
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Proton-coupled electron transfer driven by coordination-induced bond weakening at a trivalent uranium center†
I. J. Huerfano ,
Tate M. Quinn,
Ahmed Kangmennaa,
Matthias Zeller and
Suzanne C. Bart*
,
Tate M. Quinn,
Ahmed Kangmennaa,
Matthias Zeller and
Suzanne C. Bart*
H. C. Brown Laboratory, James Tarpo Jr. and Margaret Tarpo Department of Chemistry, Purdue University, West Lafayette, Indiana 47907, USA. E-mail: sbart@purdue.edu
First published on 5th May 2026
Abstract
We present the experimental determination of the N–H bond dissociation free energy (BDFE) of a uranium(III)-anilido N–H bond revealing a powerful uranium-induced bond weakening effect and establishing the competency of actinides to facilitate PCET reactions.
Proton-coupled electron transfer (PCET) reactions are of fundamental importance in both natural and artificial chemical systems relevant to energy production, synthesis, and environmental remediation.1–7 In this context, there has been a growing interest in using metals to tune the reactivity of adjacent element-hydrogen bonds through coordination-induced bond weakening effects.8 Through this influence, metal coordination provides a possible route to initiate PCET reactions and facilitate challenging chemical transformations such as ammonia and water oxidation.9,10 These particular reactions are of interest as hydrogen generated from ammonia or water oxidation may provide promising alternatives to carbon-based fuels.11–21 Beyond its direct utility, exploration of the thermochemistry and kinetics of PCET at nitrogen-based ligands also provides valuable insights into critical N–H bond making and breaking steps22 relevant to nitrogen fixation, the reverse reaction of ammonia oxidation.
While there is extensive precedence for PCET reactivity in transition metal-based systems, there have been limited investigations exploring analogous behaviour with actinides. In recent work, La Pierre and coworkers synthesized a transient homoleptic plutonium(V) imidophosphorane complex which rapidly undergoes PCET through a net hydrogen atom abstraction of the THF solvent to give the corresponding Pu(IV) with a protonated ligand.23 Similarly, Blakemore, Wilson, and coworkers demonstrated the synthesis of two polynuclear oxo-bridged neptunium complexes resulting from PCET at one oxo ligand of a neptunyl species, forming water as a byproduct.24 Despite this important precedence, to date there have been no investigations into coordination-induced bond weakening effects or how they may lead to PCET reactions at actinides. Uranium specifically provides a promising platform for deeper exploration of PCET reactions due to its high Lewis acidity hypothesized to impart a strong electron withdrawing effect on the coordinated ligand, thus facilitating proton transfer, as well as its robust redox chemistry allowing for facile electron transfer. Further, its competency in dinitrogen activation,25–29 originally demonstrated by Fritz Haber's patent for ammonia synthesis,30 makes uranium an interesting target for exploring its influence on the reactivity of N–H bonds.
Previously our group demonstrated the capacity of uranium(III)-anilido complexes to undergo a net hydrogen-atom transfer (HAT) reaction to form uranium(IV)-imido complexes upon reaction with both an oxidant and a strong base via oxidative deprotonation, highlighting a potential for PCET (Scheme 1).31 Further work from our group expanded this original observation through direct reactivity of several uranium(III)-anilido complexes, of varying steric and electronic character, with common hydrogen atom abstractors suggesting at least a modest N–H bond weakening effect.32 Herein, we present the thermodynamics of the interconversion between a uranium(III)-anilido and uranium(IV)-imido complex via PCET. Experimental determination of the N–H bond dissociation free energy (BDFE) indicates the uranium(III)-anilido contains an exceptionally weak N–H bond comparable to those seen in previously reported molybdenum analogues.22,33 Together, these experiments reveal a powerful N–H bond weakening effect by low-valent uranium and highlight its possible utility in driving PCET reactions.
