
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, reactivity and properties of versatile gem-bis[(trifluoromethyl)sulfonyl] functionalized organic molecules†
Daniel Diez-Iriepa
a,
José M. Alonso
*b and
Pedro Almendros
 *c
*c
aFacultad de Farmacia, Universidad de Castilla-La Mancha, Av. Dr José María Sánchez Ibáñez, s/n, 02071 Albacete, Spain
bGrupo de Lactamas y Heterociclos Bioactivos, Unidad Asociada al CSIC por el IQOG, Departamento de Química Orgánica, Facultad de Química, Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: josalo08@ucm.es
cInstituto de Química Orgánica General, Consejo Superior de Investigaciones Científicas, IQOG-CSIC, Juan de la Cierva 3, 28006 Madrid, Spain. E-mail: palmendros@iqog.csic.es
First published on 25th May 2026
Abstract
Incorporation of fluorinated functionalities is a valuable strategy to improve both chemical and biological properties of organic compounds. In this context, much attention is being focused on the trifluoromethanesulfonyl (triflyl) group (SO2CF3), due to its potent electron-withdrawing effect and mild lipophilicity. Consequently, different protocols have been developed over the years to prepare triflones, which are organic molecules bearing the triflyl functionality. The electron-withdrawing effect of the triflyl group also makes compounds bearing gem-bis(triflyl)methyl group (Tf2CH) strongly acidic. Several one-pot reactions, in combination with a derivation step in some cases, merged under simple reaction conditions for the straightforward formation of divergent fluorosulfonyl-decorated cyclic scaffolds from simple starting materials, providing a convenient access to an under-explored structural space. In this way, cyclobutenes, heterocycles, and non-cyclic products have been easily prepared without the requirement for either a catalyst or an additive. This article reviews reactions between differently functionalized alkynes, alkenes and allenes with Tf2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH2 (generated in situ from a shelf-stable betaine) to afford cyclic and acyclic bis(triflyl)-functionalized organic molecules with novel chemical structures.
CH2 (generated in situ from a shelf-stable betaine) to afford cyclic and acyclic bis(triflyl)-functionalized organic molecules with novel chemical structures.
 Daniel Diez-Iriepa | Daniel Diez Iriepa received his BSc degree in Chemistry from Universidad de Alcalá in 2017 with Extraordinary End-of-Degree Award, and his MSc degree in Research in Sciences from the same university in 2018. In 2023, he obtained his PhD degree in Chemistry with Cum Laude distinction from Universidad de Alcalá and Instituto de Química Orgánica General IQOG-CSIC. In 2024, he joined United Arab Emirates University as postdoctoral researcher. In 2025, he joined Universidad de Castilla-La Mancha in the Faculty of Pharmacy, Albacete, as postdoctoral. Since 2026, he has been supported by a postdoctoral fellowship for the formalization of a contract for access as a senior researcher, funded by the Regional Government of Castilla-La Mancha and co-financed by the European Social Fund. His research interests include medicinal chemistry, heterocyclic chemistry, materials chemistry and synthetic methodology. |
 José M. Alonso | José Miguel Alonso (Madrid, 1975) received his BS degree (1998) and his PhD degree (2003) from Universidad Complutense de Madrid (Prof. Benito Alcaide and Prof. Pedro Almendros). Besides a diverse experience in the industry (environmental science, polymer chemistry), he has been working as postdoctoral researcher at Universidade de Santiago de Compostela (Santiago de Compostela, Spain), Universidad Complutense de Madrid (Madrid, Spain), University of East Anglia (Norwich, UK) and IQOG-CSIC (Madrid, Spain). He has hold an Assistant Professor position in the Organic and Inorganic Chemistry Department at Universidad de Alcalá (Madrid, Spain), and he is currently Associate Professor in the Organic Chemistry Department at Universidad Complutense de Madrid. His research interests include: allene and aryne chemistry, metal-promoted heterocyclisations, and homogeneous & heterogeneous catalysis. |
 Pedro Almendros | Pedro Almendros (Albacete, 1966) earned his PhD in chemistry from Universidad de Murcia (1994, Profs. Molina and Fresneda). He was a Spanish MEC and European Marie Curie Postdoctoral Programs Fellow (1995–1998, University of Manchester, Prof. Eric J. Thomas). He joined Universidad Complutense de Madrid (UCM) in 1998 as Associate Researcher (Prof. Benito Alcaide). Subsequent appointments have included Assistant Professor at the UCM (2000–2002), Científico Titular “Tenured Scientist” (2002–2007) and Investigador Científico “Researcher Scientist” (2007–2016) at the Instituto de Química Orgánica General, CSIC, Madrid. In 2016 he was promoted to Profesor de Investigación “Researcher Professor” at the IQOG-CSIC, Madrid. His research interest includes allene and alkyne reactivity, strain rings, and triflone chemistry. |
1. Introduction
In 1976, Robert Koshar and Loren Barber were working as research scientists at the Minnesota Mining and Manufacturing Company, the former name of today's big technology group 3M. During the attempts of isolate bis(triflyl)ethene molecules from a solution containing (CF3SO2)2CHCH3, they found that treatment of the mixture with pyridine enabled the clean precipitation of the pyridinium salt 1a from the mother liquor. Once the pure adduct 1a was isolated, subsequent addition of sulfuric acid successfully promoted the C–N bond dissociation, thereby recovering the pure bis(triflyl)ethene molecule. This pure compound could be then used in chemical reactions, particularly in polymer synthesis (Scheme 1).1 Both scientists pursued long and productive careers at 3M. In particular, Koshar reported numerous patents centred on fluorine-containing advanced materials. In subsequent years, the academic community regarded the so-called Koshar zwitterion as a remarkable curiosity—a bench-stable crystalline solid that is easily prepared and handled yet encapsulates a latent bis(triflyl)ethene moiety. Nevertheless, applications in organic synthesis were almost unexplored until in the early 2010s Tokyo-based Professor Hikaru Yanai started to study Koshar-type zwitterions from a multidisciplinary perspective.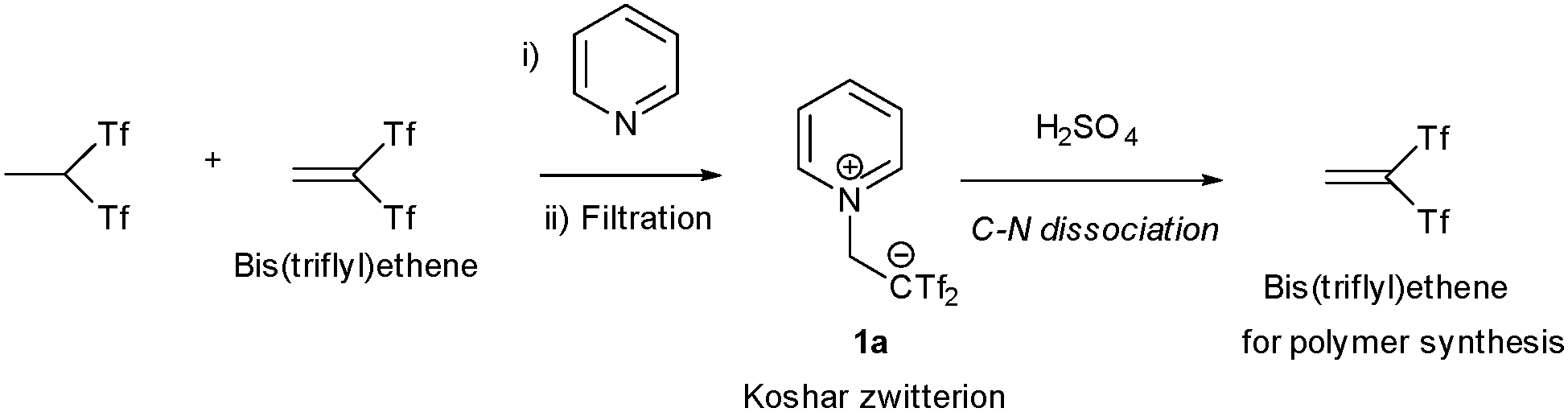 | ||
| Scheme 1 Koshar zwitterion. Preparation and use. | ||
Yanai's research group carefully determined the electronic distribution, molecular conformation, X-ray structure, and an optimized synthetic method to access Koshar-type zwitterions.2–5 They found that the multicomponent reaction of bis(triflyl)methane [triflyl = (trifluoromethyl)sulfonyl] with paraformaldehyde and pyridine in 1,2-dichloroethane (DCE) yielded the desired compounds after a simple work-up (Scheme 2a). They also identified the 2-fluoropyridine derivative 1b as the most efficient precursor of bis(triflyl)ethene, and reported pioneering synthetic applications, including nucleophilic additions leading to carbon acids and Diels–Alder cycloadditions. Interestingly, bis(triflyl)ethene can be described as an equilibrium between the olefinic and the dipolar forms I and II, respectively. The negative charge in II is delocalized by the assistance of the two triflyl (Tf) groups, exhibiting great reactivity and a high instability (Scheme 2b). Considering these aspects, Alcaide and Almendros identified Yanai's zwitterion as a promising platform for a broad range of transformations, including cycloadditions, cascade processes, and even metal-catalyzed reactions, thereby enabling exquisitely chemo-, regio-, and stereocontrolled methodologies. In addition, Koshar-type chemistry allows the incorporation of triflyl groups in final structures. The demand for diverse fluorinated heterocyclic compounds drives the development of versatile synthetic strategies, such as direct C–H fluoroalkylation, although this remains challenging.6–8 Concerns also arise from metal-catalyzed protocols, which may leave impurities in medicinal and engineering products. The trifluoromethylsulfonyl (trifyl) group, one of the strongest electron-withdrawing substituents, imparts mild lipophilicity and enhances the acidity of compounds, making them useful in developing effective acid catalysts.9–13
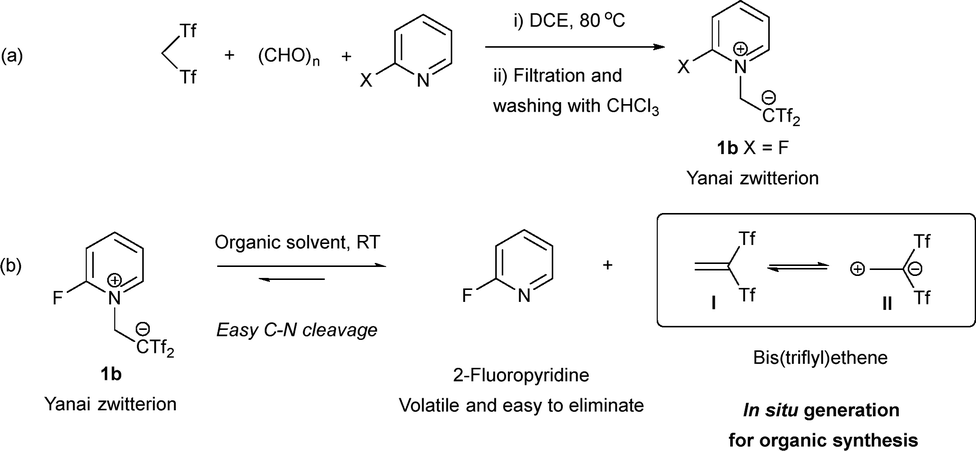 | ||
| Scheme 2 Yanai's zwitterion. Preparation and behaviour in solution. | ||
A substantial portion of this Feature Article is devoted to the synthetic applications, substrate development, and mechanistic understanding of chemical transformations involving the reagent (CF3SO2)2CCH2. A previous review article has addressed complementary aspects of its chemistry, including the pronounced C–H acidity of the (Tf2CH) motif arising from geminal substitution by two triflyl groups (ref. 5c). Other review has highlighted the distinctive ionic yet lipophilic nature of the corresponding carbanions, which enables their use as versatile “substituents” for simultaneously enhancing the aqueous solubility and lipophilicity of organic molecules (ref. 5a). In addition, a recent review has summarized progress in the field of stable fluorinated sulfonyl carbanions, encompassing their preparation methods, bonding characteristics, coordination chemistry, and emerging synthetic applications (ref. 5b).
This article reviews the synthetic applications of Koshar-type zwitterions reported over the past two decades, organized according to the target structures obtained. The synthesis of challenging four-membered rings and their subsequent synthetic transformations are discussed first. Subsequently, the generation of five- and six-membered cyclic frameworks will be presented, encompassing the synthesis of naturally occurring motifs such as triazoles, flavones, aurones, isocoumarins, and pyrans, among others. The biological evaluations of the synthesized compounds will also be discussed. Finally, the preparation of non-cyclic systems derived from readily accessible starting materials—including bis(triflyl)enones, bis(triflyl)enals, and heteroatom-based betaines—will be described.
2. Four-membered rings
2.1. Synthesis of bis(triflyl)cyclobutenes
Cyclobutene derivatives serve as both target molecules and versatile building blocks for constructing complex structures, including bioactive compounds.14–23 The traditional synthetic methods, such as [2+2] cycloaddition reactions of alkynes with unsaturated systems, often yield modest results and require harsh conditions, limiting their applicability. While recent metal-catalyzed strategies have emerged, they suffer from narrow substrate scopes and moderate selectivity, hindering their widespread use.24–26 Due to the increasing importance of the cyclobutene motif, efforts are underway to develop innovative synthetic methodologies.27 However, the efficient and selective introduction of functionality into the four-membered skeleton remains a difficult challenge, leading the need for novel synthetic approaches to synthesize functionalized cyclobutenes with high chemo- and regioselectivity. As mentioned above, Yanai reported that betaine 1b, containing both a 2-fluoropyridinium cation and a stabilized carbanion moiety, quickly equilibrates in acetonitrile to form mixtures of the betaine, (CF3SO2)2CCH2, and volatile 2-fluoropyridine. This property makes it a shelf-stable, eco-friendly, and easy-to-handle source of (CF3SO2)2CCH2.2–5 In this context, our research group has explored the potential of alkynes and betaine 1b as starting materials for the synthesis of cyclobutenes via 1,2-dipolar cycloaddition reactions. Although this synthetic approach has been hindered by the limited availability of methods to access 1,2-dipoles, the uncatalyzed reaction employing in situ generated 1,1-bis(trifluoromethylsulfonyl)ethene (3) has delivered promising results. Building on Yanai's previous investigations into the use of betaines 1 as practical precursors of dipoles 3, it was discovered that fluorine-substituted pyridinium salt at the 2-position (1b) exhibited remarkable efficiency in promoting the desired transformation under mild reaction conditions (Scheme 3).28 Thus, a variety of aliphatic, aromatic, and heteroaromatic substituted-alkynes 2 were used in the intermolecular cyclization reaction with zwitterion 1b. Interestingly, non-symmetrical di-substituted alkynes could be successfully transformed into cyclobutenes 4 in a regioselective way. The steric properties of the substituents in the acetylenic moiety did not significantly affect either yield or regioselectivity, while the electronic nature of the substituents did influence the reaction. In addition, disubstituted alkynes with electron-donating groups at R1 generally resulted in better conversions compared to those with electron-withdrawing substituents and promoted the regioselective formation of isomers 4a versus 4′. (Scheme 3). Besides, less reactive terminal alkynes were also examined (R2 = H, D). While terminal aliphatic alkynes failed to react under the normal conditions, aromatic systems smoothly underwent cycloaddition to afford the corresponding cyclobutenes 4b in satisfactory yields. Interestingly, when R1 is an electron-donating group a higher reactivity was observed compared to disubstitued alkynes. Noteworthy, the reaction of certain terminal alkynes exhibiting electron-poor substituents at R1 resulted in the formation of unexpected pyridines 5 with the participation of MeCN, highlighting the importance of electronic substitution in determining the outcome of the reaction. DFT calculations supported a stepwise reaction mechanism for the formation of compounds 4 and 5, with cyclobutene systems kinetically preferred.28
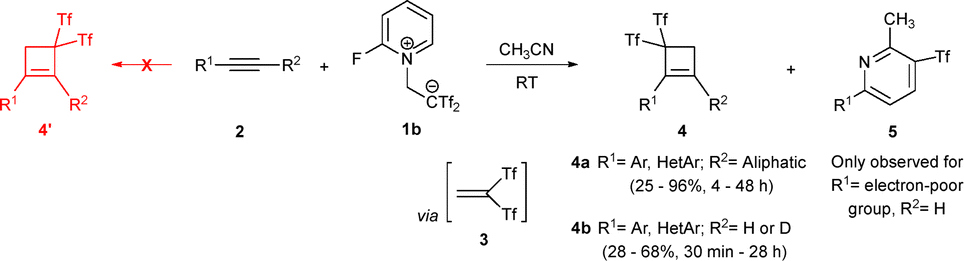 | ||
| Scheme 3 Synthesis of bis(triflyl)cyclobutenes from alkynes. | ||
To increase the family of cyclobutene-based structures that can be accessed through this methodology, ynamides 6 were studied in the presence of betaine 1b under otherwise similar reaction conditions. Interestingly, heteroatom-decorated bis(triflyl)cyclobutenes 7 were obtained, providing an easy route to aminocyclobutenes. Both aryl and alkyl substituents on the alkyne were well tolerated, delivering the desired compounds 7 with total regioselectivity and good yields (Scheme 4a). Different ynamides 8 incorporating heterocycles were also investigated, giving the expected cyclobutenes 9 with moderate to excellent yields (Scheme 4b).29,30
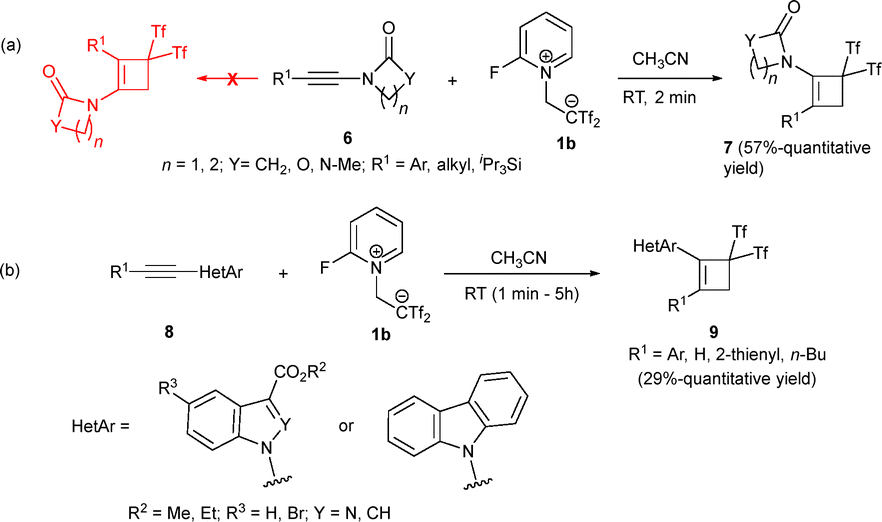 | ||
| Scheme 4 Synthesis of aminocyclobutenes. | ||
A later report explored the versatility of zwitterion 1b in the presence of sulfur, selenium and tellurium-decorated alkynes. Despite the presumed instability of chalcogen-substituted systems, triflones 11, 13, 15, 17 and 19 were obtained in high yields in most of the cases. Thus, the reactivity of sulfur-based alkynes exhibiting different oxidation states was explored, delivering the desired cyclobutenes 11, 13 and 15 (Scheme 5, top). Interestingly, a reverse regiochemistry was observed for sulfinyl- and sulfonyl-cyclobutenes 11 and 13, showing the exquisite selectivity of this transformation, influenced by subtle changes of the substitution pattern. Also, alkynyl precursors 16 and 18 featuring Se- and Te- respectively, formed polyfunctionalized cyclobutenes with moderate to excellent yields (Scheme 5, bottom).30 To gain a better understanding of betaine's reactivity with differently substituted alkynes, haloalkynes 20a and alkynyl ethers 20b were submitted to cycloaddition reaction conditions. No remarkable electronic or steric effects were observed in the formation of chloro-, bromo-, or iodo-cyclobutenes, delivering the expected structures 21a with very good yields in all cases (Scheme 6). The enhanced reactivity of alkynyl ethers 20b led to complex reaction mixtures using standard conditions. Fortunately, lowering the temperature to 0 °C allowed the synthesis and isolation of the corresponding cyclic enol ethers 21b in good yields (Scheme 6).
