC.
- C. Shao, C. Ma, L. Li, J. Liu, Y. Shen, C. Chen, Q. Yang, T. Xu, Z. Hu, Y. Kan and T. Zhang, Beilstein J. Org. Chem., 2024, 20(14), 155–161 CrossRef CAS.
- D. R. Motati, D. Uredi and E. B. Watkins, Chem. Sci., 2018, 9, 1782–1788 RSC.
- L. Zhu, R. Qiu, X. Cao, S. Xiao, X. Xu, C. T. Au and S. F. Yin, Org. Lett., 2015, 17, 5528–5531 CrossRef CAS PubMed.
- C. Xia, K. Wang, J. Xu, C. Shen, D. Sun, H. Li, G. Wang and P. Zhang, Org. Biomol. Chem., 2017, 15, 531–535 RSC.
- X. Cong and X. Zeng, Org. Lett., 2014, 16, 3716–3719 CrossRef CAS.
- A. M. Suess, M. Z. Ertem, C. J. Cramer and S. S. Stahl, J. Am. Chem. Soc., 2013, 135, 9797–9804 CrossRef CAS.
- M. Ye, G. L. Gao, A. J. F. Edmunds, P. A. Worthington, J. A. Morris and J. Q. Yu, J. Am. Chem. Soc., 2011, 133, 19090–19093 CrossRef CAS.
- M. Borgini and P. Wipf, Tetrahedron, 2022, 120, 132876 CrossRef CAS.
- X. Liu, G. Mao, J. Qiao, C. Xu, H. Liu, J. Ma, Z. Sun and W. Chu, Org. Chem. Front., 2019, 6, 1189–1193 RSC.
- S. Mondal, S. Samanta and A. Hajra, Adv. Synth. Catal., 2018, 360, 1026–1031 CrossRef CAS.
- J. Zhou, X. Li, G. Liao and B. F. Shi, Chin. J. Chem., 2018, 36, 1143–1146 CrossRef CAS.
- J. Zhou, B. Li, F. Hu and B. F. Shi, Org. Lett., 2013, 15, 3460–3463 CrossRef CAS.
- V. Botla, N. Pilli and C. Malapaka, Green Chem., 2019, 21, 1735–1742 RSC.
- S. L. James and T. Friščić, Chem. Soc. Rev., 2013, 42, 7494–7496 RSC.
- P. Baláž, M. Achimovicová, M. Baláž, P. Billik, C. Z. Zara, J. M. Criado, F. Delogu, E. Dutková, E. Gaffet, F. J. Gotor, R. Kumar, I. Mitov, T. Rojac, M. Senna, A. Streletskii and W. C. Krystyna, Chem. Soc. Rev., 2013, 42, 7571–7637 RSC.
- G. I. Lampronti, A. A. L. Michalchuk, P. P. Mazzeo, A. M. Belenguer, J. K. M. Sanders, A. Bacchi and F. Emmerling, Nat. Commun., 2021, 12, 1–9 CrossRef.
- A. Bacchi and P. P. Mazzeo, Crystallogr. Rev., 2021, 27, 102–123 CrossRef CAS.
- K. Kubota, Y. Pang, A. Miura and H. Ito, Science, 2019, 366, 1500–1504 CrossRef CAS.
- N. Fantozzi, J. N. Volle, A. Porcheddu, D. Virieux, F. García and E. Colacino, Chem. Soc. Rev., 2023, 52, 6680–6714 RSC.
- F. Cuccu, D. L. Browne and A. Porcheddu, ChemCatChem, 2023, 15, e202300762 CrossRef CAS.
- T. Stolar and K. Užarević, CrystEngComm, 2020, 22, 4511–4525 RSC.
- D. Braga, F. Grepioni, L. Maini, P. P. Mazzeo and B. Ventura, New J. Chem., 2011, 35, 339–344 RSC.
- L. Maini, P. P. Mazzeo, F. Farinella, V. Fattori and D. Braga, Faraday Discuss., 2014, 170, 93–107 RSC.