 | ||
| Scheme 1 Interconversion of U(III)-anilido and U(IV)-imido via net HAT. | ||
To investigate the ability of uranium to exert coordination-induced bond weakening effects at N–H bonds, we first synthesized our reported bis[(tris(3,5-dimethylpyrazolyl)borate]uranium(III) 4-methylanilido (1)31 and sought to interrogate its N–H bond strength. Bond energies were initially bracketed through reactions of the anilido and imido complexes with a series of HAT reagents with known BDFE values (Scheme 2).1 Consistent with our previous results, reaction of 1 with either half an equivalent of Gomberg's Dimer or one equivalent of 2,4,6-tritertbutylphenoxy radical (˙OMes*) gave clean conversion to the uranium(IV) imido (2)34 with concomitant formation of triphenylmethane (BDFE = 75.7 kcal mol−1) and 2,4,6-tri-tert-butylphenol (BDFE = 76.8 kcal mol−1), respectively. Reactions of 1 with ˙OMes* show an immediate color change from green/blue to dark red upon dropwise addition of the vibrant blue phenoxy radical. In contrast, addition of Gomberg's dimer requires an hour before complete color change is observed, indicating a slow reaction due to the equilibrium between Gomberg's Dimer and trityl radical,35 the active hydrogen atom abstractor in this reaction. The success of PCET by these reagents initially indicated at least a modest bond weakening effect by uranium of nearly 10 kcal mol−1 relative to para-toluidine (BDFE = 85 kcal mol−1).1
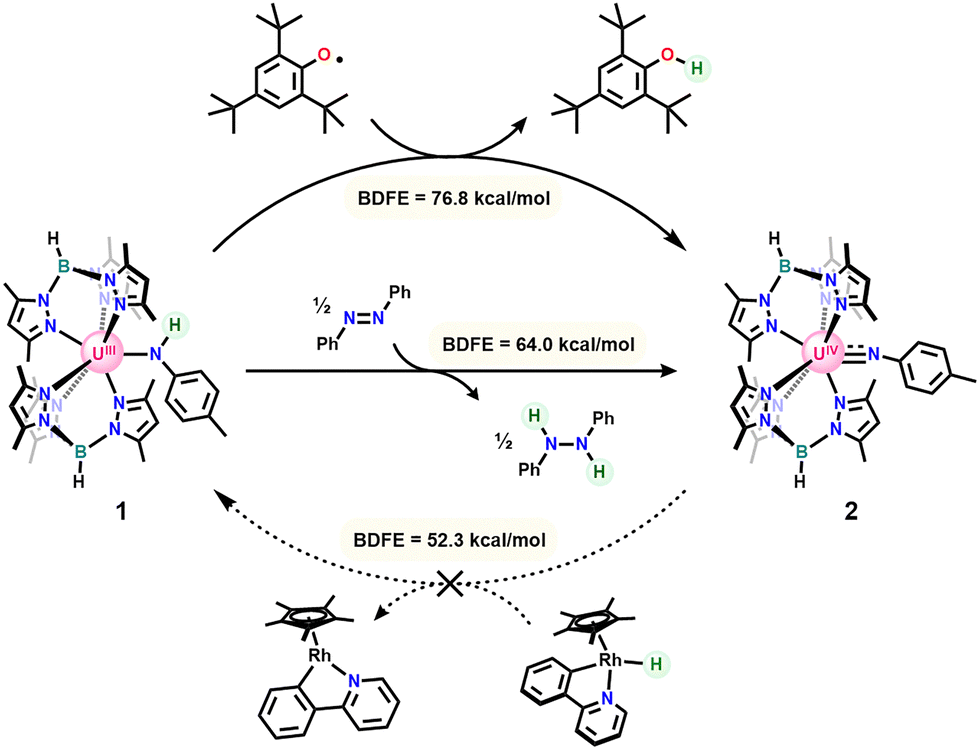 | ||
| Scheme 2 Reactions of 1 with HAT reagents to form 2. | ||
Seeking the lower limit of the N–H bond strength, HAT reagents with progressively weaker BDFE values were added to 1 and 2. Reaction of 1 with half an equivalent of azobenzene led to conversion of the anilido to 2 with formation of diphenylhydrazine (BDFE = 64.0 kcal mol−1). Upon continued reaction, the half equivalent of diphenylhydrazine, formed as a byproduct, reacted further with 2 through protonolysis of the imido ligand to generate a small amount of our previously reported uranium(IV) η2-hydrazido (3) and para-toluidine (Fig. S6 and S7).36 Independent reaction of 2 with excess diphenyl hydrazine on an NMR scale gives the same product. Attempts to react 1 with para-anthraquinone to form 2 and dihydroanthraquinone (BDFE = 54.9 kcal mol−1) lead to intractable mixtures of products consistent with uranium(III) based on chemical shifts in the 11B-NMR spectrum.37 This observation likely results from the oxophilicity of uranium and propensity for nucleophilic ligands of the bis[(tris(3,5-dimethylpyrazolyl)borate]uranium(III) system to attack electrophiles.38 The imido, 2, was next reacted with a rhodium(III) hydride with an especially weak Rh–H bond (BDFE = 52.3 kcal mol−1) originally reported by Norton and coworkers.39 Addition of the rhodium hydride to a solution of 2 in C6D6 at room temperature showed no reaction after 18 hours. Heating the reaction mixture to 80 °C for an additional 18 hours shows no conversion to 1 with only small amounts of decomposition. Altogether, these bracketing experiments indicated an N–H bond strength less than 52.3 kcal mol−1 for 1.