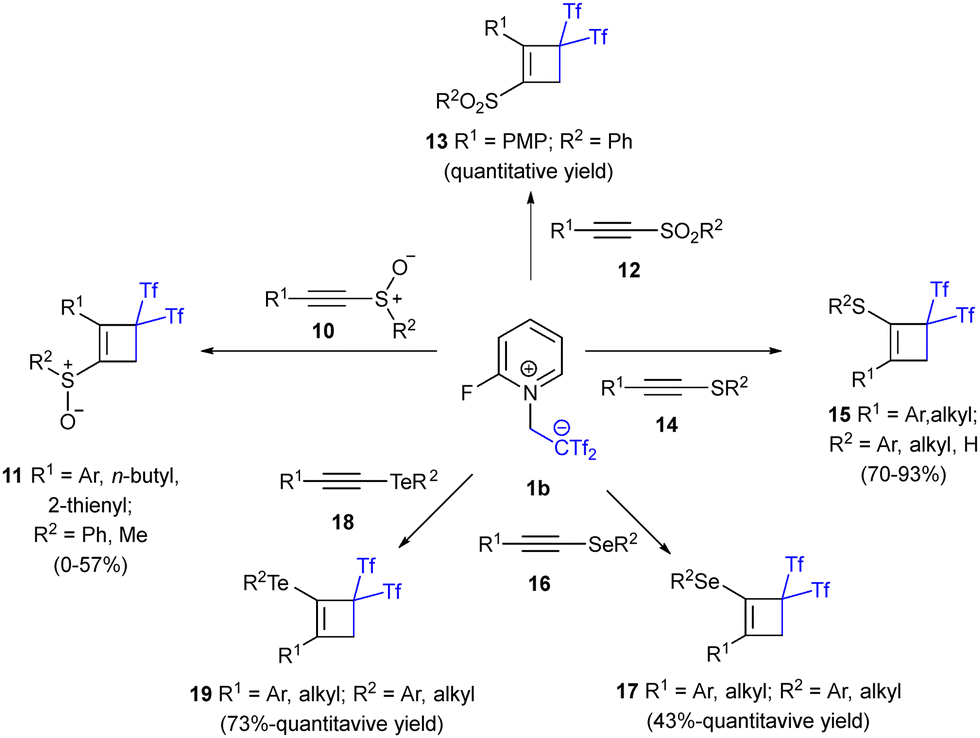 | ||
| Scheme 5 Synthesis of chalcogen-substituted bis(triflyl)cyclobutenes. | ||
 | ||
| Scheme 6 Synthesis of heteroatom-substituted bis(triflyl)cyclobutenes. | ||
Moreover, phosphorus-substituted alkynes 20c and 20d were examined as precursors of functionalized cyclobutenes 21c and 21d. In this transformation, phosphine oxides 20c yielded phosphorylcyclobutenes 21c in excellent yields and in a complete regioselective manner. In addition, thiophosphine oxide 20d produced the desired cyclobutene 21d, showing the same selectivity, although the yield was low (Scheme 6).30,31
Moses and Smedley have reported an interesting addition to the zwitterion-based synthesis of cyclobutenes using compound 1c as a source of ethene-1,1-disulfonyl difluoride (EDSF). Noteworthy, addition of sulfuric acid is needed to achieve full transformation. In these conditions, the reaction shows high efficiency in the presence of both terminal and disubstituted alkynes, and despite the presence of strong mineral acid, good group tolerance was observed (Scheme 7a).32 In a later work, alkynes 12, 14 and 18 bearing heteroatom-based substituents such as S or Te were also nicely transformed into the corresponding cyclobutene scaffolds 13-F, 15-F and 19-F (Scheme 7b).33 Taking advantage of the presence of SO2F groups in final cyclic structures, sulfur-fluoride exchange reactions (SuFEx) were examined. Thus, reaction of cyclobutenes 4-F with phenols under mild basic conditions provided substituted cyclobutenes 4-OAr in good yields (Scheme 7c).32,33
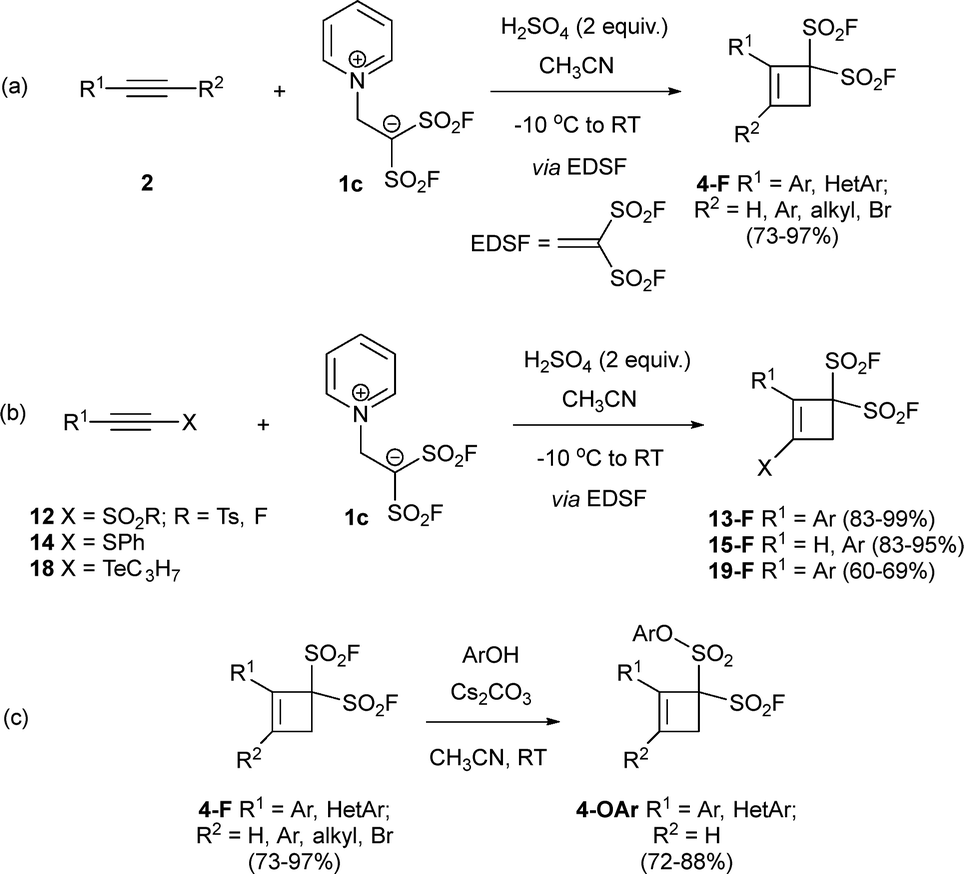 | ||
| Scheme 7 Synthesis of bis(SO2F)cyclobutenes and SuFEx transformations. | ||
2.2. Synthetic applications of bis(triflyl)cyclobutenes
The previous examples prove the versatility and efficiency of the zwitterion-based synthesis of bis(triflyl)cyclobutenes employing betaine 1b and a wide range of alkynes. Besides the inherent reactivity of the highly strained cyclobutene system, with the presence of a bis(triflyl) group embedded in the cyclic core, interesting chemical transformations are enabled. When the alkyne moiety is specifically activated by anisole groups (or electronically related motifs) on one side, and chalcogen- or ynamide-containing substrates on the opposite terminus, an unexpected reaction occurs under the standard conditions. Thus, reaction of alkynes 15, 17 or 19 with zwitterion 1b yielded cyclobutenones 22 instead of the formerly observed bis(triflyl)cyclobutenes. The formation of cyclobutenones appears to be spontaneous, but it can be facilitated by the addition of water and base during work-up (Scheme 8).30 A plausible reaction mechanism for the formation of cyclobutenones 22b, would involve two primary processes: the bis(triflyl)cyclobutene ring construction and an eventual hydrolysis. The proposal for the first process, builds upon previous DFT studies, with the regioselectivity now governed by the electronic effects of the heterocyclic amine, providing cyclobutenes 24 from the ring closing reaction of zwitterionic intermediates 23. Regarding the hydrolysis process, the presence of adventitious water in the reaction medium is essential for the double trifluoro(hydrosulfonyl)methane elimination, yielding hydrates 28. This two-fold water addition is facilitated by the resonance effect of the 4-methoxy substituent in the intermediates 25 and 27. Dehydration eventually occurs in adducts 28, leading to the formation of cyclobutenones 22b (Scheme 9).30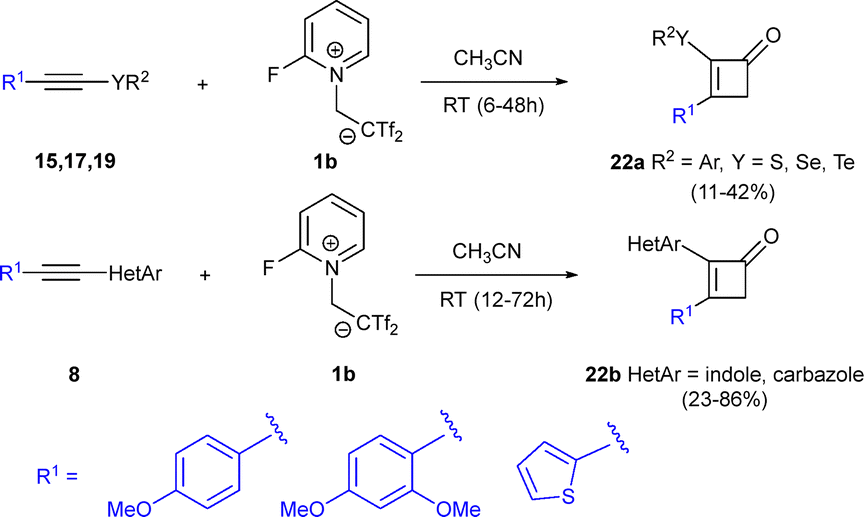 | ||
| Scheme 8 Synthesis of cyclobutenones. | ||
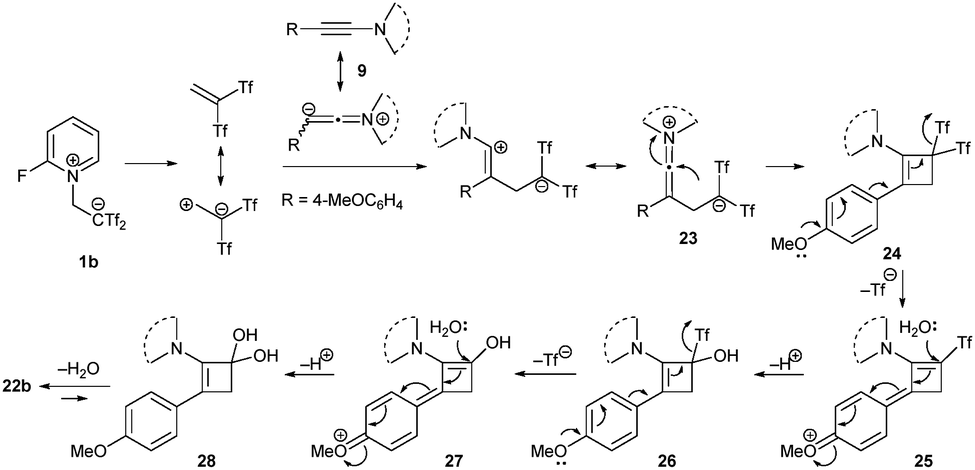 | ||
| Scheme 9 Mechanism proposal for the formation of cyclobutenones 22b. | ||
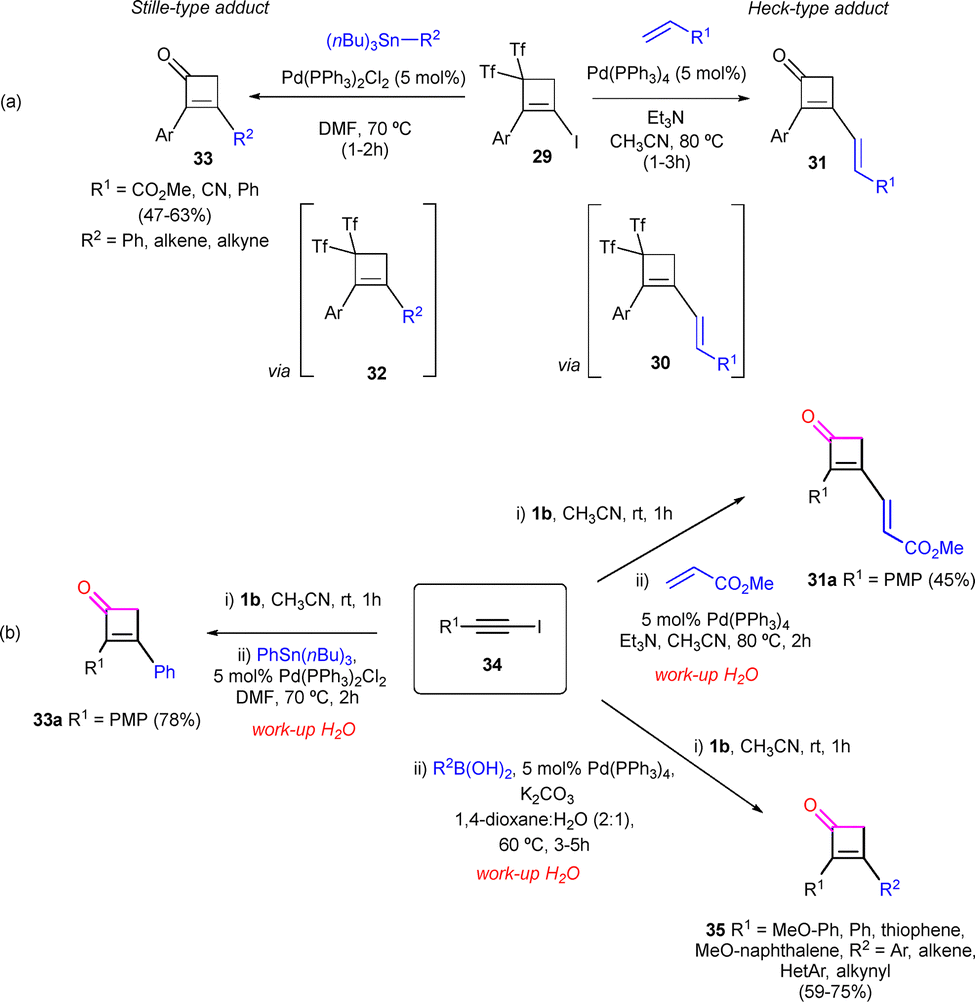
Also envisioned is the use of palladium catalysis to promote both the C–C bond formation and the subsequent ketone formation by addition of water. While iodocyclobutenes 29 exhibited limited reactivity under Negishi conditions; Heck and Stille reactions proceeded effectively, leading to cyclobutenones 31 and 33 from the non-isolated bis(triflyl)cyclobutenes 30 and 32, respectively (Scheme 10a).34 Interestingly, the one-pot version of the Heck an Stille-type reactions was succesfully developed, starting from iodoalkynes 34 and betaine 1b. The formal [2+2] cycloaddition toward the synthesis of the cyclobutene core tollerates the presence of the palladium salts, leading to the expected ketones 31a and 33a (Scheme 10b). Suzuki conditions were also examined in the cross-coupling reaction of in situ-generated bis(triflyl)iodocyclobutenes through a one-pot procedure. This approach allowed the direct synthesis of different functionalized 2,3-disubstituted-cyclobut-2-en-1-ones 35 in good yields from iodoalkynes 34 (Scheme 10b).34
 | ||
| Scheme 10 Pd-catalyzed synthesis of cyclobutenones from alkynes, step-wise and one pot methodologies. | ||
As stated, the zwitterion-alkyne methodology stands out as an efficient and straightforward tool to build bis(triflyl)cyclobutenes. Inspired by the classic Tsuji–Trost reaction, it was envisioned that metal-catalyzed tandem process to access mono(triflyl)cyclobutenes, would lead to a wider range of synthetic applications of Yanai's betaine. Thus, the combination of 5 mol% Pd(PPh3)4 and K2CO3, in 1,4-dioxane facilitated the smooth conversion of bis(triflyl)cyclobutenes 4 into (triflyl)cyclobutenes 36 (Scheme 11a). This transformation exhibited tolerance towards aryl groups with diverse electronic properties, as well as heteroaryl moieties such as indole, carbazole, and indazole. Moreover, heteroatomic substituents such as chlorine, sulfone, and phosphine oxide were effectively accommodated. Notably, the allylic substitution reaction displayed complete regioselectivity, as evidenced by the absence of isomeric products 37. Furthermore, the palladium-catalyzed hydrodetriflylation reaction of bis(triflyl)cyclobutenes 38 bearing alkyl substituents led to the formation of (triflyl)cyclobutenes 40, featuring an additional alkene functionality. This transformation resulted in the generation of 1,4-dienes 40a,b and cyclobutenal 40c through the evolution of 1,3-dienes 39 via a 1,3-H shift (Scheme 11b). Deuterium-labelling experiments and DFT calculations were conducted to elucidate the hydrodetriflylation process.35
 | ||
| Scheme 11 Triflyl-assisted metal-catalyzed transformations. | ||
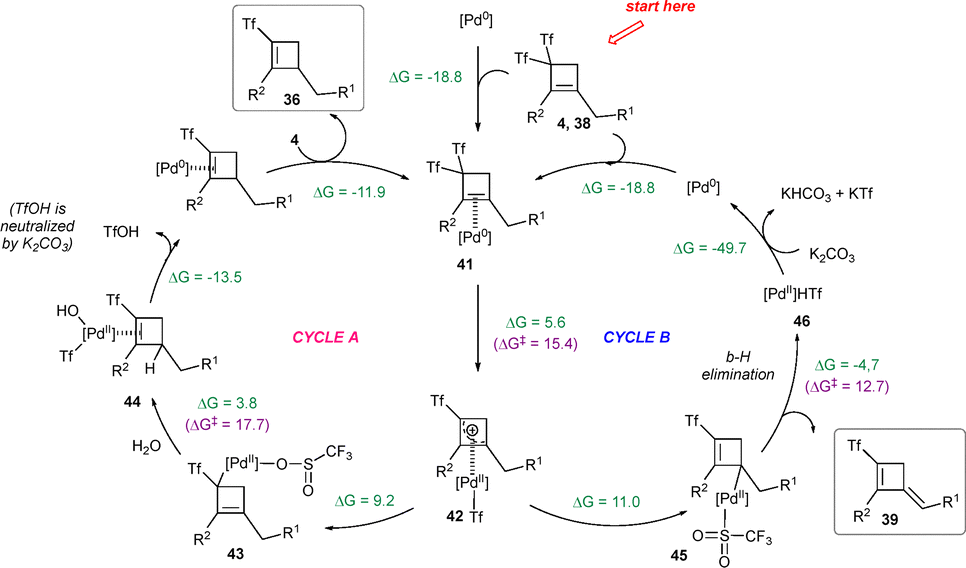
The reaction mechanism would initially start with the coordination of the Pd(0) species to the alkene moiety of bis(triflyl)cyclobutenes 4 or 38 through an exergonic process, yielding intermediate 41. Subsequent oxidative addition would lead to the formation of π-allyl Pd(II) complex 42 with a moderate activation energy. Notably, the formation of two different σ-allyl Pd(II) complexes, 43 and 45, from 42 could be feasible. The energetics suggested an equilibrium between those species in solution. Further investigations revealed distinct reaction pathways from each σ-allyl Pd(II) complex. Starting from 43, water-assisted protodemetallation to yield 44 was nearly thermoneutral, facilitated by a moderately low activation energy. In contrast, β-hydride elimination from 45 to form product 39 and palladium hydride 46 was found to be exergonic with moderately low activation energy. Interestingly, water coordination along the reaction pathways would contribute to stabilize the transition states, suggesting that the calculated activation energies may represent upper limits. Additionally, an overall assessment of the reductive elimination step assisted by K2CO3 would indicate highly favourable energetics. Based on these findings, cycle B involving β-hydrogen elimination was identified as the preferred reaction pathway due to its lower energy barrier compared to alternative cycle A. Cycle A was postulated for (triflyl)cyclobutenes 36 where β-hydride elimination was unattainable (Scheme 12).35
