
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Time-resolved IR and X-ray spectroscopy as complementary electronic structure probes of photoinduced ligand-exchange in molybdenum hexacarbonyl
Raphael M. Jay *a,
Timo Dederichsa,
Marc-Oliver Winghartb,
Debkumar Ranab,
Ru-Pan Wang†
c,
Torsten Leitner‡
a,
Robert Stefanuika,
Camelia N. Borcad,
Grigory Smolentsevd,
Ambar Banerjeee,
Michael Odeliusf,
Nils Husec,
Thomas Huthwelkerd,
Erik T. J. Nibberingb and
Philippe Werneta
*a,
Timo Dederichsa,
Marc-Oliver Winghartb,
Debkumar Ranab,
Ru-Pan Wang†
c,
Torsten Leitner‡
a,
Robert Stefanuika,
Camelia N. Borcad,
Grigory Smolentsevd,
Ambar Banerjeee,
Michael Odeliusf,
Nils Husec,
Thomas Huthwelkerd,
Erik T. J. Nibberingb and
Philippe Werneta
aDepartment of Physics and Astronomy, Uppsala University, 75120 Uppsala, Sweden. E-mail: raphael.jay@physics.uu.se; philippe.wernet@physics.uu.se
bMax Born Institute for Nonlinear Optics and Short Pulse Spectroscopy, 12489 Berlin, Germany. E-mail: erik.nibbering@mbi-berlin.de
cDepartment of Physics and Center for Free-Electron Laser Science, University of Hamburg, 22761 Hamburg, Germany
dPaul Scherer Institute, CH-5232 Villigen PSI, Switzerland
eResearch Institute for Sustainable Energy (RISE), TCG Centres for Research and Education in Science and Technology (TCG-CREST), Kolkata 700091, India
fDepartment of Physics, Stockholm University, AlbaNova University Center, 106 91 Stockholm, Sweden
First published on 30th March 2026
Abstract
Photoinduced ligand-exchange dynamics in molybdenum hexacarbonyl have been studied using time-resolved IR and X-ray spectroscopy. We find that the energies of the unoccupied molybdenum 4d-derived orbitals that are accessed through X-ray transitions correlate with red-shifts in CO stretching modes. This provides complementary electronic structure information from the metal and the ligand perspective.
Photoinduced ligand-exchange plays a pivotal role as the starting point of a wide range of photocatalytic reactions.1 To spectroscopically study the underlying ligand dissociation and association dynamics, transition metal carbonyls have long served as popular model systems.2,3 By leveraging the bright IR-active CO stretching marker modes, time-resolved IR spectroscopy has been particularly successful in studies of metal carbonyls. The method has been key in identifying intermediates and measuring timescales in a wide range of ligand-exchange reactions from classic model systems such as iron pentacarbonyl and group 6 hexacarbonyls4–7 to transition metal carbonyl catalysts used in photochemical C–H bond activation.8–11
In recent years, time-resolved X-ray absorption spectroscopy (XAS) has emerged as a complementary probe of photo-induced ligand-exchange dynamics.12–16 At metal L-edges, the underlying strong dipole-allowed metal 2p → nd transitions provide unique access to the electronic structure locally around the metal center.17–19 In selected cases, the interpretation of the observed X-ray spectroscopic signatures has been found to be fully consistent with previously observed shifts in the ligand marker modes and their interpretation in terms of changing metal–ligand interactions.15,20,21 However, a direct and comprehensive comparison of X-ray and IR spectroscopic fingerprints has thus far not been attempted.
Here, we use time-resolved IR spectroscopy and time-resolved XAS at the metal L-edge to systematically establish the complementarity of the two methods in terms of their sensitivity to changes in the valence electronic structure. Specifically, we follow the photoinduced ligand-exchange dynamics in molybdenum hexacarbonyl (Mo(CO)6) in 1-pentanol solution. It has been established that UV excitation of Mo(CO)6 leads to ultrafast dissociation of a CO ligand followed by the rapid association of a 1-pentanol solvent molecule (see photochemical pathway shown in Scheme 1).22,23 Here, we now observe with time-resolved IR and X-ray absorption spectroscopy in the tender range,15,17,19,24 how metal–ligand covalency changes upon CO to 1-pentanol ligand-exchange in Mo(CO)6 as well as the ensuing interconversion of CH to OH-coordinated reaction products from Mo(CO)5-R-OH to Mo(CO)5-OH-R.