In the absence of accessible HAT reagents with lower BDFE values, thermodynamics of PCET were further interrogated through investigation of individual proton- and electron-transfer steps which can be directly related to N–H BDFE through the Bordwell equation (eqn (1)).1 As the overall thermodynamics of a chemical transformation are ultimately independent of the pathway, the Bordwell equation combines terms for the free energies of proton-transfer, calculated from pKa, and electron-transfer, calculated from oxidation potential, as well as a solvent-dependent constant (Cg). As such, the Bordwell equation provides a convenient route to accurately calculate BDFE values if electrochemical and pKa data are obtainable.
| BDFE = 1.37(pKa) + 23.06(E°) + Cg | (1) |
Cyclic voltammetry (CV: Fig. 1) reveals that 1 undergoes an irreversible oxidation with a peak potential around −1.2 V (vs. Fc/Fc+) assigned as a U(III) to U(IV) process. While a reduction feature is observed with a peak potential around −2.3 V (vs. Fc/Fc+), the peak-to-peak separation and significant decrease in peak current between redox events indicates irreversibility, preventing accurate determination of the half-wave potential (E1/2). This irreversibility suggests the cationic U(IV) anilido is chemically unstable under the experimental conditions. To more accurately evaluate the oxidation potential, differential pulse voltammetry (DPV: Fig. S1) was utilized, giving an Ep value of −1.55 V (vs. Fc/Fc+).
 | ||
| Fig. 1 CV of 1 showing an irreversible redox process assigned as a U(III) to U(IV) oxidation event. 4 mM 1; 0.1 M TBA(BPh4)/THF; Glassy Carbon Working Electrode/Platinum Coil Counter Electrode; Ag/AgCl Pseudo-Reference Electrode. TBA = tetrabutylammonium. | ||
In an attempt to isolate the oxidized U(IV) anilido, 1 was treated with one equivalent of silver tetraphenylborate (AgBPh4), in toluene at room temperature leading to a colour change from blue/green to deep red with precipitation of a brown/black solid consistent with silver metal. Work-up of the crude reaction mixture led to isolation of a mixture of 2 and our previously reported U(IV) terminal oxo.40 Compound 2 was hypothesized to result from the Brønsted-Lowry acidity of the targeted cationic U(IV) anilido and low solubility of AgBPh4 resulting in deprotonation by leftover 1 in solution due to slow reaction. To test a weaker, more soluble oxidant, 1 was reacted with cobaltocenium tetrakis(3,5-(bis)trifluoromethylphenyl)borate (CoCp2BArF24: E1/2 = −1.3 V)41 in THF at −35 °C. Upon addition of 1 to oxidant, there is a notable colour change to red-brown with conversion to a new uranium product, 4. This new product was assigned as a cationic U(IV) anilido based on the observation of a broad resonance at −81.4 ppm and a sharp signal at −6.6 ppm in the 11B-NMR spectrum, consistent with a tris(3,5-dimethylpyrazolyl)borate (Tp*) ligand bound to U(IV) and a BArF24 counterion, respectively. Notably, clean formation of 4 requires addition of 1 to CoCp2BArF24, as the reverse addition results in significant conversion to 2 and a blue solid identified as [Tp*2U]BArF24 (5: see SI). This observation supports our initial hypothesis that the anilido ligand of 4 is readily deprotonated by the anilido ligand of 1 when both species are simultaneously present in solution to generate 2, 5, and para-toluidine (Scheme 3), accounting for the irreversibility in the CV. As a result, small amounts of the imido are nearly always present in isolated samples of 4.