 | ||
| Scheme 12 Suggested catalytic reaction pathway for triflyl-assisted transformations. | ||
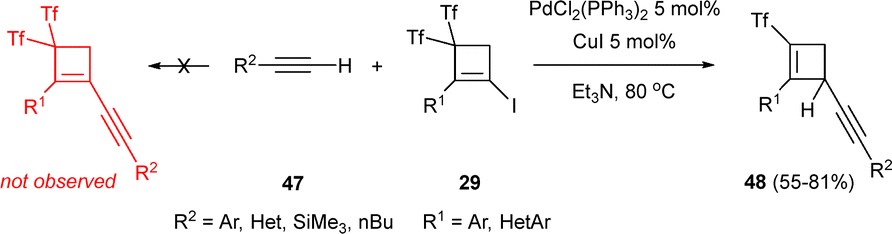
Halocyclobutenes 29 and terminal alkynes 47 were reacted with a Pd–Cu bimetallic system under Sonogashira conditions, leading to adducts 48 through a tandem reaction involving a hydride addition with loss of triflyl group (Scheme 13).36
 | ||
| Scheme 13 Palladium-catalyzed reactions of bis(triflyl)iodocyclobutenes with terminal alkynes. | ||
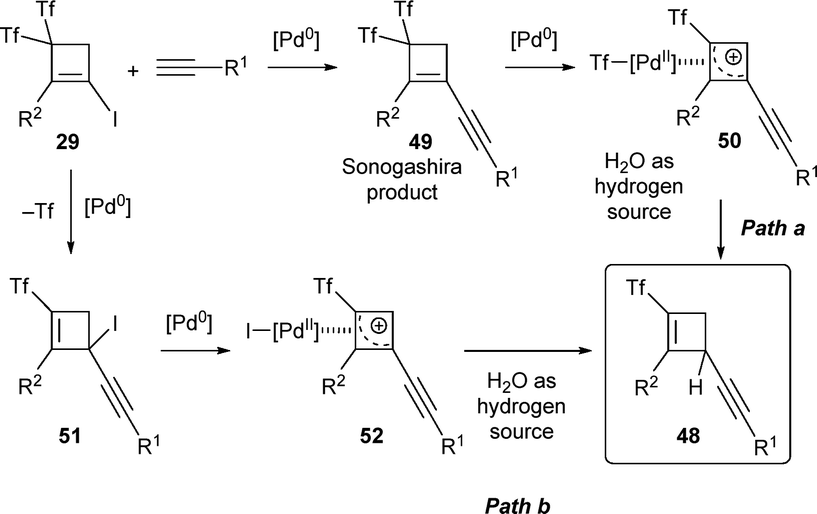
Two plausible mechanisms for the Pd-catalyzed alkynylation reaction were proposed, with experimental and computational data favouring path b (through iodopalladium intermediate 52) over the path a (through triflylpalladium complex 50). Further investigations using isotopic labelling experiments provided insights into the reaction mechanism, highlighting the involvement of water in the process (Scheme 14).36
 | ||
| Scheme 14 Possible reaction pathways for the reaction of iodocyclobutenes with alkynes. | ||
A metal-free approach to the mono(triflyl)cyclobutene skeleton was found using highly activated alkynes and zwitterion 1b in the presence of water as nucleophile. Under those conditions, mono(triflyl)-decorated cyclobutenols have been described through a one-pot strategy. For instance, the reaction of ynamides 6a with 1b yielded cyclobutenols 53a through a cyclization/hydroxylation sequence. The transformation was extended to ynamides bearing heterocycles, such as furan, thiophene, and indole, with moderately electron-rich rings showing better performance. Interestingly, enantiopure chiral ynamides 6b, despite their steric hindrance, afforded aminocyclobutenols 53b with full retention of chirality and good yields. Those results showcase the significant impact of the electronic properties of the starting alkyne and the versatility of the zwitterion-based cyclization methodology. Moreover, they reveal an interesting tolerance of Yanai's zwitterion to the presence of equimolecular quantities of water (Scheme 15).29
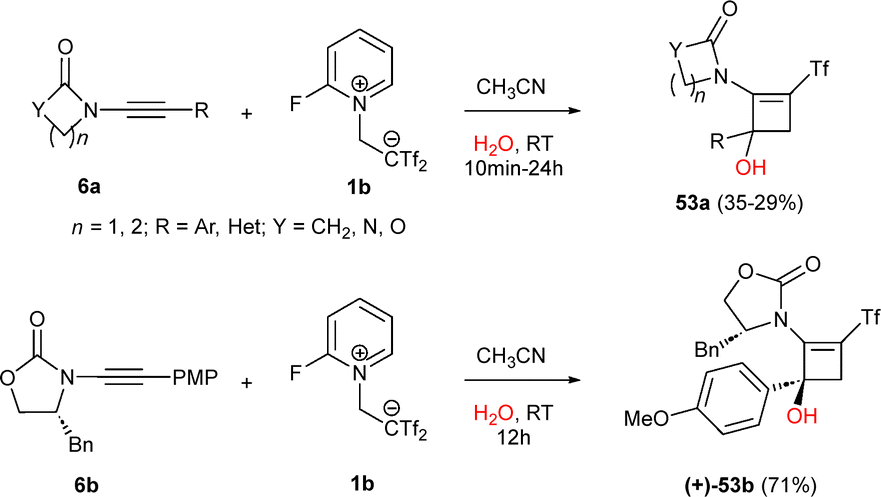 | ||
| Scheme 15 Synthesis of 2-amino-3-(trifluoromethylsulfonyl)cyclobut-2-enols. | ||
The cyclobutanone motif has emerged as a fruitful building block, giving rise to a variety of structures through different reactions. Cyclobutenones 35 were submitted to thermal ring-opening conditions in the presence of trapping reagents such as alcohols or amines. Thus, β,γ-unsaturated ester 54 and amide 55 were respectively obtained in very good yields and full regioselectivity (Scheme 16a).34 Noteworthy, cyclobutenone 35a has been submitted to a wide range of chemical transformations. For instance, a three-step route led to triazole-decorated cyclobutene 56, or β-lactam derivative 57 in one single reaction step. Also, phtalazine 1,4-diol 58 was obtained through after a four-step sequence. The chemical versatility of cyclobutenones 35 was illustrated with the preparation of conjugated ketones 59 or cyclohexa-2,5-dien-1-one 60 (Scheme 16b).34
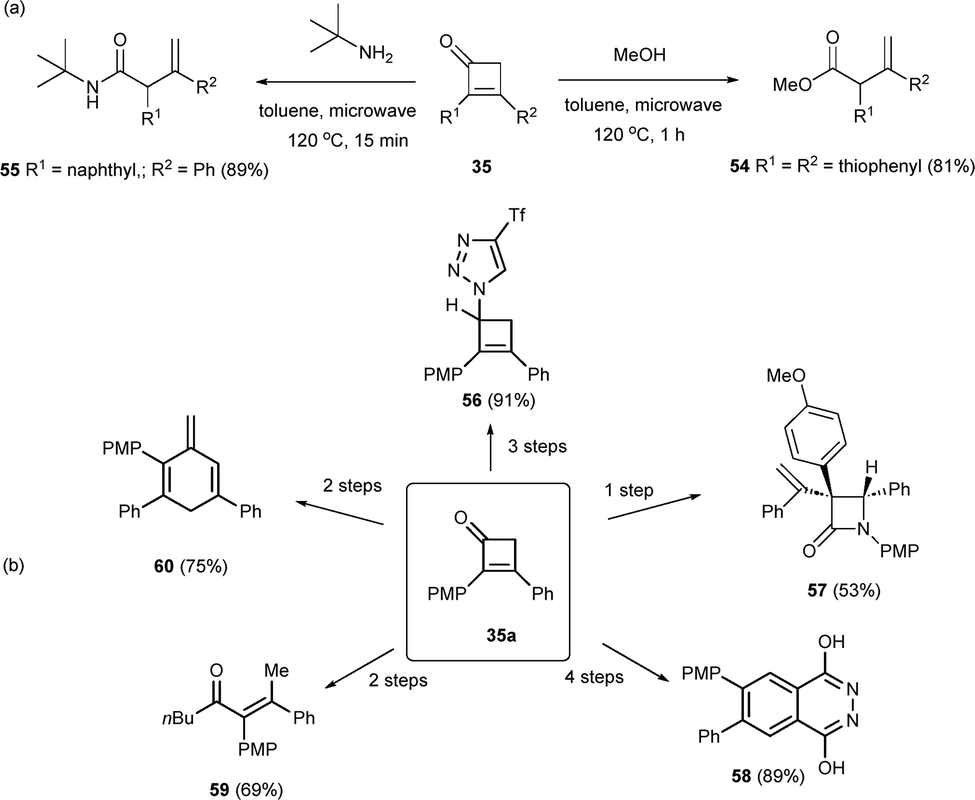 | ||
| Scheme 16 Synthetic transformations of cyclobutenones. | ||
In a recent work, Yanai's research group has presented a desulfonylation methodology of bis(triflyl)cyclobutenes promoted by HFIP and nucleophiles, leading to mono(triflyl)cyclobutene systems.37 Opposite to the examples shown above, Yanai's contribution allows the destriflylation step in the absence of metal species or external nucleophiles and under very mild conditions. Activated arenes are needed to promote the destriflylation reaction. Thus, starting from biaryl-alkynes 61 and 2-fluoropyridinium salt 1b, gem-bis(triflyl)cyclobutenes 62 were obtained in yields from 87 to 96%. Subsequent desulfinative spirocyclization step mediated by HFIP allowed the preparation of spirocyclic compounds 63 (Scheme 17a). In a parallel approach, our group has reported an alternative metal-free synthesis of mono(triflyl)cyclobutenes using Yanai's salt as a synthon for CF3SO2CHCH2. Treatment of differently substituted alkynes with zwitterion 1b under standard conditions, followed by TBAF-assisted hydrodesulfonylation allows the preparation of compounds 65 with good to excellent yields. This innovation permits the synthesis of mono(triflones) instead of the usual bis(triflyl) derivatives using a one-pot strategy, tolerant to a wide number of substituents and functionalities. Interestingly, whereas all other methods to access monotriflones from bis(triflyl)cyclobutenes involve double-bond migration leading to the conjugated triflyl motif, no isomerization was observed in adducts 65 using the TBAF-assisted methodology (Scheme 17b).38
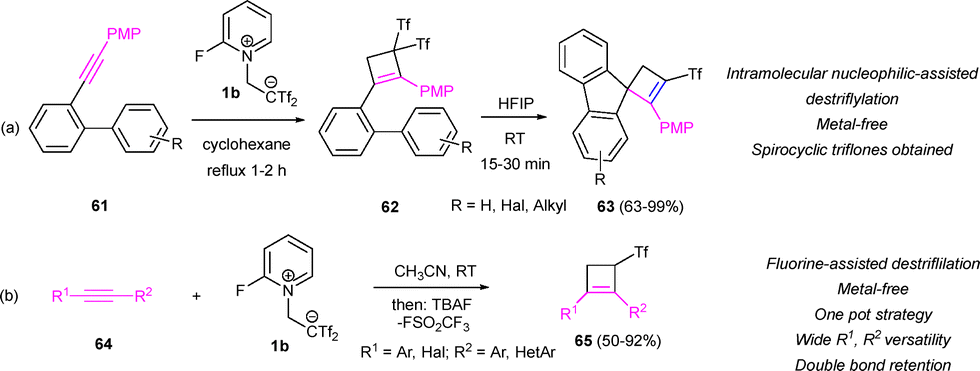 | ||
| Scheme 17 Metal-free desulfonylation of bis(triflyl)cyclobutenes. | ||
In a recent collaboration, Yanai and Almendros have described an interesting application of the bis(triflyl)cyclobutene system towards the direct synthesis of fused [4-7-6] tricycles 68. Readily accessible bisalkynes 66 were treated with betaine 1b yielding monocyclobutenes 67 exhibiting complete chemo- and regioselectivity. The presence of a nucleophile in molecules 67 allowed a further 7-exo-dig heterocyclization reaction catalyzed by gold salts, leading to tricycles 68 as sole reaction products and in a complete stereoselective manner. The regioselectivity in the cyclobutene formation step is influenced by the aryl substituent on the lower alkyne moiety. This substituent stabilizes the carbocation formed during the attack of the alkyne on the highly polar olefin Tf2CCH2, leading to the exclusive formation of cyclobutenes 67 rather than their regioisomer 69. Importantly, the transformation allows the one pot methodology, revealing an orthogonal [2+2] formal cycloaddition/Au-catalyzed heterocyclization reaction. The compatibility observed between gold metal catalysts and betaine 1b constitutes a promising precedent for future transformations (Scheme 18).39
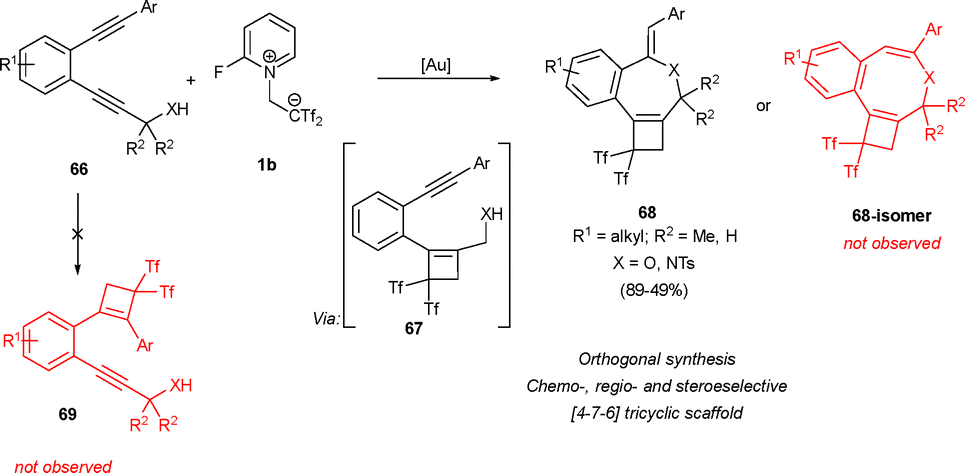 | ||
| Scheme 18 Synthesis of fused [4-7-6] tricycles through a tandem [2+2] cyclization/gold-catalyzed 7-exo-dig heteroannulation. | ||
3. Synthesis of five and six-membered rings
Five- and six-membered cyclic and heterocyclic scaffolds are crucial in bioactive compounds, functional dyes, and advanced materials. Enhancing their molecular functions often involves adding new functionalities, with fluorine-containing groups particularly valued for their unique physicochemical and pharmacological properties.40–42 Efficient organic synthesis aims to transform readily available precursors into target molecules with minimal steps, and waste. In order to explore the reaction of zwitterion 1b with organic azides to access C-triflyl 1,2,3-triazoles via a mild metal-free protocol, differently substituted azides 70 were submitted to diverse reaction conditions to avoid undesired side reactions (Scheme 19a).43 Solvent optimization was critical due to the insolubility of zwitterion 1b in several solvents, leading to heterogeneous conditions. Acetonitrile was found to be the optimal solvent, yielding the desired C-triflyl triazoles 72 in excellent yields and at room temperature. This method also showed high efficiency on a large scale, producing 72 in similar yields.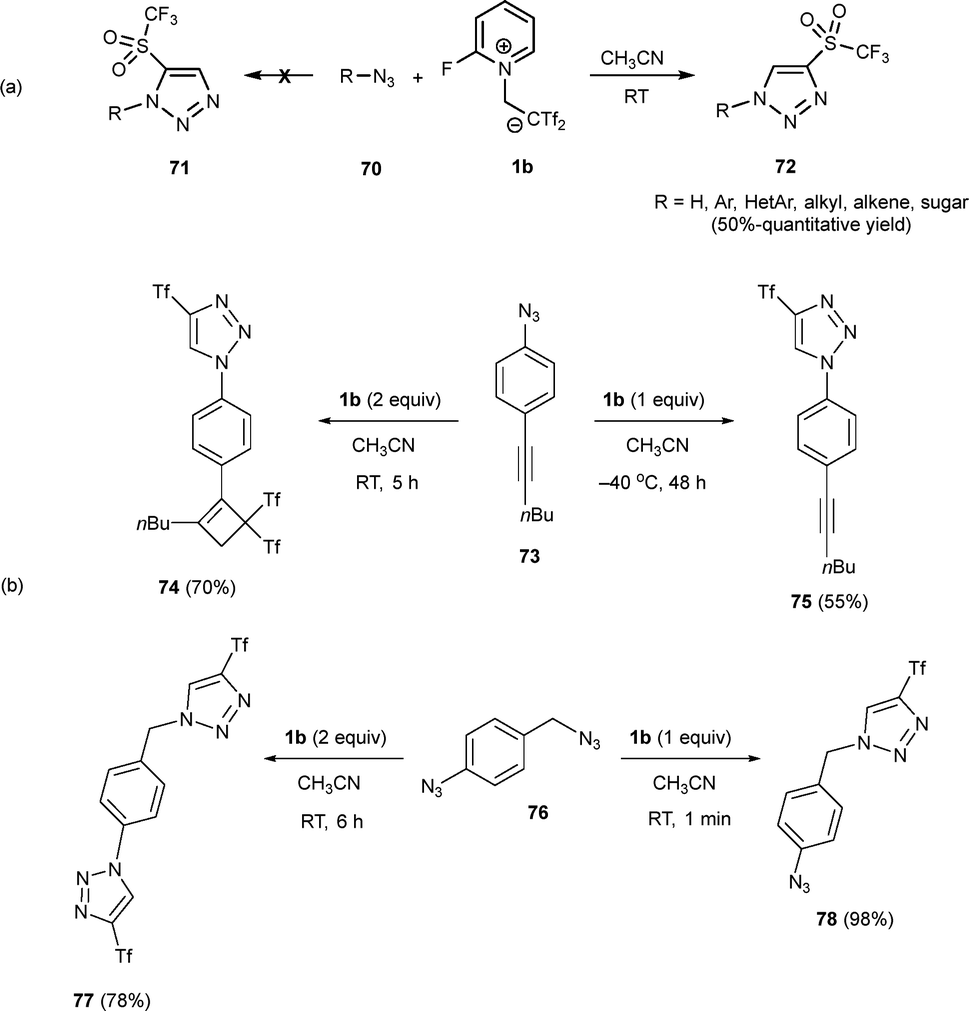 | ||
| Scheme 19 Synthesis of C-triflyl 1,2,3-triazoles. | ||