 | ||
| Scheme 1 Pathways of photoinduced ligand-exchange in Mo(CO)6 in an alcohol solution. | ||
Fig. 1 shows our time-resolved IR data of Mo(CO)6 in 1-pentanol photoexcited at 266 nm (see SI for experimental details). The transient difference spectra in Fig. 1a show a strong depletion of the CO stretching bands of the Mo(CO)6 parent complex at 1985 cm−1 concomitant with the emergence of transient absorption in the range of 1870 cm−1 to 1975 cm−1. The transient absorption bands at 1895 cm−1 and 1940 cm−1 fully develop over several 100 ps. Since these bands are absent in our measurements of Mo(CO)6 in pentane (see Supplementary Information), we assign them to CO stretching vibrations of the final Mo(CO)5-OH-R species, analogously to previous time-resolved IR measurements on Cr(CO)6.5
 | ||
| Fig. 1 (a) Transient difference IR spectra of Mo(CO)6 in 1-pentanol measured at different pump–probe delays following photoexcitation. Calculated energies and relative intensities of CO stretching frequencies of Mo(CO)6, Mo(CO)5-OH-R and the 5 possible Mo(CO)5-R-OH species are shown as vertical lines. (b) Delay traces measured at different wavenumbers and fitted with a biexponential function yielding time constants of (94 ± 9) ps and (280 ± 20 ps) ps. | ||
Transient intensity in the range of 1910 cm−1 to 1930 cm−1 as well as at 1965 cm−1 can instead be assigned to intermediate Mo(CO)5-R-OH species.7 In the 1-pentanol solution addressed here, we hence observe Mo(CO)5-R-OH to Mo(CO)5-OH-R interconversion on timescales of several 100 ps. For comparison, in methanol solution, where an extended carbon chain is absent, the Mo(CO)5-OH-R product bands directly appear within the vibrational cooling timescales of about 10 ps7 (see SI). The timescale of interconversion from CH to OH-coordinated products can be deduced from the delay traces shown in Fig. 1b. The data is fitted globally with a biexponential function. A fast time constant of 94 ± 9 ps is assigned to the decay of CO stretching excitations, which have previously been reported to relax independently of the initial vibrational energy redistribution in hot ligand-exchanged photoproducts as well as energy dissipation into the solvent.7,14 In alkanes, relaxation timescales of 160 ps have been reported for the decay of these CO stretching excitations.7 In alcohols, hydrogen bonding leads to a more anharmonic CO stretching potential, which enhances coupling to other modes and rationalizes the shorter timescale observed here. The second time constant of 280 ± 20 ps, reflected in the decay of the transient absorption at 1965 cm−1 with concomitant rise of absorption bands at 1895 cm−1 and 1940 cm−1, is assigned to the interconversion of CH to OH-bound species. Mo(CO)5-R-OH to Mo(CO)5-OH-R migration observed here is faster than Cr(CO)5-R-OH to Cr(CO)5-OH-R interconversion reported earlier.5 This difference may be related to the larger spatial extent and thus higher covalency of Mo compared to Cr.25 The resulting stronger metal-OH bonds in Mo compared to Cr may then induce a pull-effect that accelerates the migration towards the final OH-bound photoproduct. Our assignments of transient absorption bands are supported by our calculations of CO stretching frequencies shown in Fig. 1a. Besides some small relative shifts between calculated and experimental vibrational frequencies, the transient absorption bands at 1895 cm−1 and 1940 cm−1 can be assigned to CO stretching vibrations of the final Mo(CO)5-OH-R product. Transient intensity in the range of 1910 cm−1 to 1930 cm−1 as well as at 1965 cm−1 can instead be clearly attributed to the CO stretching vibrations of the intermediate Mo(CO)5-R-OH species. Overall, we hence find that CO to R-OH and OH-R ligand-exchange splits and red-shifts the quasi-degenerate CO stretching bands in the Mo(CO)5-R-OH and Mo(CO)5-OH-R species with respect to the Mo(CO)6 parent complex.