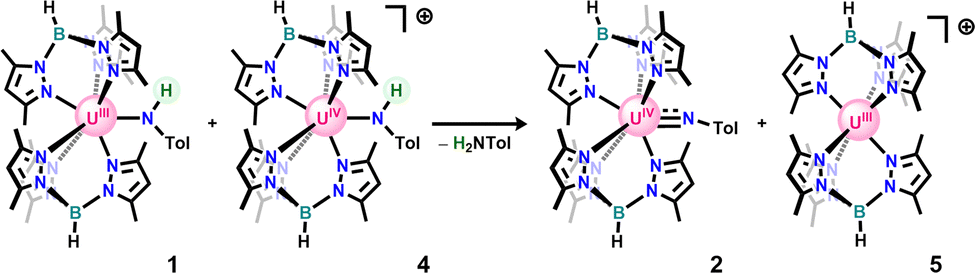 | ||
| Scheme 3 Reaction of 1 and 4 when both species are present in solution. | ||
The identity of 4 was confirmed by single-crystal X-ray diffraction revealing a cationic bis(Tp*) uranium anilido complex with an outer sphere BArF24 counter anion (Fig. 2). Compound 4 exhibits a U–N bond length of 2.182(5) Å which is intermediate between the U–N bond lengths of 1 and 2 (2.351 and 2.011 Å respectively) with a U–N–C angle of 144.3(1)°, comparable to that of the anilido, 1 (145.6°), and significantly more bent than the imido, 2 (163.6°). These metrics are consistent with the assignment of a U(IV)-anilido complex, and the N–H proton was freely refined. This hydrogen atom is also observed by FTIR spectroscopy exhibiting an N–H stretch at 3432 cm−1, shifted 73 cm−1 relative to 1 (Fig. S26). The electronic structure of 4 was further investigated by electronic absorption spectroscopy in THF at room temperature for wavelengths ranging from 300–1600 nm (Fig. S27). The visible region exhibits two charge transfer bands with absorptions at 436 nm (ε = 2200 M−1 cm−1) and 500 nm (ε = 1300 M−1 cm−1), with the latter broad signal tailing out to nearly 700 nm. This spectrum is consistent with the observed brown-red color of 4. Several poorly defined absorbances corresponding to f–f transitions (ε < 65 M−1 cm−1) are observed in the near-infrared region of the spectrum and are consistent with the assignment of a U(IV) oxidations state based on our previous work.32 Notably, 4 is the first cationic U(IV) complex supported by the bis(Tp*) system to our knowledge.
 | ||
| Fig. 2 ORTEP representation of the molecular structure of 4 (top). Thermal ellipsoids are shown at 30% probability. Hydrogen atoms bound to carbon, the BArF24 anion, and co-crystallized solvent molecules are omitted for clarity. Structural overlays for comparison of U–N bond lengths and U–N–C bond angles of 4 with 1 (left; blue) and 2 (right: red). | ||
With the successful synthesis and isolation of 4, proton transfer was investigated through reactivity with a series of organic bases with known pKaH values (Scheme 4).42 These bracketing experiments reveal that 4 shows no reactivity with either triethylamine or (−)-sparteine but is readily deprotonated by 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) to form 2, giving an experimentally determined pKaH range of 14.5–16.9 in THF. Substituting these values into the Bordwell equation along with the experimentally determined oxidation potential gives an N–H BDFE range for 1 of 44.0–47.3 kcalmol−1 (THF). This range represents an immense uranium-induced bond weakening effect of nearly 40 kcalmol−1 relative to the N–H bonds of para-toluidine and is comparable to previously reported molybdenum-based systems.10,22
 | ||
| Scheme 4 Stepwise oxidative deprotonation of 1 using cobaltocenium and bases of varying pKaH values. | ||
Given the relatively modest acidity of 4 and very negative oxidative potential of 2, this bond weakening is likely driven thermodynamically by the favorability of electron transfer to convert U(III) to the more thermodynamically stable U(IV).