An interesting chemoselectivity was revealed regarding the reactivity of alkynyl azide 73. Under optimized reaction conditions, cyclobutenyl triazole 74 was obtained from alkynyl azide 73 with good yields as sole reaction product when two equivalents of 1b were employed. Nevertheless, performing the reaction at a lower temperature (−40 °C) and controlling the addition of zwitterion 1b exclusively produced alkynyl triazole 75, showing the higher reactivity of azides versus alkynes. On the other hand, the selective monofunctionalization of diazide 76, containing both aromatic and aliphatic azide moieties, resulted in 4-triflyl triazole 78 using one equivalent of 1b, while a two-fold reaction yielded bis(triazole) 77. The mildness of the protocol allowed for chemocontrol and differentiation in the reactivity of the alkyl azide (faster reaction) versus the aryl azide in 76. This method effectively resolved chemoselectivity issues that are typically challenging with traditional methods (Scheme 19b).43
The formation of 4-trifluoromethanesulfonyl 1,2,3-triazoles 72 from 1b and organic azides 70 would likely involves the initial generation of 1,1-bis(trifluoromethylsulfonyl)ethene from zwitterion 1b. This is followed by a stepwise [3+2] cycloaddition between azides 70 and the in situ generated dipole, forming the zwitterionic species 79. Intermediate 79 would then undergo ring-closure reaction to produce the cycle 80. This intermediate rapidly eliminates trifluoro(hydrosulfonyl)methane, yielding the final 4-triflyl 1,2,3-triazoles 72, driven by the gain in aromaticity associated with triazole formation (Scheme 20).43
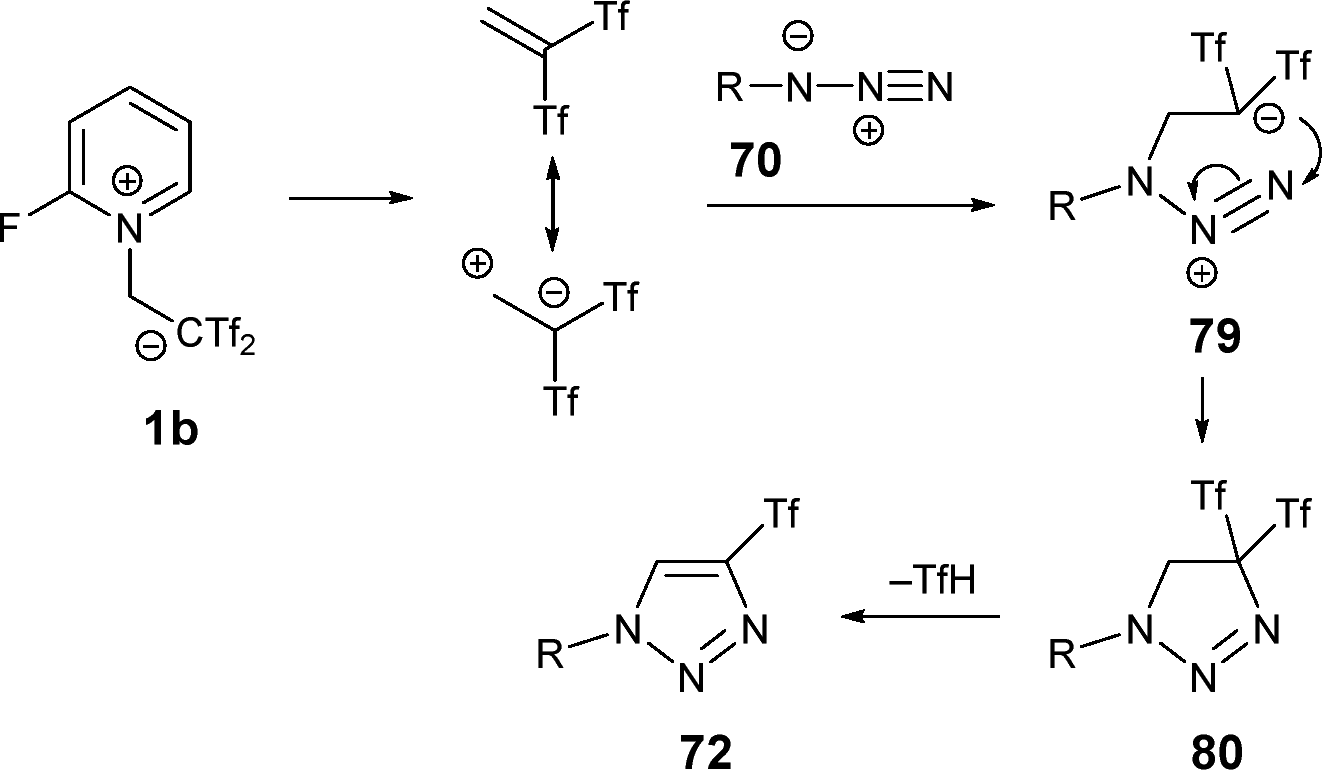 | ||
| Scheme 20 Mechanism proposal for the formation of 4-trifluoromethanesulfonyl 1,2,3-triazoles. | ||
The enhanced reactivity of the allene system compared to the alkyne moiety normally leads to complex reaction mixtures upon exposure of allenes to Yanai's zwitterion 1b. Nevertheless, some examples have been reported when the starting material is carefully designed under the appropriate reaction conditions. Thus, starting with aniline-tethered allenols 81 and zwitterion 1b, indolines 82 have been prepared exhibiting moderate to good yields. The methodology tolerated both aromatic and aliphatic substituents at the allene moiety, and diverse functional groups were well accommodated at the aromatic ring (Scheme 21a). Indeed, allenols 81 behave as activated alkenols, with only the inner double bond of the allene participating in the reaction. With this result in hand, the scope of the transformation was successfully extended to aniline-tethered alkenols 83. When anilide 83 with an electronically unbiased allylic alcohol was exposed to the standard conditions, the desired indoline 84 was obtained, showing similar functional group tolerance and yields (Scheme 21b). The structures of compounds 84 were determined unambiguously by single-crystal X-ray diffraction analysis.44 Tricyclic indolines were submitted to further synthetic transformations, showing the applicability of the protocol. For instance, nucleophilic addition of amines and thiols to 82a occurred in one pot, yielding functionalized derivatives 85 and 86. Interestingly, conjugate diene 87 also served as an excellent dienophile in the Diels–Alder reaction with 2,3-dimethylbuta-1,3-diene, forming 88 stereoselectively, a compound with the tetracyclic core of indole sesquiterpene polyveoline (Scheme 22).44
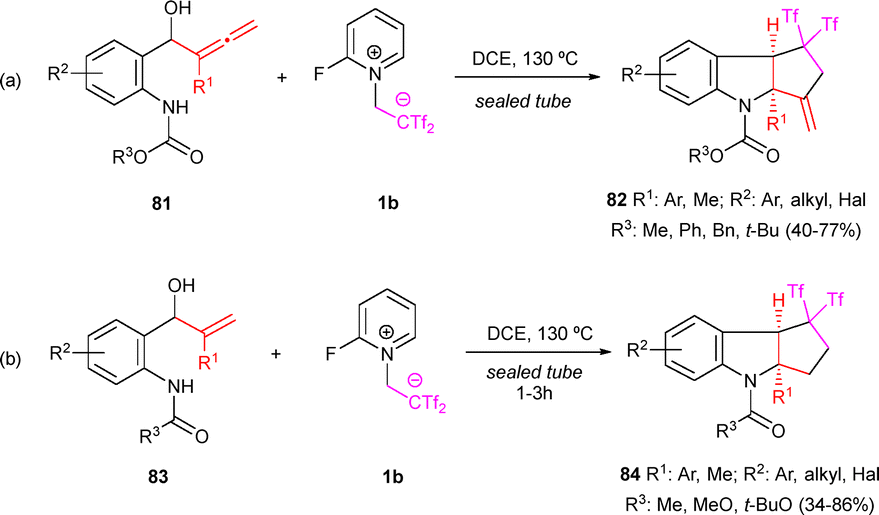 | ||
| Scheme 21 Synthesis of bis(triflyl)-decorated tricyclic indolines. | ||
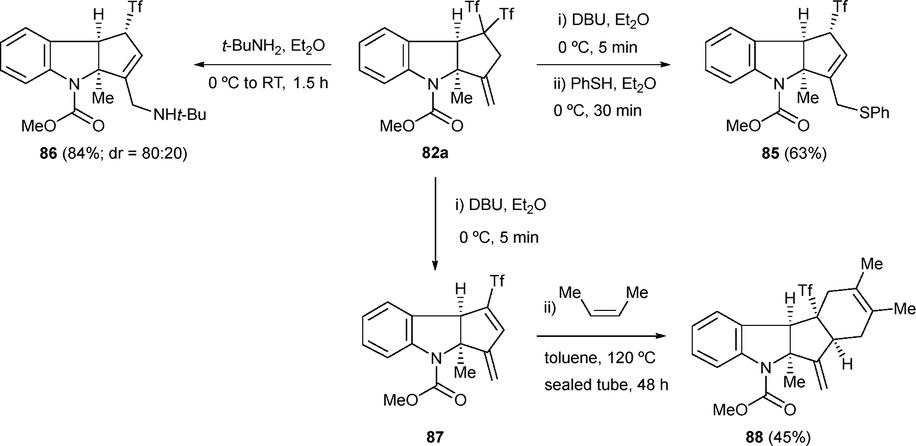 | ||
| Scheme 22 Synthetic transformations of indolines. | ||
The formation of indolines 82 and 84 would start with the nucleophilic attack of the β-carbon from the alkenol (or allenol) moiety to the Tf2CCH2 unit, forming a zwitterionic intermediate 89. This intermediate could cyclize to generate 90, which, after protonation and dehydration, is proposed to form the 2H-indol-1-ium intermediate 92. 92 then would undergo an intramolecular ionic carbocyclization, resulting in gem-bis(triflyl)indolines 82 and 84. The proposed reaction mechanism is supported by DFT simulations, showing a low activation barrier for key steps and explaining the selective formation of cis-fused tricyclic indolines (Scheme 23).44
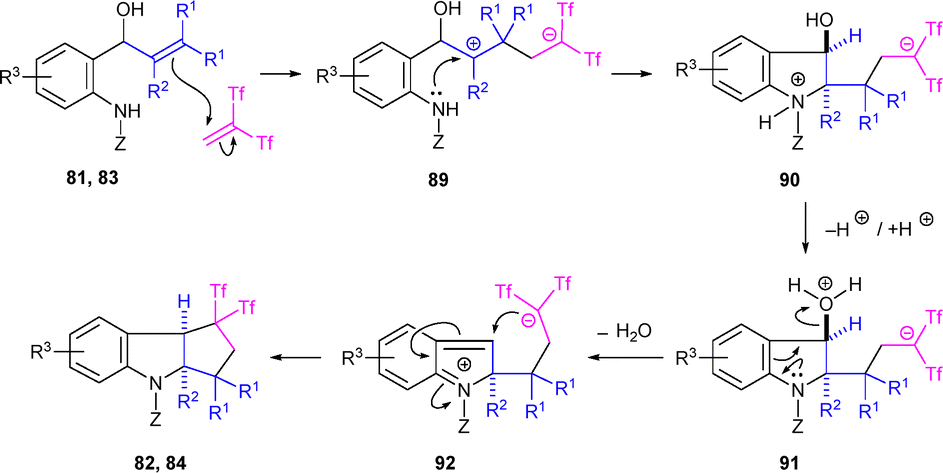 | ||
| Scheme 23 Proposed mechanism for the formation of indolines. | ||
Interestingly, a divergent reactivity was observed for precursor 93 changing the substitution pattern of precursors, resulting in the formation of novel tetrahydroquinoline (THQ) structures 94 through a [4+2] reaction, instead of the indoline structure previously observed. Different substituents at both the amine and aromatic moieties were well tolerated, leading to compounds 94 with good to excellent yields in most cases. Also, diverse alkene substituents (R2) were successfully accommodated (Scheme 24).45
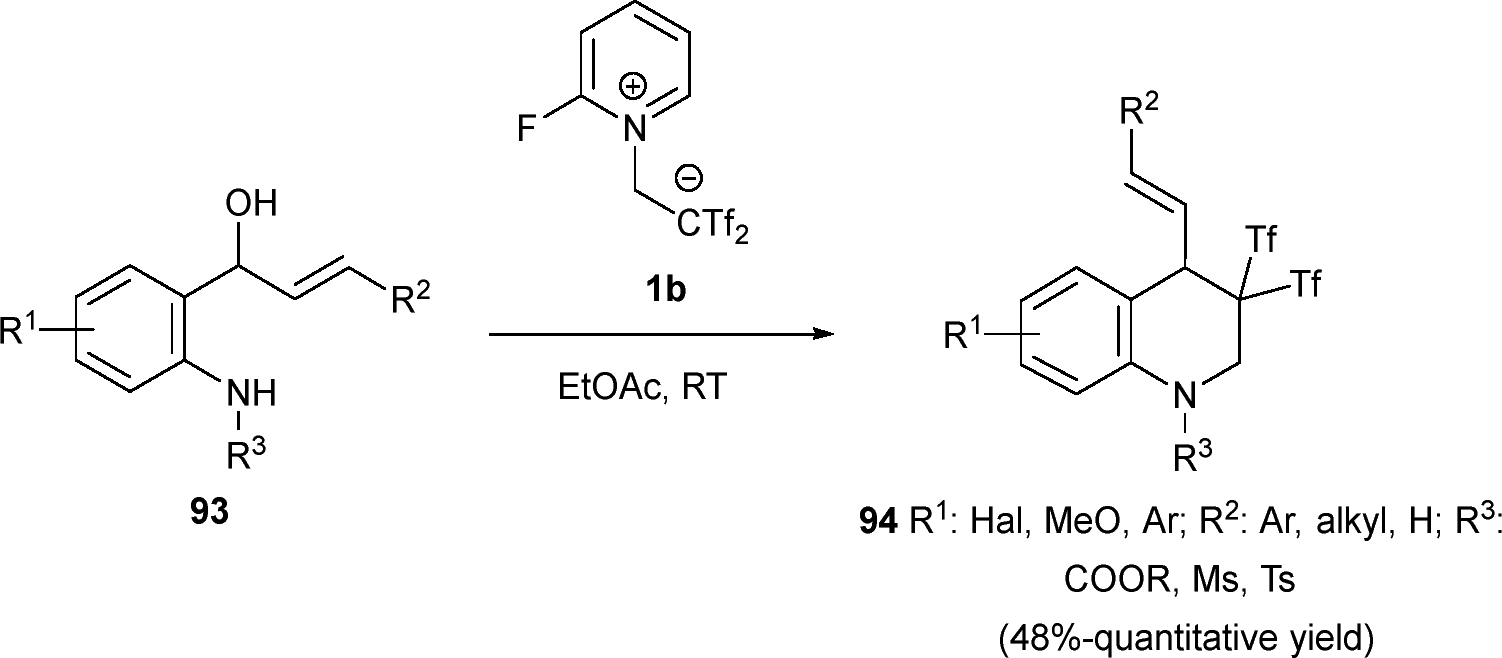 | ||
| Scheme 24 Synthesis of tetrahydroquinolines. | ||
Interestingly, allylic alcohols 95 and allenyl substrates 96 exhibited similar behaviour as 93, yielding THQs 94 and 97 (Scheme 25a and b). However, tertiary allylic alcohol 98 significantly altered the reactivity, resulting in the exclusive formation of bis(triflyl)ethylated dihydroquinoline 99 (Scheme 25c).45
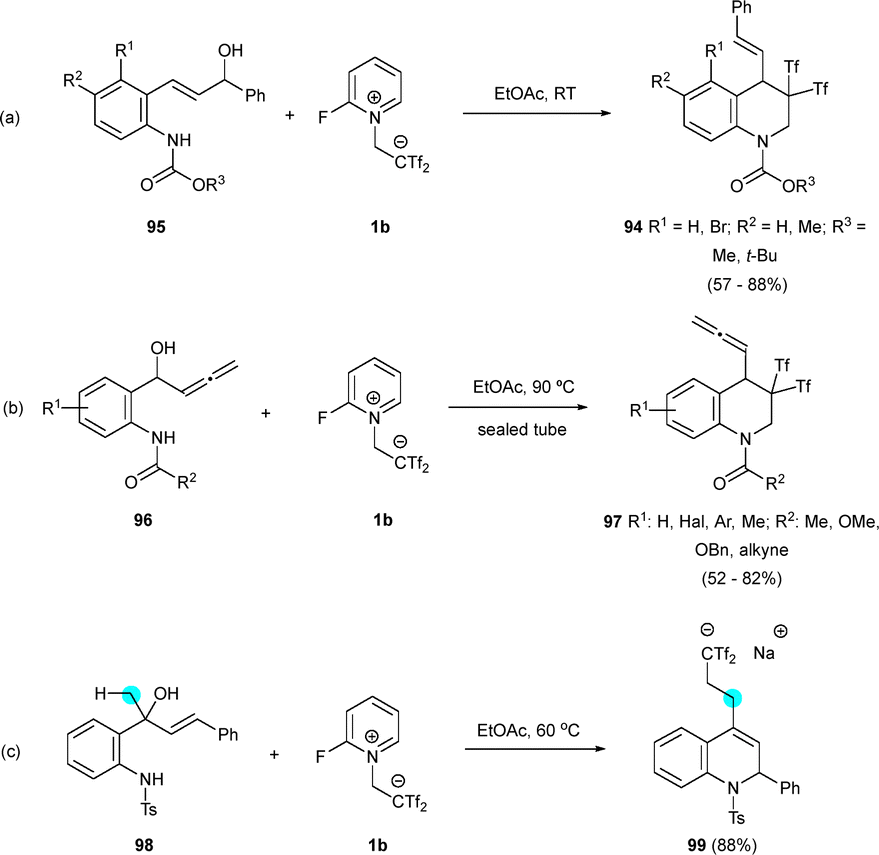 | ||
| Scheme 25 Synthesis of bis(triflyl)ated tetrahydroquinolines and dihydroquinolines. | ||
A possible pathway for the formation of bis(triflyl) tetrahydroquinolines 94 and 97 would start with the generation of aza-ortho-quinone methide intermediate 100 via a dehydration process catalyzed by bis((trifluoromethyl)sulfonyl)methane, which may be formed in situ from betaine 1b in the presence of water. Subsequently, a zwitterionic intermediate 101 could be formed by the nucleophilic attack of the imine double bond in 100 to one molecule of Tf2CCH2. Finally, ring process would lead to the formation of the experimentally observed THQs 94 and 97 (Scheme 26).45
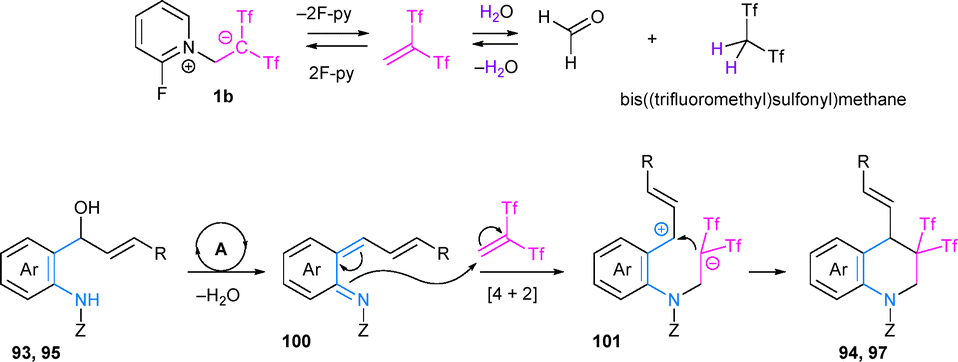 | ||
| Scheme 26 Proposed reaction pathway for the formation of bis(triflyl) tetrahydroquinolines. | ||
The generation of six-membered rings using zwitterion 1b has a smart precedent in a preliminary work from Yanai et al. Bis(triflyl)ethene obtained in situ from the dissociation in solution of betaine 1b reacts effectively with differently substituted dienes through a [4+2] cycloaddition. Thus, bis(triflyl) cyclohexenes 102 can be prepared in good yields in a straightforward manner (Scheme 27).3
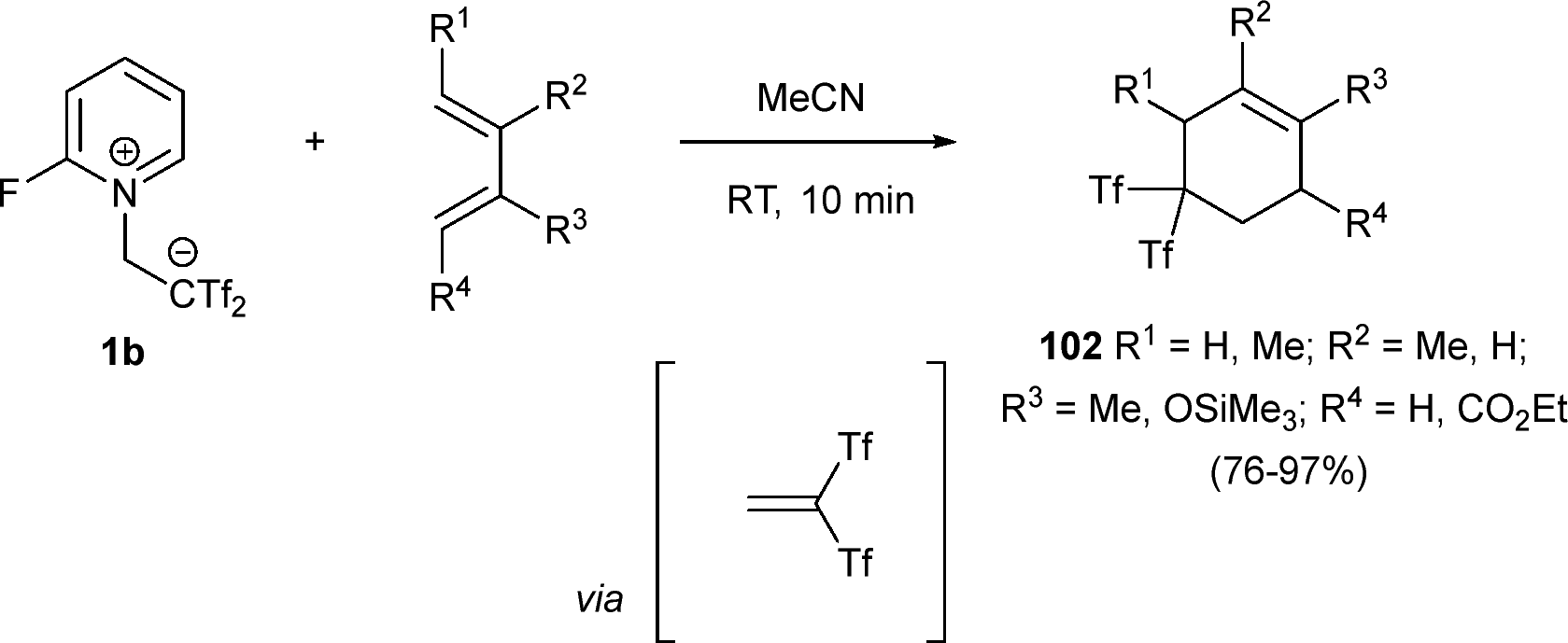 | ||
| Scheme 27 [4+2] cycloaddition reaction of bis(triflyl)ethene and dienes. | ||
In a different approach, bis(triflyl)ethene has been employed as electrophile promoter in intramolecular carbocyclizations, leaving the bis(triflyl) moiety at an exocyclic position in the final structures. Thus, a straightforward synthesis of bis(triflyl)ethyl-linked carbazoles 104 has been described from easily available indolyl alkynols 103 under mild reaction conditions. Interestingly, the strong acidic nature of the proton directly connected to both triflyl groups allows the isolation of carbazoles 104 as sodium or triethylammonium salts, depending on the conditions used during purification. The scope of the bis(triflyl)ethylation/benzannulation reaction was investigated, showing that substituents of different electronic nature and steric hindrance successfully yielded carbazole molecules 104 (Scheme 28a).46 Different starting materials were investigated for alternative electrophile-mediated carbocyclization reactions, such as propargyl ether 105a, leading to 2H-chromene 106 in 95% yield. However, dialkyl alkyne 105b showed significantly lower reactivity, even under harsh reaction conditions (Scheme 28b).46
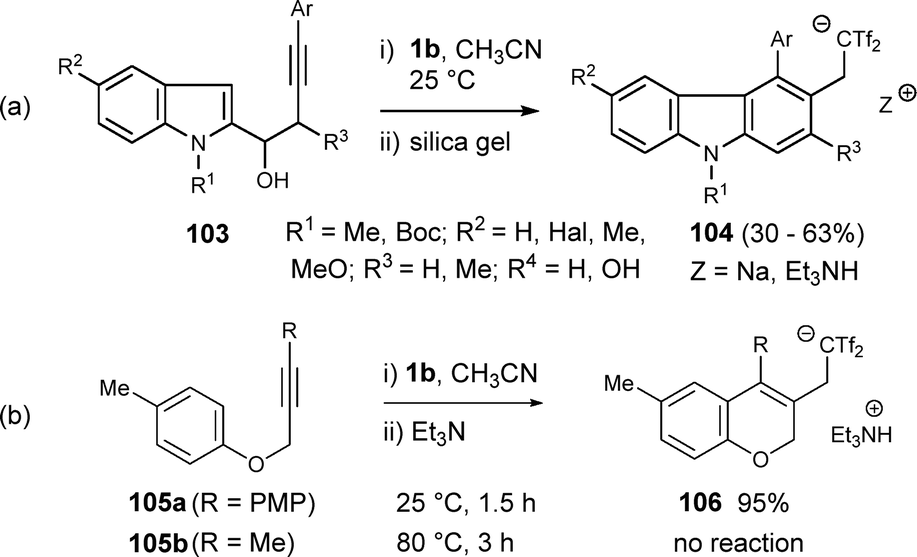 | ||
| Scheme 28 Synthesis of bis(triflyl)ethylcarbazoles and related structures. | ||
A plausible reaction pathway for the formation of carbazoles would start with the reaction of the alkyne moiety of 103 with Tf2CCH2, resulting in the formation of the zwitterionic vinyl-type carbocation 108. Then, a spirocyclization could take place yielding spirocyclic indolinium species 109. Subsequently, fused tricyclic intermediate 110 is formed through 1,2-alkenyl migration within the spirocyclic nucleus of 109. Further aromatization, involving deprotonation and dehydration of 111, would lead to carbazoles 104 (Scheme 29).46