Our time-resolved XAS data of Mo(CO)6 in 1-pentanol photoexcited at 266 nm is shown in Fig. 2 (see SI for experimental details). Upon ligand-exchange, strong depletion of the Mo(CO)6 main edge at 2525 eV can be observed concomitant with the emergence of a transient pre-edge centered at 2522.5 eV. At the earlier measured time delay of 0.3 ns, at which the IR data suggests that a mix of Mo(CO)5-R-OH and Mo(CO)5-OH-R species is present, the pre-edge is slightly higher in intensity than at later timescales (>160 ns), when all photo-products have interconverted into the final OH-coordinated Mo(CO)5-OH-R product.26,27 A delay trace measured at 2521.4 eV in the rising edge of the transient pre-edge is shown in the inset in Fig. 2a. While the signal-to-noise ratio of the delay trace does not allow for a robust extraction of a time constant, the data is consistent with the 280 ps timescale of interconversion extracted from the IR data (accordingly, a fit with a fixed time constant of 280 ps agrees well the XAS data).
 | ||
| Fig. 2 (a) Steady-state Mo L3-edge absorption spectrum of Mo(CO)6 in 1-pentanol and transient difference spectra measured at different pump–probe delays. Difference intensities are displayed relative to the maximum of the steady-state absorption spectrum. The inset shows a delay trace measured at the onset of the transient pre-edge at 2521.4 eV. To guide the eye, a fit with a fixed time constant of 280 ps is additionally shown. (b) Calculated L-edge absorption spectrum of Mo(CO)6 (scaled to the measured spectrum of Mo(CO)6) as well as calculated difference spectra of the Mo(CO)5-OH-R and the 5 possible Mo(CO)5-R-OH species with respect to the Mo(CO)6 parent complex (single X-ray transitions are displayed as sticks, the Mo(CO)5-OH-R difference spectrum is scaled to match the pre-edge intensity of the experimental difference spectrum at delays >160 ns and the Mo(CO)5-R-OH spectra are scaled accordingly). A spectrum representing a mix of Mo(CO)5-R-OH and Mo(CO)5-OH-R contributions is shown additionally. | ||
The experimentally observed emergence of a transient pre-edge XAS peak and its varying intensity depending on CH or OH coordination is reproduced by our calculations shown in Fig. 2b (see SI for computational details). The theoretical overestimation of transient intensities in the pre-edge and the main-edge depletion in the spectrum at 0.3 ns are due to the fact that at this timescale a majority of species (∼66% based on the time-resolved IR data) have already interconverted to the OH-coordinated configuration. To simulate the expected spectral behavior at 0.3 ns delay, an additional calculated difference spectrum is shown in Fig. 2b, which is a mixture of Mo(CO)5-R-OH and Mo(CO)5-OH-R species and reproduces the measured difference spectrum at 0.3 ns well. This good agreement between experiment and theory therefore allows for a robust assignment and characterization of the underlying transitions.
In agreement with our earlier analysis of the L-edge absorption spectrum of Cr(CO)6,14 the main absorption line in Mo(CO)6 at 2525 eV is dominated by substantially mixed transitions of Mo 2p electrons into unoccupied Mo 4d(eg)-derived orbitals as well as into unoccupied CO π*-derived orbitals of t2g symmetry28 (see Supplementary Information for plots of molecular orbitals). The latter adopt substantial 4d character through backdonation from the occupied Mo 4d orbitals with the same symmetry. Upon ligand-exchange and breaking of Oh symmetry, the manifold of CO π* orbitals splits. This splitting is reflected by a broader distribution of single X-ray transitions in the Mo L3 main edge absorption in the CH and OH-coordinated species compared to the Mo(CO)6 parent complex (see sticks in Fig. 2b). Importantly, upon removal of a CO ligand, one CO π* orbital originating from t1u manifold in Oh symmetry is substantially stabilized and acquires substantial Mo d character, an effect observed before,14,29 and consistent with fundamental notions of metal–ligand orbital interactions.30 Analogous to previous XAS-based observations in the ligand-exchange dynamics of Cr(CO)6 and W(CO)6,6,14 this orbital mixing enables Mo 2p excitations into this CO π*-hybridized 4d orbital (see Supplementary Information for plots of molecular orbitals) and leads to marked transient pre-edge intensity in the Mo L-edge absorption spectrum at 2520–2523 eV.