As N–H bonds with BDFE values less than half that of dihydrogen (BDFE = 104 kcal mol−1) can be susceptible to spontaneous hydrogen evolution, 1 was allowed to sit in C6D6 at both room temperature and 80 °C for one week under a nitrogen atmosphere. Neither sample showed evidence for imido formation with only retention of 1 and modest decomposition. To further test the possibility of hydrogen generation, a degassed solution of 1 in C6D6 was left under static vacuum for 72 hours to determine if the reaction could be driven through liberation of H2 gas into the evacuated headspace,10 but no conversion was observed. Finally, a C6D6 solution of 1 was photolyzed at 350 nm for 18 hours to see if UV light could facilitate H2 loss, which proved unsuccessful. Together, these experiments indicate the lack of an accessible mechanism for hydrogen-hydrogen coupling to generate H2, a kinetic limitation, despite favorable thermodynamics. This observation may potentially result from a combination of the 7-coordinate ligand environment of 1 and the rarity of terminal uranium hydrides43 preventing H2 formation as metal hydrides are proposed intermediates in previous transition metal systems.44
In conclusion, we demonstrate uranium's capability in exerting a powerful coordination-induced N–H bond weakening effect on par with molybdenum. Initial BDFE and pKa bracketing experiments of uranium anilido complexes combined with electrochemical studies reveal an N–H bond weakening of nearly 40 kcal mol−1 likely driven thermodynamically by the highly reducing nature of U(III). While efforts to generate H2 from the U(III)-anilido were unsuccessful, this work establishes the competency of actinides to facilitate PCET reactions and especially highlights the potential utility of uranium for important chemical transformations involving net hydrogen atom transfer. Beyond applications in small molecule activation, such as ammonia or water oxidation, the ability of actinides to readily engage in PCET also may provide a route to synthesize new complexes with actinide-ligand multiple bonds for exploring covalency.
I. J. Huerfano: conceptualization, formal analysis, investigation, methodology, project administration, supervision, validation, visualization, writing – original draft. T. M. Quinn: investigation, validation, writing – review & editing. A. Kangmennaa: investigation, writing – review & editing. M. Zeller: supervision, validation, writing – review & editing. S. C. Bart: conceptualization, funding acquisition, methodology, project administration, resources, supervision, writing – review & editing.
Conflicts of interest
There are no conflicts to declare.Data availability
The data for this article has been included as part of the supplementary information (SI). See DOI: https://doi.org/10.1039/d6cc01484d.CCDC 2530055 (4) contains the supplementary crystallographic data for this paper.45
Acknowledgements
This work was supported by a grant from the Chemical Synthesis Program, National Science Foundation (CHE1665170).References
- R. G. Agarwal, S. C. Coste, B. D. Groff, A. M. Heuer, H. Noh, G. A. Parada, C. F. Wise, E. M. Nichols, J. J. Warren and J. M. Mayer, Chem. Rev., 2022, 122, 1–49 CrossRef CAS PubMed.
- J. L. Dempsey, J. R. Winkler and H. B. Gray, Chem. Rev., 2010, 110, 7024–7039 CrossRef CAS PubMed.
- C. L. Ford, Y. J. Park, E. M. Matson, Z. Gordon and A. R. Fout, Science, 2016, 354, 741–743 CrossRef CAS.