 | ||
| Scheme 29 Proposed reaction mechanism for the formation of carbazoles. | ||
Among cyclic compounds, polycyclic aromatic hydrocarbons (PAHs), have unique photophysical and electronic properties.47–51 Various syntheses for these compounds, including ring-closing reactions of biaryl-alkynes, have been explored. The bis(triflyl)ethene-mediated carbocyclization has also been extended to the formation of PAHs exhibiting two main advantages; milder reaction conditions with no metal catalysts needed, and the incorporation of bis(triflyl) units in the final compounds, providing good solubility in organic solvents. Despite having electronically neutral aromatic rings, substrates 112 led to cyclobutenes 113 in a complex reaction mixture. More activated systems such as compounds 114 effectively provided carbocycles 115 and 116. Interestingly, polar solvents such as MeCN and 1,1,1,3,3,3-hexafluoropropan-2-ol (HFIP) improved selectivity towards the formation of bis(triflyl)-decorated PAHs, with HFIP providing best results, as it likely suppresses the carbanion nucleophilicity (Scheme 30).52
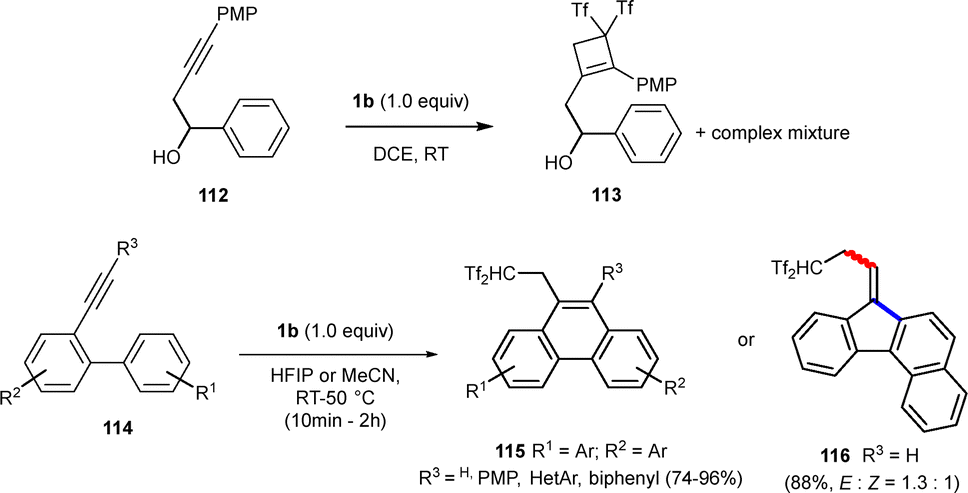 | ||
| Scheme 30 Bis(triflyl)ethene-promoted synthesis of triflyl-decorated PAHs. | ||
Bis(triflyl)ethene-mediated cyclization reactions employing zwitterion 1b have also been described in heterocyclization tandem processes. Thus, 1-[2-(methylthio)phenyl]-3-phenylprop-2-yn-1-one 117 was treated with 1b in acetonitrile at room temperature, obtaining bis(triflyl)thioflavone 118 as sole reaction product, with no evidence of cyclobutene formation or Friedel–Crafts-type bis(triflyl)alkylation adducts in the reaction mixture. This example constitutes a mild formation of a carbon–sulfur bond, present in many natural products and bioactive molecules. Additionally, the organofluorine substituent was incorporated in the same step under metal-free conditions via dual functionalization of the alkyne moiety. The scope of MeS-functionalized ynones 117 was examined to identify those susceptible to thioflavone generation. Various substituents at both the aromatic ring and the terminal alkyne were well tollerated, including heteroaromatic and ferrocene rings. Aliphatic and highly deactivating substituents at the alkyne terminus did not yield the corresponding thioflavones 118, giving only complex reaction mixtures or unreacted starting materials (Scheme 31).53
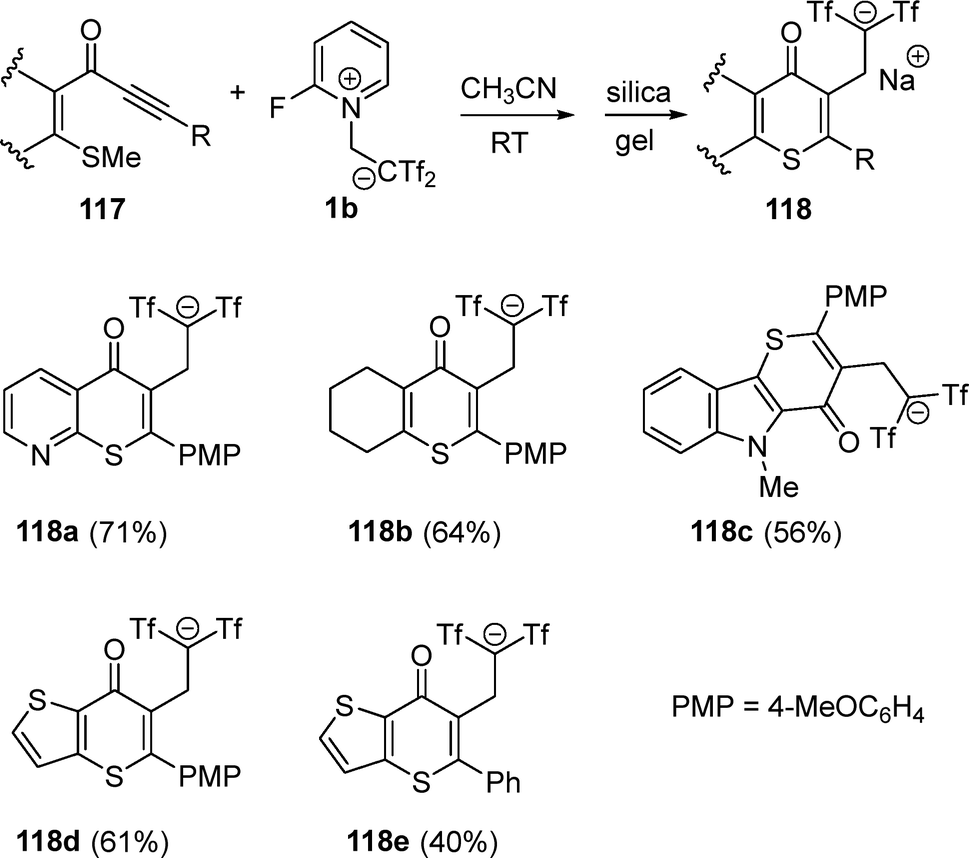 | ||
| Scheme 31 Synthesis of bis(triflyl)thioflavones. | ||
Given the rich chemistry and significant biological properties of organoselenium compounds, selenium-decorated alkynones 119 were also explored as suitable precursor of selenoflavones 121 in the presence of zwitterion 1b. The expected heterocyclic structures 121 were obtained as sole reaction products, exhibiting complete chemo- and regioselectivity, in moderate yields (Scheme 32a).53 The scope of heterocyclization reactions of alkynones with Yanai's reagent was extended to oxy- and aza-derivatives. Both (methoxy)-alkynones and (hydroxy)-alkynones 120 were found to be suitable cyclization precursors. The synthesis of bis(triflyl)flavones 122 was achieved with yields ranging from 56% to 84% (Scheme 32b). By contrast, compounds 123 containing amide functionalities were unreactive under the given conditions, failing to produce triflylated quinolin-4-ones 124 (Scheme 32b).53
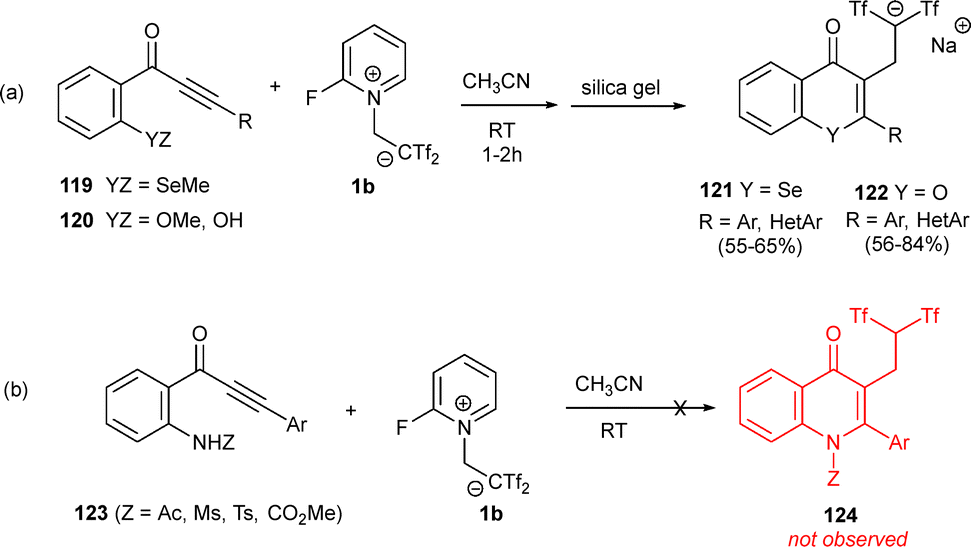 | ||
| Scheme 32 Synthesis of bis(triflyl)flavones. | ||
The above-mentioned heteroatom-decorated alkynones exhibited an interesting dual behaviour in the presence of betaine 1b. While room temperature experiments built the flavone skeleton through an electrophile-mediated heterocyclization, higher temperature experiments revealed a divergent reaction pattern towards unexpected structures. Thus, alkynone 117a (R = Ph) in the presence of zwitterion 1b in acetonitrile at 80 °C, yielded a mixture (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) of the already described bis(triflyl)thioflavone 118a (R = Ph) and novel pyran system 125a (R = Ph). Fortunately, adding zwitterion 1b to a boiling solution of alkynone 117a in toluene exclusively yielded tricycle 125a, proving that simple changes in temperature and solvent can lead to the formation of distinct triflylated heterocyclic cores from a common precursor. Noteworthy, this domino process enables direct, metal-free access to a tricyclic framework, simultaneously forming C–S, C–O, and C–C bonds. Various MeS-alkynones 117 with different functional groups and tethers were subjected to cyclization, resulting in the selective formation of fused thieno[3,2-b]pyrans 125 with moderate to good yields (Scheme 33a). This transformation was successfully extended to selenyl-alkynones 119, giving selenium-containing pyrans 126 with good to excellent yields (Scheme 33b).53
:1) of the already described bis(triflyl)thioflavone 118a (R = Ph) and novel pyran system 125a (R = Ph). Fortunately, adding zwitterion 1b to a boiling solution of alkynone 117a in toluene exclusively yielded tricycle 125a, proving that simple changes in temperature and solvent can lead to the formation of distinct triflylated heterocyclic cores from a common precursor. Noteworthy, this domino process enables direct, metal-free access to a tricyclic framework, simultaneously forming C–S, C–O, and C–C bonds. Various MeS-alkynones 117 with different functional groups and tethers were subjected to cyclization, resulting in the selective formation of fused thieno[3,2-b]pyrans 125 with moderate to good yields (Scheme 33a). This transformation was successfully extended to selenyl-alkynones 119, giving selenium-containing pyrans 126 with good to excellent yields (Scheme 33b).53
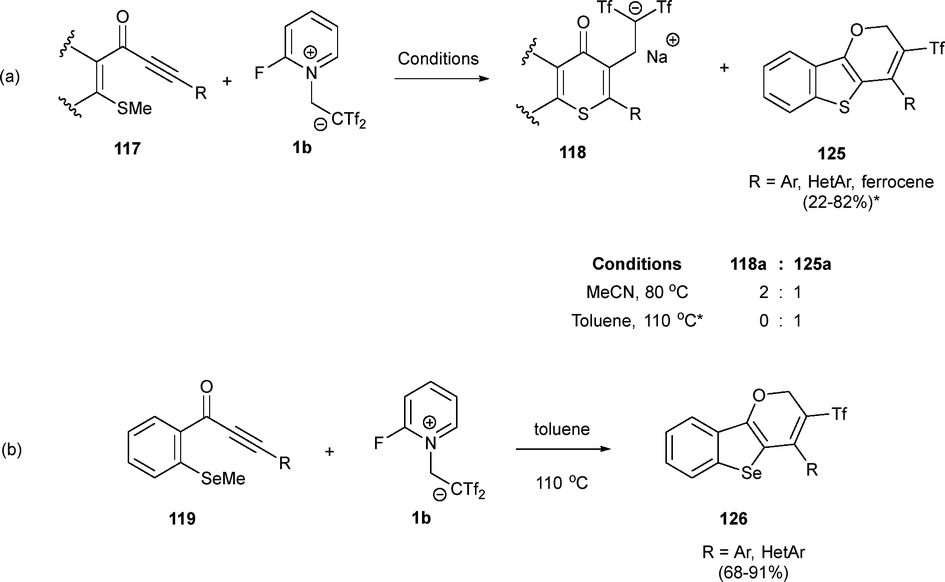 | ||
| Scheme 33 Synthesis of tricyclic triflyl-benzothienopyrans. | ||
Interestingly, treating hydroxy-alkynones 120 with zwitterion 1b in boiling toluene did not directly yield the expected tricycles due to the formation of several unstable products, with spirocyclic cyclobutenes 127 being the only isolated compounds in low yields (Scheme 34a). Nevertheless, hydroxy-alkynones 120 efficiently yielded (triflyl)vinyl aurone-type products 129 in the presence of a base, likely enhancing the nucleophilicity of the hydroxyl group (Scheme 34b). This unexpected result could tentatively be explained by considering bond lengths. The C–S and C–Se bonds are larger than the C–O bond, which may disfavor the final cyclization in this last case. An intriguing result was found when cyclobutene 127 bearing a 4-methoxynaphthyl substituent was heated in acetonitrile at 110 °C in a sealed tube, giving compound 129 in quantitative yields, and pointing to spirocyclic cyclobutene species as plausible reaction intermediates (Scheme 34c).53
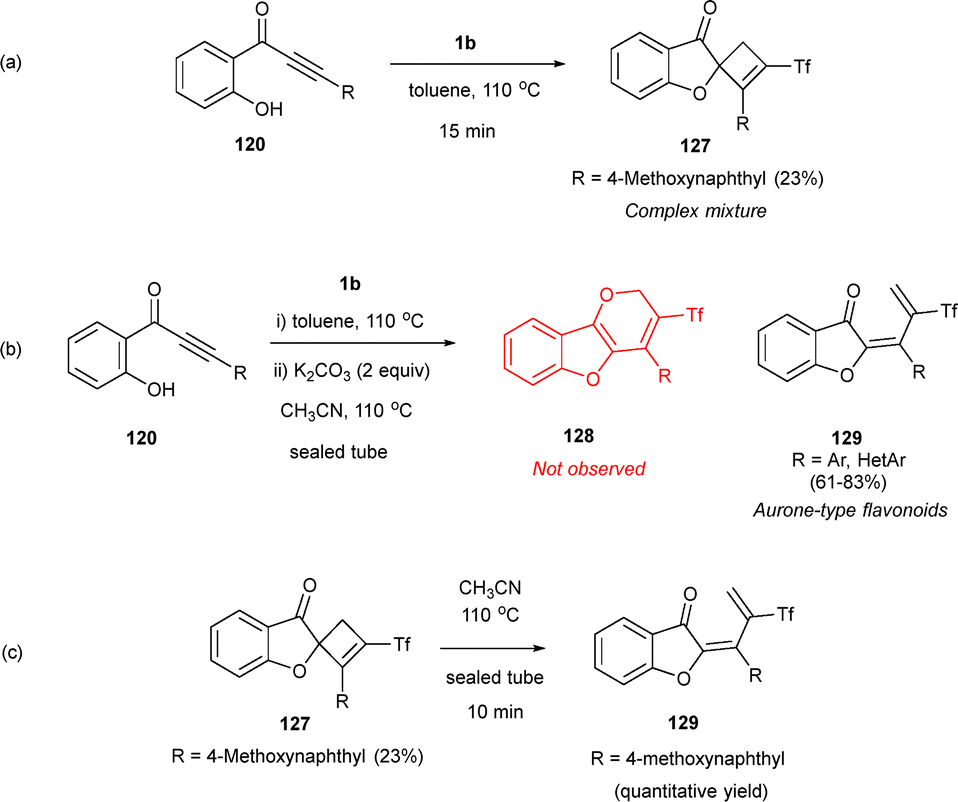 | ||
| Scheme 34 Synthesis of triflyl-allylidenebenzofuranones and spirocyclic cyclobutene. | ||
To understand the divergent formation of the tricyclic pyran skeleton versus bicyclic isoflavone molecules, we attempted the conversion of isoflavone 118a into fused tricycle 125a under thermal conditions. Nevertheless, heating a toluene solution of bicycle 118a at 110 °C did not result in any reaction (Scheme 35a). Notwithstanding, when thioalkynones 118 were submitted to the thermal rearrangement during short reaction times, novel cyclobutenones 130 were observed instead. Interestingly, treating an acetonitrile solution of 130d and 130e in a sealed tube at 110 °C led to complete conversion to pyrans 125d and 125e (Scheme 35b). This suggests that cyclobutenes 130, rather than bicycles 118, are intermediates in the formation of fused pyrans 125.