- F. Farinella, L. Maini, P. P. Mazzeo, V. Fattori, F. Monti and D. Braga, Dalton Trans., 2016, 45, 17939–17947 RSC.
- G. Cagossi, P. P. Mazzeo, A. Bacchi and P. Pelagatti, RSC Mechanochem., 2025, 2, 475–481 RSC.
- P. F. M. De Oliveira, R. M. Torresi, F. Emmerling and P. H. C. Camargo, J. Mater. Chem. A, 2020, 8, 16114–16141 RSC.
- C. Gazzurelli, M. Solzi, F. Cugini, P. Pio Mazzeo, A. Bacchi and P. Pelagatti, Inorg. Chim. Acta, 2022, 539, 121010 CrossRef CAS.
- C. Gazzurelli, A. Migliori, P. P. Mazzeo, M. Carcelli, S. Pietarinen, G. Leonardi, A. Pandolfi, D. Rogolino and P. Pelagatti, ACS Sustainable Chem. Eng., 2020, 8, 14886–14895 CrossRef CAS.
- R. Mocci, S. Murgia, L. De Luca, E. Colacino, F. Delogu and A. Porcheddu, Org. Chem. Front., 2018, 5, 531–538 RSC.
- F. Cuccu, L. De Luca, F. Delogu, E. Colacino, N. Solin, R. Mocci and A. Porcheddu, ChemSusChem, 2022, 15, e202200362 CrossRef CAS PubMed.
- P. P. Mazzeo, G. I. Lampronti, A. A. L. Michalchuk, A. M. Belenguer, A. Bacchi and F. Emmerling, Faraday Discuss., 2023, 241, 289–305 RSC.
- F. Mele, A. M. Constantin, A. Porcheddu, R. Maggi, G. Maestri, N. Della Ca' and L. Capaldo, Beilstein J. Org. Chem., 2025, 21(33), 458–472 CrossRef CAS.
- P. Ying, J. Yu and W. Su, Adv. Synth. Catal., 2021, 363, 1246–1271 CrossRef CAS.
- A. M. Constantin, F. Mele, M. Lanzi, G. Maestri, R. Maggi, N. Della Ca' and L. Capaldo, ChemSusChem, 2025, 2500211 Search PubMed.
- K. K. Jha, B. Gruza, P. Kumar, M. L. Chodkiewicz and P. M. Dominiak, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2020, 76, 296–306 CrossRef CAS.
- N. K. Hansen and P. Coppens, Acta Crystallogr., Sect. A, 1978, 34, 909–921 CrossRef.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339–341 CrossRef CAS.
- F. Kleemiss, O. V. Dolomanov, M. Bodensteiner, N. Peyerimhoff, L. Midgley, L. J. Bourhis, A. Genoni, L. A. Malaspina, D. Jayatilaka, J. L. Spencer, F. White, B. Grundkötter, G. Grundkötter-Stock, S. Steinhauer, D. Lentz, H. Puschmann and S. Grabowsky, Chem. Sci., 2021, 12, 1675–1692 RSC.
- M. L. Chodkiewicz, S. Migacz, W. Rudnicki, A. Makal, J. A. Kalinowski, N. W. Moriarty, R. W. Grosse-Kunstleve, P. V. Afonine, P. D. Adams and P. M. Dominiak, J. Appl. Crystallogr., 2018, 51, 193–199 CrossRef CAS.
- K. K. Jha, B. Gruza, A. Sypko, P. Kumar, M. L. Chodkiewicz and P. M. Dominiak, J. Chem. Inf. Model., 2022, 62, 3752–3765 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Adv., 2015, 71, 3–8 CrossRef PubMed.
- (a) CCDC 2388814: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2l5rj2; (b) CCDC 2388841: Experimental Crystal Structure Determination, 2025, DOI:10.5517/ccdc.csd.cc2l5sdz.
| This journal is © The Royal Society of Chemistry 2026 |