- P. Hutchison, L. E. Smith, C. L. Rooney, H. Wang and S. Hammes-Schiffer, J. Am. Chem. Soc., 2024, 146, 20230–20240 CrossRef CAS PubMed.
- P. J. Jabalera-Ortiz, A. M. Rodriguez-Jimenez and P. Garrido-Barros, ChemSusChem, 2025, 18, e202402630 CrossRef CAS PubMed.
- J. M. Moore, T. J. Miller, M. Mu, M. N. Peñas-Defrutos, K. L. Gullett, L. S. Elford, S. Quintero, M. García-Melchor and A. R. Fout, J. Am. Chem. Soc., 2025, 147, 8444–8454 CrossRef CAS PubMed.
- S. Y. Reece and D. G. Nocera, Annu. Rev. Biochem., 2009, 78, 673–699 CrossRef CAS PubMed.
- N. G. Boekell and R. A. Flowers, Chem. Rev., 2022, 122, 13447–13477 CrossRef CAS PubMed.
- M. J. Bezdek, S. Guo and P. J. Chirik, Science, 2016, 354, 730–733 CrossRef CAS PubMed.
- C. C. Almquist, T. Rajeshkumar, W. Zhou, L. Maron and W. E. Piers, J. Am. Chem. Soc., 2025, 147, 40374–40388 CrossRef CAS PubMed.
- J. G. Chen, R. M. Crooks, L. C. Seefeldt, K. L. Bren, R. M. Bullock, M. Y. Darensbourg, P. L. Holland, B. Hoffman, M. J. Janik, A. K. Jones, M. G. Kanatzidis, P. King, K. M. Lancaster, S. V. Lymar, P. Pfromm, W. F. Schneider and R. R. Schrock, Science, 2018, 360, eaar6611 CrossRef PubMed.
- W. I. F. David, J. W. Makepeace, S. K. Callear, H. M. A. Hunter, J. D. Taylor, T. J. Wood and M. O. Jones, J. Am. Chem. Soc., 2014, 136, 13082–13085 CrossRef CAS.
- P. L. Dunn, B. J. Cook, S. I. Johnson, A. M. Appel and R. M. Bullock, J. Am. Chem. Soc., 2020, 142, 17845–17858 CrossRef CAS PubMed.
- O. Elishav, B. Mosevitzky Lis, E. M. Miller, D. J. Arent, A. Valera-Medina, A. Grinberg Dana, G. E. Shter and G. S. Grader, Chem. Rev., 2020, 120, 5352–5436 CrossRef CAS PubMed.
- W.-C. Hsu and Y.-H. Wang, ChemSusChem, 2022, 15, e202102378 CrossRef CAS PubMed.
- A. Kazemi, F. Manteghi and Z. Tehrani, ACS Omega, 2024, 9, 7310–7335 CAS.
- M. Kondo, H. Tatewaki and S. Masaoka, Chem. Soc. Rev., 2021, 50, 6790–6831 RSC.
- K. E. Lamb, M. D. Dolan and D. F. Kennedy, Int. J. Hydrogen Energy, 2019, 44, 3580–3593 CrossRef CAS.
- N. V. Rees and R. G. Compton, Energy Environ. Sci., 2011, 4, 1255–1260 RSC.
- F. Schüth, R. Palkovits, R. Schlögl and D. S. Su, Energy Environ. Sci., 2012, 5, 6278–6289 RSC.
- D. N. Stephens and M. T. Mock, Eur. J. Inorg. Chem., 2024, e202400039 CrossRef CAS.
- M. J. Bezdek and P. J. Chirik, Angew. Chem., Int. Ed., 2018, 57, 2224–2228 CrossRef CAS PubMed.
- K. S. Otte, J. E. Niklas, C. M. Studvick, C. L. Montgomery, A. R. C. Bredar, I. A. Popov and H. S. La Pierre, J. Am. Chem. Soc., 2024, 146, 21859–21867 CrossRef PubMed.
- E. R. Mikeska, R. E. Wilson and J. D. Blakemore, Chem. – Eur. J., 2025, 31, e202402963 CrossRef CAS.