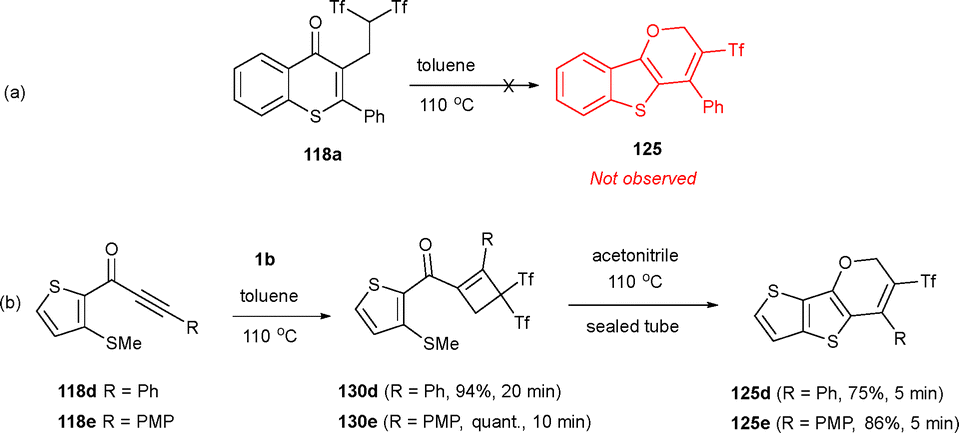 | ||
| Scheme 35 Thermally-induced experiments towards a mechanistic proposal for the synthesis of tricyclic pyrans. | ||
A plausible reaction mechanism which may explain those results would start with the nucleophilic attack of propynones 117, 119 or 120 to one molecule of 1,1-bis((trifluoromethyl)sulfonyl)ethene, generating zwitterionic species 131, which may evolve through two divergent reaction pathways. In one hand, selective intramolecular heterocyclization would form isoflavone bicycles 118, 121 or 122 (path A). Alternatively, a different mechanistic pathway involving carbocyclization to generate cyclobutenes 132 may operate (path B). Subsequent regioselective nucleophilic addition with loss of Tf group could yield spirocyclic cyclobutenes 127, which upon ring-opening would form dienone intermediates 129. Finally, 6π-electrocyclic ring-closure produces tricyclic triflones 125 or 126 (Scheme 36).53
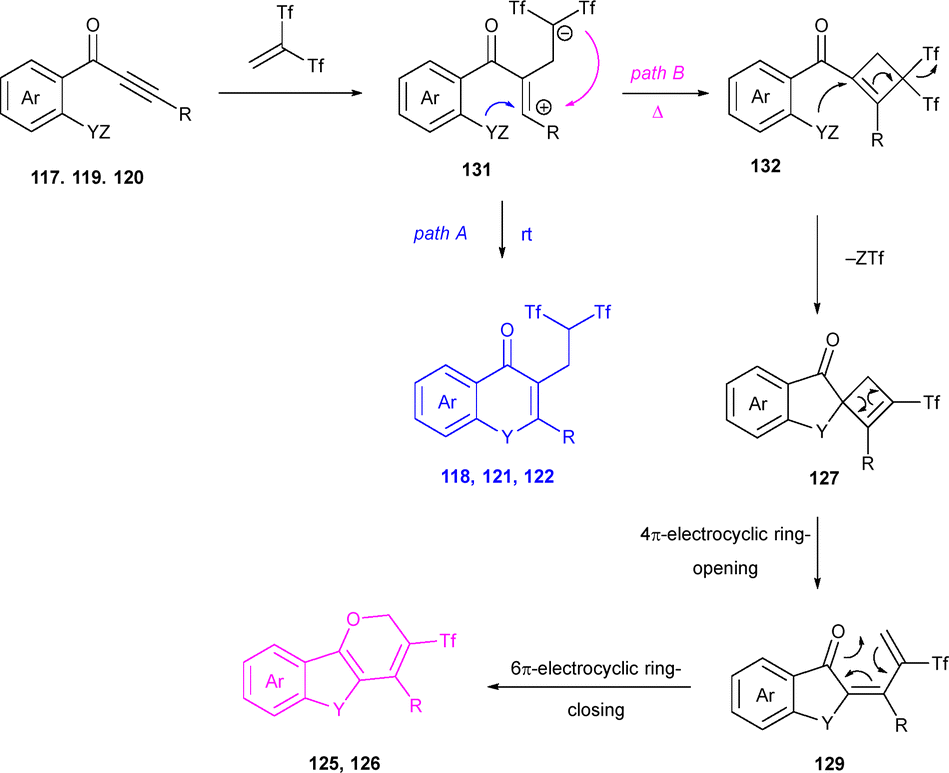 | ||
| Scheme 36 Mechanistic proposal for the synthesis of the isoflavone and pyran skeletons from alkynones. | ||
Flavone derivatives are known to be potent anticancer drugs.54,55 Thus, structures 118, 121, 122 were tested as novel anticancer drugs. The inhibitory nature of the synthetic flavones and bioisosteric analogues against P-gp was confirmed. The rhodamine 123 accumulation assay showed a dose-dependent inhibition of P-gp by these compounds. Notably, some compounds (with R = 2-methoxynaphthalene) exhibited potent inhibitory activity, surpassing verapamil. These findings suggest that the flavone derivatives and bioisosteric analogues can be effective P-gp efflux pump inhibitors for MDR cancer treatment. Additionally, all tested compounds showed promising properties as adjuvants in cancer therapy due to their P-gp efflux pump-modulating activity.56
In view to the above-presented results, alkynyl esters 133 were envisioned as readily available precursors of the isocoumarin skeleton, a recurring organic motif with a wide range of biological applications, bearing an extra bis(triflyl) moiety. Thus, reaction of 2-ethynylbenzoates 133 with 1b smoothly provided the expected heterocyclic compounds 134 with moderate to excellent yields, exhibiting complete chemo- and regioselectivity. Different substitution at the arene ring was examined, showing that electron-donating groups provided better yields under milder reaction conditions. Also, steric effects were minimal and only the 6-endo-dig oxycyclization occurred. Interestingly, substitution at the alkyne terminus showed more dramatic influence in the reaction outcome. While activating groups at R2 position efficiently yielded the expected isocoumarins, electron-withdrawing groups, such as deactivated arenes, quenched the oxycyclization process, giving cyclobutenes 135 as sole reaction products (Scheme 37).57
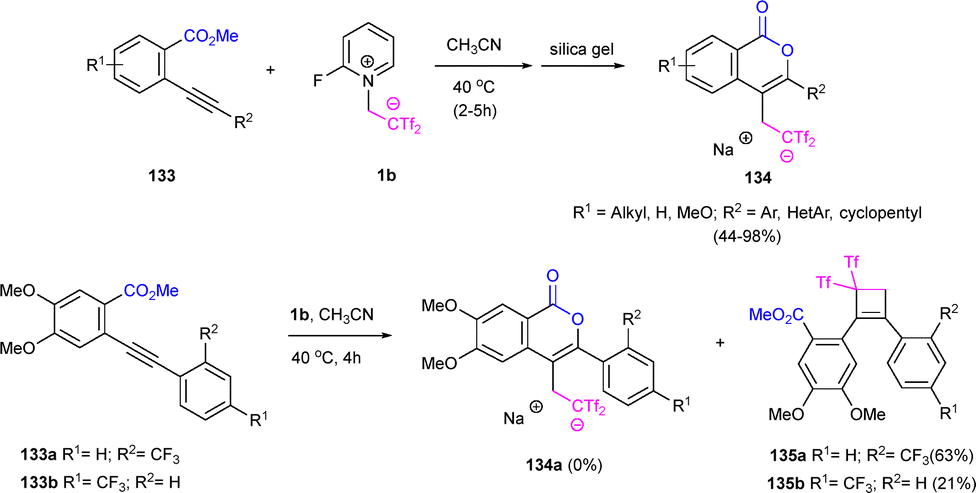 | ||
| Scheme 37 Oxycyclization versus carbocyclization of alkynyl esters. | ||
Given the significance of nitrogen heterocycles in natural products and pharmaceuticals, related 2-ethynylbenzamides 136 and 2-ethynylbenzenesulfonamides 138 were examined as cyclization precursors. Benzamide-derived arylethylenes 136 successfully formed isoquinolin-1(2H)-ones 137 through a 6-endo-dig azacyclization/functionalization cascade. Notwithstanding, benzenesulfonamides 138 underwent intermolecular [2+2] cyclization, producing cyclobutenes 139, presumably due to the lower nucleophilicity of the sulfonamide group. These results underscore the unpredictable yet highly selective reactivity between betaine 1b and functionalized alkynes (Scheme 38).57
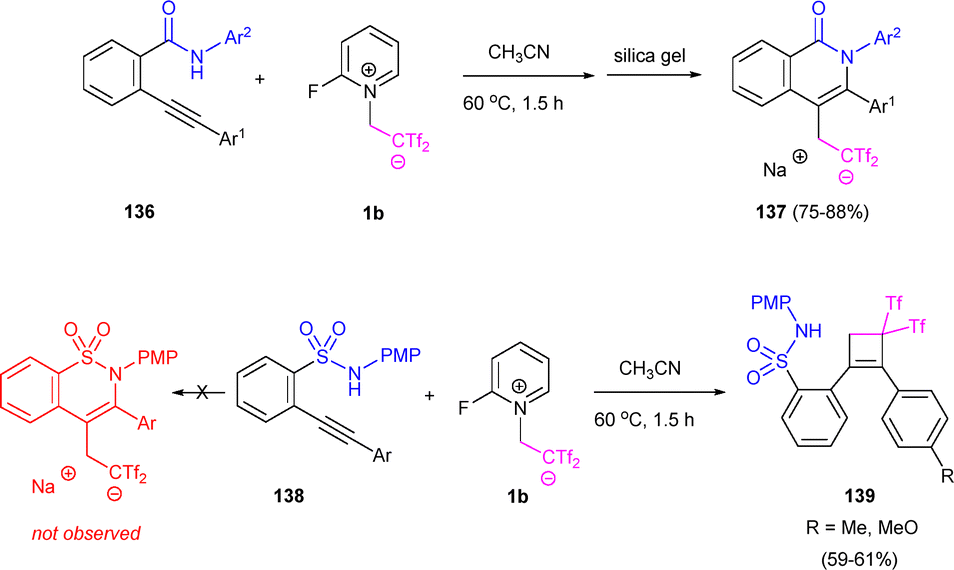 | ||
| Scheme 38 Aza-cyclization versus carbocyclization of alkynyl amides. | ||
A possible reaction mechanism would start with the nucleophilic attack of the alkyne moiety in compounds 133 to the highly polar olefin Tf2CCH2, generating zwitterionic species 140. Then, annulation across the carbonyl oxygen, facilitated by methoxy group conjugation, would yield intermediate 141, stabilized by resonance with species 142. Subsequent water addition at the carbonyl carbon could lead to species 143, which may eventually evolve into 3,4-disubstituted isocoumarins 134-H by releasing methanol. The regioselectivity towards the 6-endo-dig path is attributed to the preferential formation of intermediate 140, with the positive charge closer to the more activating aromatic moiety R2, supressing a plausible 5-exo-dig path (Scheme 39).
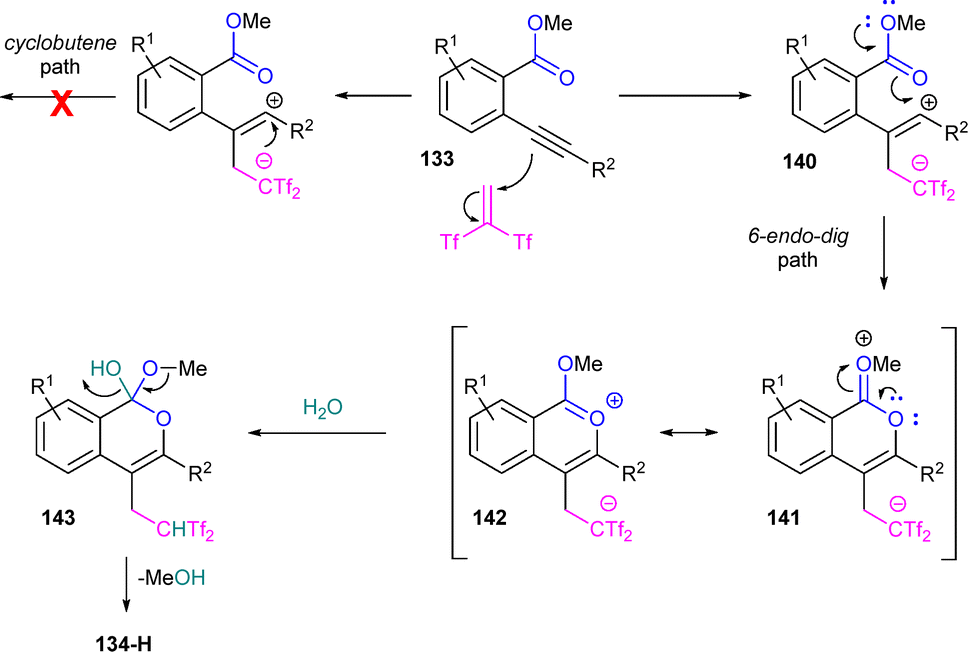 | ||
| Scheme 39 Proposed reaction mechanism for the functionalization-oxycyclization reaction of alkynyl esters and Yanai's reagent. | ||
The biological potential of the isocoumarin family, particularly in neurological disorders, has been extensively studied.58,59 Recently, 4-isochromanones with fluoroaryl groups have shown promising activity as Alzheimer's disease inhibitors by interacting with human acetylcholinesterase (hAChE).60 It was hypothesized that compounds 134, featuring flexible fluoroalkyl substituents, could serve as alternative hAChE inhibitors for docking studies. Molecular docking of compounds 134a and 134b revealed distinct binding modes within hAChE. In mode I, 134a exhibited higher affinity for the catalytic active site (CAS) due to interactions with key amino acids, while in mode II, interaction with the peripheral anionic site (PAS) is preferred. Interestingly, the more sterically demanding 134b adopted a conformation akin to 134a in mode II, favoring PAS interaction over CAS. This suggests that less substituted compounds, like 134a, may have better access to the active site (Fig. 1).57
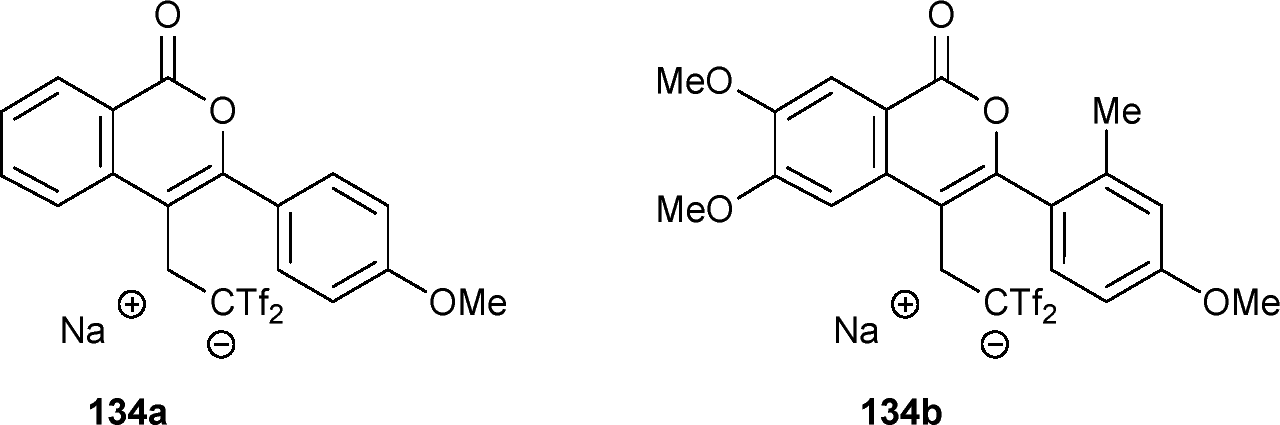
 | ||
| Fig. 1 Amino acids at the binding site of hAChE interact with ligand 134a in mode I (a) and ligand 134b (b). | ||
4. Non-cyclic products
The utility of zwitterion 1b extends beyond the formation of cyclic products, as it enables the addition of the Tf2CCH2 group to carbon- and heteroatom-based nucleophiles, providing access to diverse functionalization. In this context, Yanai's research group has invested great effort building the pillars of the zwitterion chemistry regarding alkyltriflylation procedures and ene reactions. Those transformations allow the synthesis of different molecular structures exhibiting the group CHTf2, with a super acidic proton conferring interesting properties to final compounds. In a preliminary work, zwitterion 1b was tested as an efficient trifluorosulfonyl-alkylating agent in the presence of p-cresol (144a) as nucleophile. A remarkable 92% yield was observed in final compound 145a, compared to an 8% yield using X = H derivative 1a (Scheme 40a).61 This result encouraged the group to prepare a family of carbon acid structures from electronically activated arenes 144 (Scheme 40b), ketones 146 (Scheme 40c) or Grignard reagents 148 (Scheme 40d).3
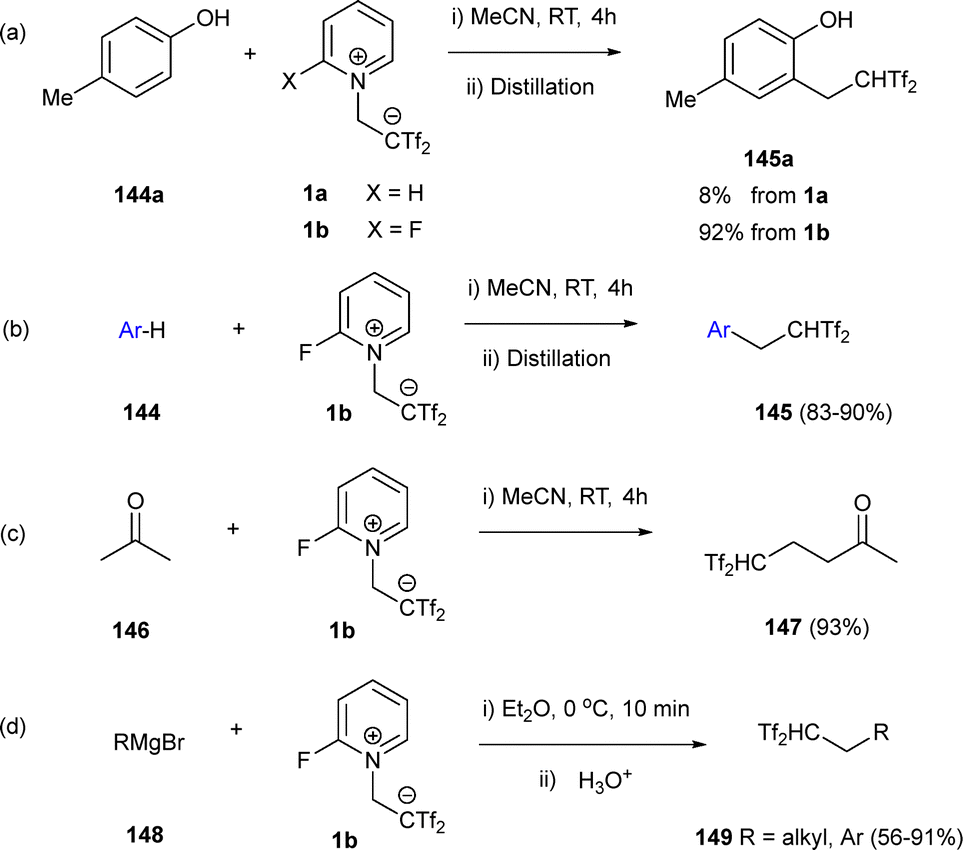 | ||
| Scheme 40 Synthesis of carbon acids through nucleophilic addition reactions with zwitterion 1b. | ||
The same group has reported the first example of a reaction between betaine 1b and alkene systems 150. Alkenes exhibiting hydrogen atoms at β-position proceed through an ene rearrangement in the presence of 1b to yield carbon acid structures 151. Both aromatic and aliphatic alkenes are suitable for this transformation, expanding the family of molecules showing the CHTf2 group (Scheme 41).62
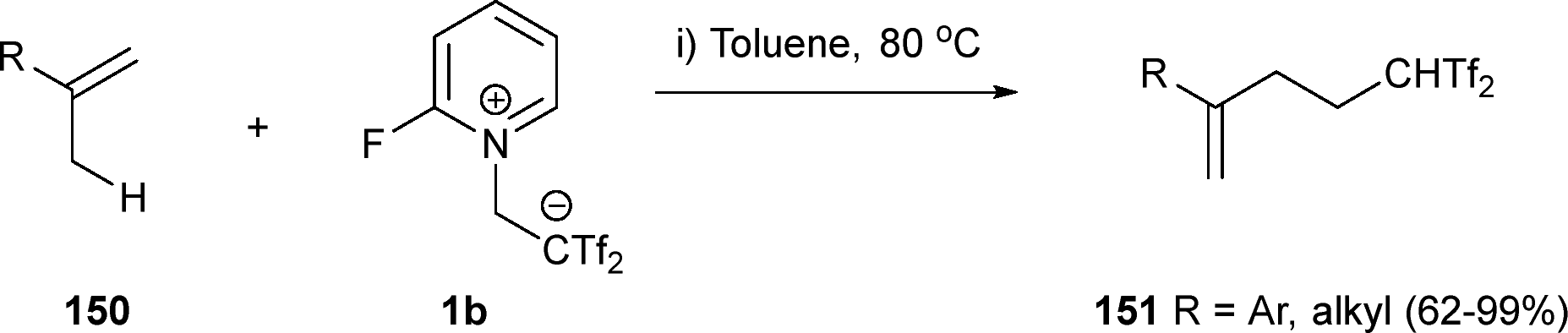 | ||
| Scheme 41 Alkene-ene reaction with zwitterion 1b. | ||
The application of zwitterion chemistry towards the synthesis of carbon acids has recently expanded into polymer and materials science, where it has been employed in post-polymerization modifications to introduce bis(triflyl)methyl groups onto material surfaces, enabling the development of superacid-functionalized membranes with catalytic activity.63 In a recent report, it has been presented an approach that utilizes a naked carbanion as counteranion in bis(trifyl)-based polybetaines and preliminarily unveiled their antifouling properties against human whole blood.64 Boron-dipyrromethenes (BODIPYs) 152, fluorescein 154 and aminocoumarin 156 have been successfully submitted to 2,2-bis(triflyl)ethylation reaction using betaine 1b providing carbanionic dyes. Notably, compounds 153, 155, and 157 showed an unusual balance of high hydrophilicity and lipophilicity, while maintaining significant fluorescent activity (Scheme 42).65 Moreover, Yanai and co-workers have developed structural variations of the reagent, such as 4-substituted 2-fluoropyridinium zwitterions, which not only allow efficient in situ generation of Tf2CCH2 but also facilitate recovery of the fluoropyridine byproduct through multiple purification strategies, thus broadening its synthetic utility and operational practicality.66
 | ||
| Scheme 42 Bis(triflyl)ethylation of organic dyes. | ||
In the context of a collaboration between Yanai's and Almendros’ research groups, different heterocyclic scaffolds 158 were selected to react with in situ generated bis(triflyl)ethene olefin delivering bis(triflyl)-decorated molecules 159 with moderate to excellent yield and high selectivity (Scheme 43a).67 In a later contribution, triphenylphosphine and triphenylarsine have been studied as nucleophiles in the presence of zwitterion 1b. Thus, treatment of 1b with Ph3P and Ph3As yielded the heteroatom-alkylated betaines 162 and 163, respectively, in excellent yields under mild conditions (Scheme 43b). In addition, reactions of salt 1b with phosphorus ylides allowed the access to 1,4-carbabetaines. For instance, treatment of both unstabilized and substituted ylides 164 with fluoropiridinium salt 1b afforded compounds 165a and 165b in good yields (Scheme 43c). Spectroscopic and computational analyses confirmed that these compounds exhibit pronounced carbabetaine character, opening pathways for their application in organocatalysis and beyond.68
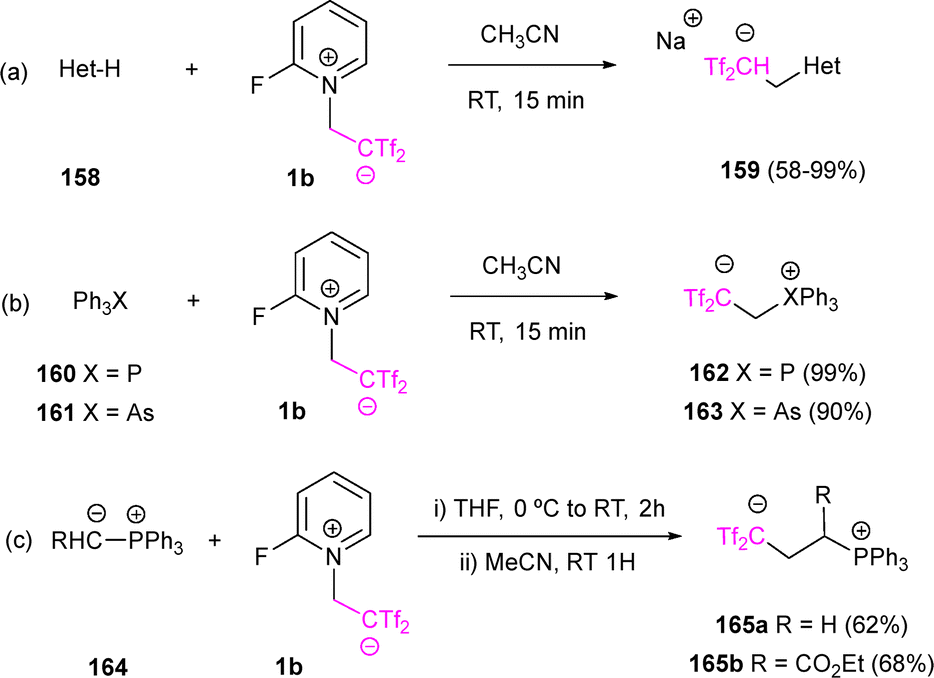 | ||
| Scheme 43 Synthesis of 1,3- and 1,4-carbabetaines. | ||
In a different transformation, the reaction of α-allenols 166 with reagent 1b in acetonitrile at room temperature resulted in the unexpected formation of (E)-α,β-unsaturated ketones 167, with no cyclization observed. This outcome likely arises from the electrophilic attack of Tf2CCH2 on the terminal sp2-hybridized carbon of the starting allene. Interestingly, the (Z)-isomer was detected as a minor isomer only in some cases. Due to the acidic nature of the hydrogen in the Tf2CH group, products 167 were isolated as sodium salts after purification by column chromatography. The scope of the reaction was explored with differently substituted allenols at R1 and R2 positions. Allenols with both aromatic and aliphatic substituents were effectively converted into bis(triflyl)enones 167 in good yields. However, allenols tethered to heterocycles, such as indole and oxindole, yielded complex mixtures due to side reactions between the arene moiety and Tf2CCH2. Remarkably, allenols with quaternary carbinolic centers also performed well, delivering stereoselective (E)-isomers of bis(triflyl)enones in most cases (Scheme 44a).69
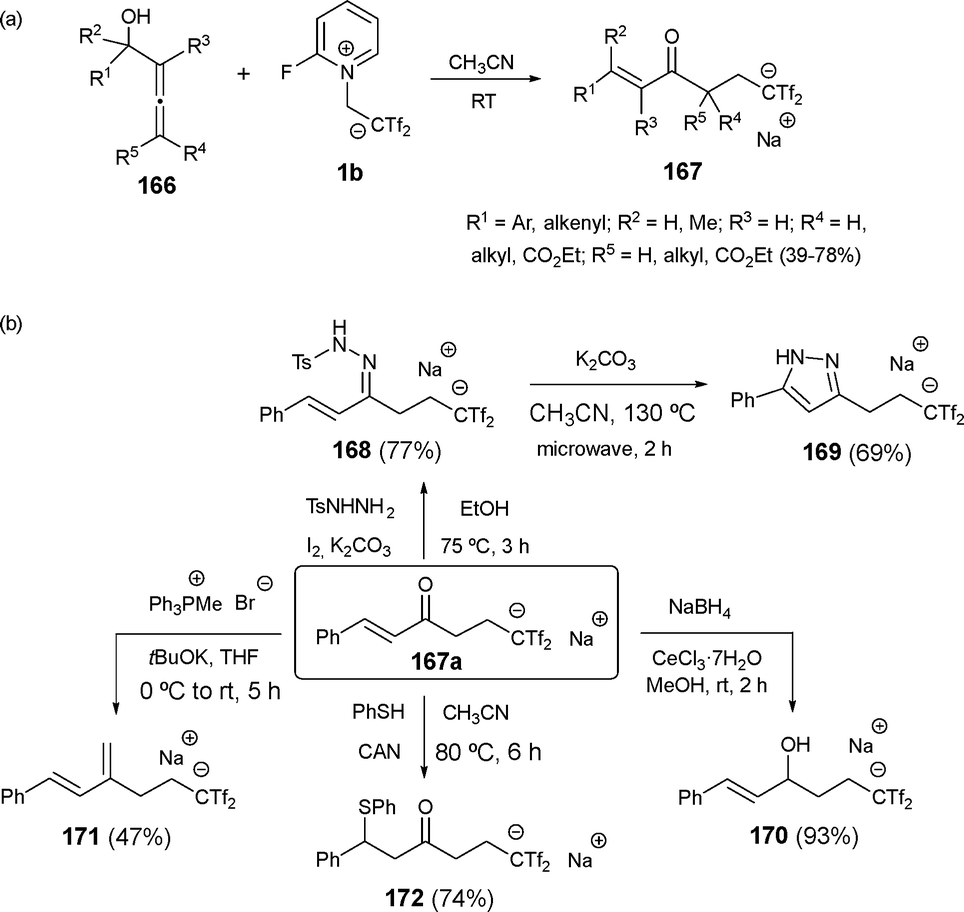 | ||
| Scheme 44 Synthesis of bis(triflyl)-functionalized enones and synthetic transformations. | ||
Functionalized enones 167, containing versatile groups, provide an excellent platform for further derivatization. For example, α,β-unsaturated ketone 167a was transformed into bis(triflyl)propyl-decorated pyrazole 169 via N-tosylhydrazone intermediate 168 in a two-step process. The chemoselective reduction of the carbonyl group in 167a afforded bis(triflyl)allylic alcohol 170, while Wittig olefination provided the bis(triflyl)ated 1,3-diene 171. Additionally, a radical 1,4-addition of benzenethiol to enone 167a produced thioether 172, among other chemical transformations (Scheme 44b).69
As previously noted, zwitterion 1a and the in situ generated bis(triflyl)ethene exhibit chemical compatibility with gold salts, suggesting their potential in tandem transformations. Moreover, a separate transformation involving allenols 166, betaine 1b, and gold catalysts unveiled an unexpected reaction pathway, affording sterically encumbered β-methylene-δ,δ-bis(triflyl)pentanals 173. Different allenols 166 exhibiting diverse substitution patterns were examined. Electron-donating groups like methoxy or methyl on the aromatic ring were well tolerated, as were halogens (Br and Cl). However, steric effects were evident, as ortho-substituted allenols showed slightly lower yields compared to its meta- and para-substituted counterparts. Additional studies with other electron-deficient alkenes, such as ethenesulfonyl fluoride and (ethene-1,1-diyldisulfonyl)dibenzene, resulted in messy reactions, highlighting the necessity of both Tf groups in betaines 1b for successful reactivity (Scheme 45).70
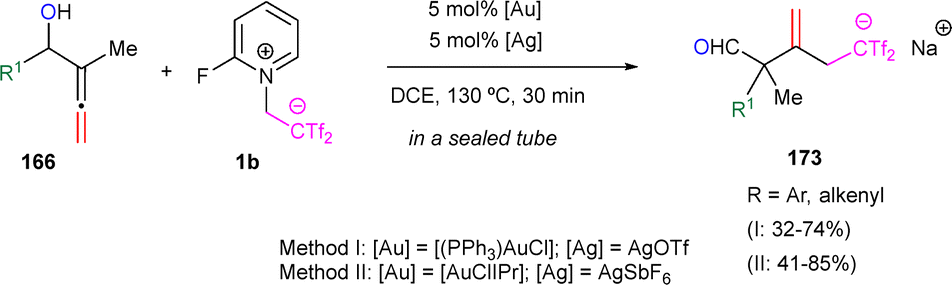 | ||
| Scheme 45 Synthesis of bis(triflyl)enals. | ||
The proposed mechanism for the formation of bis[(trifluoromethyl)sulfonyl]ethylated enals 173 from allenols 166 under gold catalysis is shown in Scheme 46. Initially, allenols form π-activated complexes 174 in the presence of gold salts. Addition of the proximal allenol double bond to the electrophile 1b generates zwitterionic intermediates 175, which undergo a 1,2-aryl migration to form species 176. These intermediates would then experience deprotonation of the aldehyde moiety followed by protonation of the resulting carbanion, leading to neutral gold complexes 177. The final products 173 are obtained after deaurative elimination, with regeneration of the gold catalyst. This pathway, where Au(I) acts as a π-Lewis base, involves an unusual allene activation, strongly supported by DFT calculations (Scheme 46).70
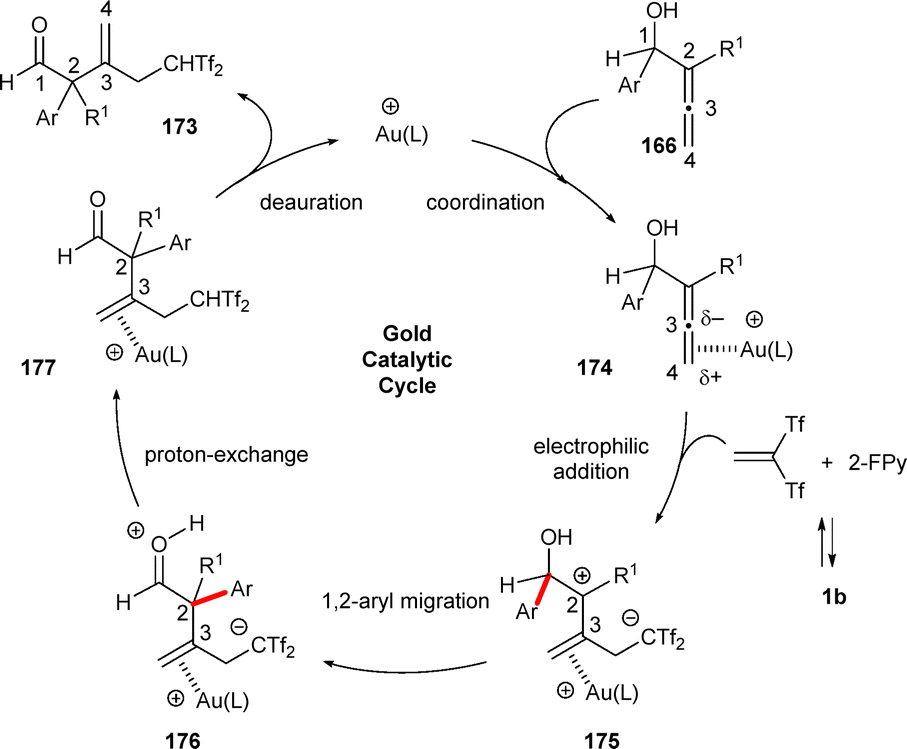 | ||
| Scheme 46 Proposed mechanism for the formation of β-methylene-δ,δ-bis(triflyl)pentanals. | ||
5. Conclusions
Pyridinium-based zwitterions 1 constitute a shelf-stable and efficient source of highly reactive bis(triflyl)ethene molecules. Beyond their industrial utility in polymer synthesis and the earliest theoretical studies, betaines 1 have increasingly featured in a wide range of synthetic applications. Specifically, 2-fluoropyridine derivative 1b has shown the greatest efficiency promoting diverse reaction pathways from different starting materials.Highly strained cyclobutenes are easily synthesize from readily available alkyne systems through a stepwise [2+2] cycloaddition. Opposite to recurring cyclobutene synthesis, the transformation proceeds in one reaction step, under mild conditions and in the absence of metal catalysts. Fluorine atoms have been incorporated not only into the design of new pharmaceuticals aimed at improving human health, but also into the construction of innovative molecular frameworks that broaden chemical space. In this context, the combination of strained carbocyclic motifs with fluorosulfonyl substituents constitutes an attractive approach for the diversification of biologically relevant scaffolds, which may provide access to unprecedented pharmacophores and distinctive privileged architectures.