- M. S. Batov, H. T. Partlow, L. Chatelain, J. A. Seed, R. Scopelliti, I. Zivkovic, R. W. Adams, S. T. Liddle and M. Mazzanti, Nat. Chem., 2025, 17, 1425–1433 CrossRef CAS PubMed.
- L. Chatelain, E. Louyriac, I. Douair, E. Lu, F. Tuna, A. J. Wooles, B. M. Gardner, L. Maron and S. T. Liddle, Nat. Commun., 2020, 11, 337 CrossRef CAS PubMed.
- N. Jori, M. Keener, T. Rajeshkumar, R. Scopelliti, L. Maron and M. Mazzanti, Chem. Sci., 2023, 14, 13485–13494 RSC.
- N. Jori, T. Rajeshkumar, R. Scopelliti, I. ĭvković, A. Sienkiewicz, L. Maron and M. Mazzanti, Chem. Sci., 2022, 13, 9232–9242 RSC.
- P. Wang, I. Douair, Y. Zhao, S. Wang, J. Zhu, L. Maron and C. Zhu, Angew. Chem., Int. Ed., 2021, 60, 473–479 CrossRef CAS.
- F. Haber, Verfahren zur Herstellung von Ammoniak durch katalytische Vereinigung von Stickstoff und Wasserstoff, zweckmäßig unter hohem Druch, 1909.
- D. Perales, S. A. Ford, S. R. Salpage, T. S. Collins, M. Zeller, K. Hanson and S. C. Bart, Inorg. Chem., 2020, 59, 11910–11914 CrossRef CAS PubMed.
- D. Perales, N. J. Lin, M. R. Bronstetter, S. A. Ford, M. Zeller and S. C. Bart, Organometallics, 2022, 41, 606–616 CrossRef CAS.
- H. D. A. C. Jayaweera, C. C. Almquist, T. Rajeshkumar, W. Zhou, L. Maron and W. E. Piers, Inorg. Chem., 2025, 64, 1860–1874 CrossRef CAS PubMed.
- C. J. Tatebe, M. Zeller and S. C. Bart, Inorg. Chem., 2017, 56, 1956–1965 CrossRef CAS PubMed.
- T. T. Tidwell, in Stable Radicals, 2010, pp. 1–31.
- J. J. Kiernicki, R. F. Higgins, S. J. Kraft, M. Zeller, M. P. Shores and S. C. Bart, Inorg. Chem., 2016, 55, 11854–11866 CrossRef CAS PubMed.
- D. Perales, R. Bhowmick, M. Zeller, P. Miro, B. Vlaisavljevich and S. C. Bart, Chem. Commun., 2022, 58, 9630–9633 RSC.
- E. M. Matson, W. P. Forrest, P. E. Fanwick and S. C. Bart, J. Am. Chem. Soc., 2011, 133, 4948–4954 CrossRef CAS PubMed.
- Y. Hu and J. R. Norton, J. Am. Chem. Soc., 2014, 136, 5938–5948 CrossRef CAS PubMed.
- S. J. Kraft, J. Walensky, P. E. Fanwick, M. B. Hall and S. C. Bart, Inorg. Chem., 2010, 49, 7620–7622 CrossRef CAS PubMed.
- N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS.
- S. Tshepelevitsh, A. Kütt, M. Lõkov, I. Kaljurand, J. Saame, A. Heering, P. G. Plieger, R. Vianello and I. Leito, Eur. J. Org. Chem., 2019, 6735–6748 CrossRef CAS.
- M. G. Kaumini, I. Del Rosal, P. Rungthanaphatsophon, S. P. Kelley, L. Maron and J. R. Walensky, Inorg. Chem., 2025, 64, 17809–17816 CrossRef CAS PubMed.
- M. J. Bezdek, I. Pelczer and P. J. Chirik, Organometallics, 2020, 39, 3050–3059 CrossRef CAS.
- CCDC 2530055: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qxqp2.
Footnote |
| † Dedicated in memory of Professor Kenneth G. Caulton, a “hero” of organometallic chemistry. |
| This journal is © The Royal Society of Chemistry 2026 |