Also, formal [3+2] cycloaddition between 1b and azides allows the preparation of the triazole scaffold. Besides formal cycloadditions, Koshar-type zwitterions can promote intramolecular nucleophilic addition with incorporation of a bis(triflyl) unit. Thus, different molecular motifs, including flavones, isocoumarins, and pyrans, as well as open-chain systems, have been accessed. Remarkably, an intriguing chemo-, regio- and stereoselectivity is normally observed. In addition, first examples of orthogonal tandem reactions in the presence of metal catalysts reveal a promising chemical compatibility of zwitterion 1b with metallic salts, giving light to complex reaction mechanisms.
Despite the significant advances described above, many chemical avenues remain largely unexplored. In contrast to the established alkyne chemistry, the application of various carbon-nucleophiles—such as allenes, allenamides, or activated alkenes—is currently understudied. The judicious design of these starting materials, along with the proper selection of substituents, will be crucial for achieving novel reaction outcomes and discovering new reaction mechanisms.
Furthermore, the initial reports of tandem methodologies employing gold catalysis point toward the development of sophisticated new transformations that combine metal catalysis, zwitterions 1, and suitable substrates. Since concerted [2+2] processes are thermally forbidden, zwitterions 1 are known to undergo stepwise cycloaddition mechanisms. Therefore, the engagement of Koshar-type zwitterions in photochemical reactions would open a new, exciting avenue for methodologies proceeding through previously undescribed pathways.71 It is also expected that flow synthesis integrated with informatics-driven optimization platforms can be employed in a near future to accelerate reaction development in the synthesis of gem-bis[(trifluoromethyl)sulfonyl] functionalized organic molecules.
Recent advances in synthetic methodologies have enabled the isolation of a broad range of fluorinated sulfonyl carbanion-containing salts, thereby allowing detailed investigations into their chemical bonding features and providing deeper insight into their stabilization modes and coordination properties. On the basis of these fundamental studies, stable bis(Tf)-based carbanions have progressively evolved from purely academic curiosities into versatile functional motifs with broad utility in molecular engineering and materials science. In the near future, these frameworks may emerge as valuable platforms not only for anionic species but also for radical-based systems. Although the currently reported examples have not yet exploited the (Tf2C) fragment as a chiral scaffold or as a ligand framework for transition-metal coordination, continuous progress in synthetic methodologies is expected to considerably broaden its applicability as a modular substituent. The stability of organic compounds incorporating the (Tf2C) moiety is highly dependent on the stereoelectronic characteristics of the neighboring substituents. Accordingly, expanding the structural diversity of stable bis(triflyl)-substituted organic compounds and elucidating their intrinsic properties through molecular-level theoretical studies remain highly desirable goals.
Moreover, the recent successful incorporation of the bis[(trifluoromethyl)sulfonyl]methyl unit into polymers through straightforward post-polymerization modification strategies, together with the preparation of polybetaines derived from (Tf2C)-based carbanions, is expected to significantly broaden the scope of polymer chemistry and open new opportunities for advanced material applications involving polybetaines. Future developments may ultimately enable the preparation of organic membranes bearing strongly acidic carbon-centered functionalities, thus paving the way toward innovative applications, including ion-exchange membranes and highly efficient immobilized organocatalysts.
Finally, the reactions presented herein, combined with these promising perspectives, establish a solid pillar for zwitterion-based chemistry and anticipate the near future arrival of even more exciting results in this field.
Author contributions
Daniel Diez-Iriepa: visualization, investigation and writing – original draft, review; José Miguel Alonso: visualization, supervision, investigation and writing – original draft, review; and Pedro Almendros: conceptualization, supervision, writing – original draft, review.Conflicts of interest
There are no conflicts of interest to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This work was supported by MICIU/AEI/10.13039/501100011033/FEDER (Project PID2021-122183NB-C21) and CSIC (Project 2025AEP049).Notes and references
- L. L. Barber Jr and R. J. Koshar, US Pat., 3962342, 1976 Search PubMed.
- H. Yanai, Y. Takahashi, H. Fukaya, Y. Dobashi and T. Matsumoto, Chem. Commun., 2013, 49, 10091–10093 RSC.
- H. Yanai, R. Takahashi, Y. Takahashi, A. Kotani, H. Hakamata and T. Matsumoto, Chem. – Eur. J., 2017, 23, 8203–8211 CrossRef CAS PubMed.
- H. Yanai, T. Suzuki, F. Kleemiss, H. Fukaya, Y. Dobashi, L. A. Malaspina, S. Grabowsky and T. Matsumoto, Angew. Chem., Int. Ed., 2019, 58, 8839–8844 CrossRef CAS PubMed.
- (a) H. Yanai, Chem. Rec., 2023, 23, e202300076 CrossRef CAS PubMed; (b) H. Yanai, Coord. Chem. Rev., 2026, 554, 217522 CrossRef CAS; (c) H. Yanai, M. Fujita, A. Takahashi, M. Zhang, M. Mishima, A. Kotani, T. Matsumoto and T. Taguchi, Molecules, 2013, 18, 15531–15540 CrossRef CAS PubMed.
- T. Li, C. Zhou, X. Yan and J. Wang, Angew. Chem., Int. Ed., 2018, 57, 4048–4052 CrossRef CAS PubMed.
- D. Zhang, K. Qiao, J. Hua, Z. Liu, H. Qi, Z. Yang, N. Zhu, Z. Fang and K. Guo, Org. Chem. Front., 2018, 5, 2340–2344 RSC.
- M. Nagase, Y. Kuninobu and M. Kanai, J. Am. Chem. Soc., 2016, 138, 6103–6106 CrossRef CAS PubMed.
- F. Wang, X. Yu, Z. Qi and X. Li, Chem. – Eur. J., 2016, 22, 511–516 CrossRef CAS PubMed.
- L. A. Smyth, E. M. Phillips, V. S. Chan, J. G. Napolitano, R. Henry and S. Shekhar, J. Org. Chem., 2016, 81, 1285–1294 CrossRef CAS PubMed.
- H. Yanai, H. Ogura, H. Fukaya, A. Kotani, F. Kusu and T. Taguchi, Chem. – Eur. J., 2011, 17, 11747–11751 CrossRef CAS PubMed.
- H. Yanai, T. Yoshino, M. Fujita, H. Fukaya, A. Kotani, F. Kusu and T. Taguchi, Angew. Chem., Int. Ed., 2013, 52, 1560–1563 CrossRef CAS PubMed.
- Y. Sumii, Y. Sugita, E. Tokunaga and N. Shibata, ChemistryOpen, 2018, 7, 204–211 CrossRef CAS PubMed.
- X. N. Wang, E. H. Krenske, R. C. Johnston, K. N. Houk and R. P. Hsung, J. Am. Chem. Soc., 2014, 136, 9802–9805 CrossRef CAS PubMed.
- H. Yanai, S. Hoshikawa, K. Kawai and T. Matsumoto, Chem. – Asian J., 2025, 20, e70414 CrossRef CAS PubMed.
- M. J. Ralph, D. C. Harrowven, S. Gaulier, S. Ng and K. I. Booker-Milburn, Angew. Chem., Int. Ed., 2015, 54, 1527–1531 CrossRef CAS PubMed.
- Y. P. Wang and R. L. Danheiser, Tetrahedron Lett., 2011, 52, 2111–2114 CrossRef CAS PubMed.
- M. Shi, L. P. Liu and J. Tang, J. Am. Chem. Soc., 2006, 128, 7430–7431 CrossRef CAS PubMed.
- J. C. Namyslo and D. E. Kaufmann, Chem. Rev., 2003, 103, 1485–1537 CrossRef CAS PubMed.
- V. M. Dembitsky, J. Nat. Med., 2008, 62, 1–33 CAS.
- R. Liu, M. Zhang, T. P. Wyche, G. N. Winston-Mcpherson, T. S. Bugni and W. Tang, Angew. Chem., Int. Ed., 2012, 51, 7503–7506 CrossRef CAS PubMed.
- Y. Wang, M. E. Muratore, Z. Rong and A. M. Echavarren, Angew. Chem., Int. Ed., 2014, 53, 14022–14026 CrossRef CAS PubMed.
- J. Hartung and R. H. Grubbs, Angew. Chem., Int. Ed., 2014, 53, 3885–3888 CrossRef CAS PubMed.
- A. Nishimura, M. Ohashi and S. Ogoshi, J. Am. Chem. Soc., 2012, 134, 15692–15695 CrossRef CAS PubMed.
- S. Cañellas, J. Montgomery and M. Pericàs, J. Am. Chem. Soc., 2018, 140, 17349–17355 CrossRef PubMed.
- W. Tam, J. Goodreid and N. Cockburn, Curr. Org. Synth., 2009, 6, 219–238 CrossRef CAS.
- M. Yamada, Beilstein J. Org. Chem., 2024, 20, 125 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, I. Fernández and C. Lázaro-Milla, Chem. Commun., 2015, 51, 3395–3398 RSC.
- B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem. – Eur. J., 2016, 22, 8998–9005 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros and C. Lázaro-Milla, Adv. Synth. Catal., 2017, 359, 2630–2639 CrossRef CAS.
- The transformation of alkynylsilanes into silicon-substituted cyclobutenes proved to be challenging in the presence of zwitterion 1b, and attempts to access tin-substituted cyclobutenes from alkynylstannanes were unsuccessful. See ref. 30.
- C. J. Smedley, M.-C. Giel, T. Fallon and J. E. Moses, Angew. Chem., Int. Ed., 2023, 62, e202303916 Search PubMed.
- C. J. Smedley, Asian J. Org. Chem., 2025, 14, e00525 Search PubMed.
- B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem. – Eur. J., 2019, 25, 7547–7552 CrossRef CAS PubMed.
- C. Lázaro-Milla, M. T. Quirós, D. J. Cárdenas and P. Almendros, Chem. Commun., 2020, 56, 6070–6073 RSC.
- C. Lázaro-Milla, M. T. Quirós, D. J. Cárdenas and P. Almendros, Chem. Commun., 2021, 57, 8456–8459 RSC.
- (a) S. Hoshikawa, H. Yanai and T. Matsumoto, Chem. – Eur. J., 2022, 28, e202200704 CrossRef CAS PubMed; (b) H. Yanai, S. Kurogi, S. Hoshikawa and T. Matsumoto, Chem. – Eur. J., 2024, 30, e202400843 CrossRef CAS PubMed.
- A. S. Petcu, C. Lázaro-Milla, J. M. Alonso and P. Almendros, Org. Lett., 2024, 26, 4560–4565 CrossRef CAS PubMed.
- S. Hoshikawa, J. M. Alonso, T. Matsumoto, H. Yanai and P. Almendros, Adv. Synth. Catal., 2024, 366, 3683–3688 CrossRef CAS.
- L. K. San, S. N. Spisak, C. Dubceac, S. H. M. Deng, I. V. Kuvychko, M. A. Petrukhina, X. Bin Wang, A. A. Popov, S. H. Strauss and O. V. Boltalina, Chem. – Eur. J., 2018, 24, 1441–1447 CrossRef CAS PubMed.
- D. L. Orsi and R. A. Altman, Chem. Commun., 2017, 53, 7168–7181 RSC.
- E. P. Gillis, K. J. Eastman, M. D. Hill, D. J. Donnelly and N. A. Meanwell, J. Med. Chem., 2015, 58, 8315–8359 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros and C. Lázaro-Milla, Chem. Commun., 2015, 51, 6992–6995 RSC.
- C. Lázaro-Milla, H. Yanai and P. Almendros, Org. Lett., 2021, 23, 2921–2926 CrossRef PubMed.
- C. Lázaro-Milla and P. Almendros, Chem. – Eur. J., 2021, 27, 13534–13538 CrossRef PubMed.
- I. Martín-Mejías, C. Aragoncillo, H. Yanai, S. Hoshikawa, Y. Fujimoto, T. Matsumoto and P. Almendros, Chem. Commun., 2020, 56, 1795–1798 RSC.
- M. Zhao, A. G. Barrado, K. Sprenger, C. Golz, R. A. Mata and M. Alcarazo, Org. Lett., 2020, 22, 4932–4937 CrossRef CAS PubMed.
- W. Yang, R. Bam, V. J. Catalano and W. A. Chalifoux, Angew. Chem., Int. Ed., 2018, 57, 14773–14777 CrossRef CAS PubMed.
- I. Takahashi, T. Fujita, N. Shoji and J. Ichikawa, Chem. Commun., 2019, 55, 9267–9270 RSC.
- Y. Segawa, T. Maekawa and K. Itami, Angew. Chem., Int. Ed., 2015, 54, 66–81 CrossRef CAS PubMed.
- M. Grzybowski, K. Skonieczny, H. Butenschön and D. T. Gryko, Angew. Chem., Int. Ed., 2013, 52, 9900–9930 CrossRef CAS PubMed.
- S. Hoshikawa, H. Yanai, I. Martín-Mejías, C. Lázaro-Milla, C. Aragoncillo, P. Almendros and T. Matsumoto, Chem. – Eur. J., 2021, 27, 16112–16116 CrossRef CAS PubMed.
- B. Alcaide, P. Almendros, C. Lázaro-Milla and P. Delgado-Martínez, Chem. – Eur. J., 2018, 24, 8186–8194 CrossRef CAS PubMed.
- M. Singh, M. Kaur and O. Silakari, Eur. J. Med. Chem., 2014, 84, 206–239 CrossRef CAS PubMed.
- K. Nakagawa-Goto, K. Yamada, S. Nakamura, T. H. Chen, P. C. Chiang, K. F. Bastow, S. C. Wang, B. Spohn, M. C. Hung, F. Y. Lee, F. C. Lee and K. H. Lee, Bioorg. Med. Chem. Lett., 2007, 17, 5204–5209 CrossRef CAS PubMed.
- M. A. Marć, A. Kincses, B. Rácz, M. J. Nasim, M. Sarfraz, C. Lázaro-Milla, E. Domínguez-Álvarez, C. Jacob, G. Spengler and P. Almendros, Pharmaceuticals, 2020, 13, 453 CrossRef PubMed.
- A. S. Petcu, C. Lázaro-Milla, F. J. Rodríguez, I. Iriepa, Ó. M. Bautista-Aguilera, C. Aragoncillo, J. M. Alonso and P. Almendros, J. Org. Chem., 2023, 88, 7373–7380 CrossRef CAS PubMed.
- G. Shabir, A. Saeed and H. R. El-Seedi, Phytochemistry, 2021, 181, 112568 CrossRef CAS PubMed.
- Z. Zhao, K. Kang, J. Yue, X. Ji, H. Qiao, P. Fan and X. Zheng, Eur. J. Med. Chem., 2021, 210, 113073 CrossRef CAS PubMed.
- C. Wang, Z. Wu, H. Cai, S. Xu, J. Liu, J. Jiang, H. Yao, X. Wu and J. Xu, Bioorg. Med. Chem. Lett., 2015, 25, 5212–5216 CrossRef CAS PubMed.
- H. Yanai, Y. Takahashi, H. Fukaya, Y. Dobashi and T. Matsumoto, Chem. Commun., 2013, 49, 10091–10093 RSC.
- H. Yanai, R. Sano, S. Hoshikawa and T. Matsumoto, Asian J. Org. Chem., 2025, 14, e202400651 CrossRef CAS.
- R. Kakuchi, T. Oguchi, M. Kuroiwa, Y. Hirashima, M. Omichi, N. Seko and H. Yanai, Chem. Sci., 2024, 15, 19322–19327 RSC.
- R. Kakuchi, T. Oguchi, M. Omichi, N. Seko, G. V. Dizon, Y. Chang and H. Yanai, Macromolecules, 2026, 59, 2698–2705 CrossRef CAS.
- H. Yanai, S. Hoshikawa, Y. Moriiwa, A. Shoji, A. Yanagida and T. Matsumoto, Angew. Chem., Int. Ed., 2021, 60, 5168–5172 CrossRef CAS PubMed.
- H. Yanai, S. Hoshikawa, H. Watanabe, H. Kaneko, H. Nakaminami and T. Matsumoto, Chem. Pharm. Bull., 2024, 72, 884–889 CrossRef CAS PubMed.
- P. Almendros, H. Yanai, S. Hoshikawa, C. Aragoncillo, C. Lázaro-Milla, M. Toledano-Pinedo, T. Matsumoto and B. Alcaide, Org. Chem. Front., 2018, 5, 3163–3169 RSC.
- H. Yanai, P. Almendros, S. Takahashi, C. Lázaro-Milla, B. Alcaide and T. Matsumoto, Chem. – Asian J., 2018, 13, 1956–1961 CrossRef CAS PubMed.
- C. Lázaro-Milla, J. Macicior, H. Yanai and P. Almendros, Chem. – Eur. J., 2020, 26, 8983–8989 CrossRef PubMed.
- M. Toledano-Pinedo, T. Martínez Del Campo, H. Yanai and P. Almendros, ACS Catal., 2022, 12, 11675–11681 CrossRef CAS.
- For the photopromoted synthesis of triflyl-cyclobutanes through intermolecular [2+2] cyclization of alkenyl triflones and olefins, see: Z. Gao, X. Lu, R. Zhang, D. Shen and Z. Liu, Org. Lett., 2026, 28, 1499–1504 CrossRef CAS PubMed.
Footnote |
| † Dedicated to Prof. José Luis Marco on the occasion of his retirement. |
| This journal is © The Royal Society of Chemistry 2026 |