To deduce a comprehensive picture of the electronic structure changes in Mo(CO)6 upon ligand exchange from both the Mo and the ligand perspectives with an emphasis on changes in bond covalency, we turn to the fragment charge decomposition analysis shown in Table 1. This analysis provides calculated values of the total electronic charge transfer between the (CO)5 fragment and the Mo-L fragments (where L = CO, R-OH or OH-R) as well as between the Mo(CO)5 and the L fragments. A schematic summary of the associated changes in covalency upon photo-induced ligand-exchange is shown in Scheme 2. Exchange of the strongly π-accepting CO ligand by a weakly π-accepting alcohol (in both CH and OH-coordinated geometries) leads to an increase in π-backdonation onto the remaining CO ligands (see in Table 1 the increasing charge transfer from Mo-L to (CO)5, when exchanging CO for R-OH and OH-R). This increase in backdonation compared to CO is more pronounced for OH-R than R-OH due to how σ-donation by OH-R and R-OH ligands varies compared to CO. While CO is the strongest σ-donor, net charge transfer onto Mo(CO)5 is minimal (see Table 1) because donation and backdonation act in different directions and are balanced in magnitude. For the (almost) exclusively σ-donating R-OH and OH-R ligands, effective charge transfer onto the Mo(CO)5 fragment is found (Table 1). This charge transfer is larger in Mo(CO)5-OH-R compared to Mo(CO)5-R-OH because the OH-R ligand is a stronger σ-donor than the R-OH one. The larger charge transfer from the OH-R ligand to Mo(CO)5 compared to the R-OH and CO ligands, in turn, enables the increased Mo-OH-R to (CO)5 backdonation compared to Mo(CO)5-R-OH and Mo(CO)6.
| Species | Mo-L → (CO)5 | L → Mo(CO)5 |
|---|---|---|
| Mo(CO)6 | 0.19 | −0.04 |
| Mo(CO)5-R-OH | 0.37 | 0.11 |
| Mo(CO)5-OH-R | 0.53 | 0.15 |
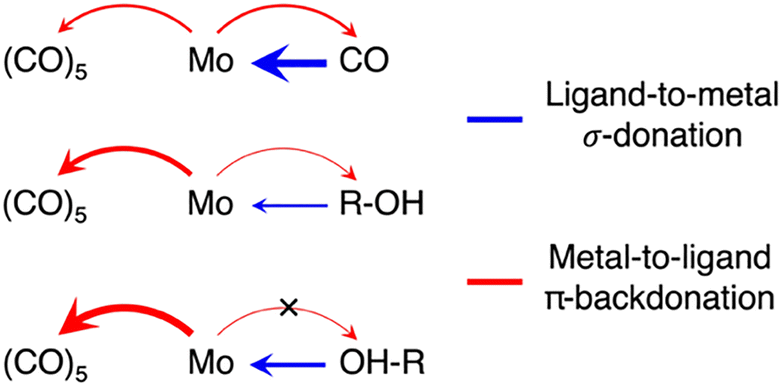 | ||
| Scheme 2 Schematic depiction of donation and backdonation interactions in the three Mo(CO)5-L species (L = CO, R-OH, OH-R). | ||
This theoretical prediction of unbalanced donation and backdonation in Mo(CO)5-CH and Mo(CO)5-OH compared to Mo(CO)6 is consistent with the observed spectral trends in our time-resolved IR and X-ray absorption data. Within the framework of the Tolman electronic parameter,31 an increase in metal-to-CO π-backdonation along the series from Mo(CO)6 to Mo(CO)5-R-OH and Mo(CO)5-OH-R (see Scheme 2) increasingly weakens the CO bonds consistent with the observed increasingly red-shifted CO stretching bands (see Fig. 1a). The concomitant decrease in ligand-to-metal σ-donation when exchanging the strong σ-donor CO by an alcohol (see Scheme 2) is reflected in the appearance of the low-energy pre-edge peak in the Mo L-edge absorption spectra of Mo(CO)5-R-OH and Mo(CO)5-OH-R compared to Mo(CO)6 (see Fig. 2). This pre-edge peak is due to excitations of Mo 2p electrons into the anti-bonding combination of the Mo 4d(eg)-derived molecular orbital representing the metal–ligand σ-donation interaction. For R-OH and OH-R ligands and compared to CO, σ-donation is smaller, the anti-bonding 4d(eg)-derived orbital is stabilized (its energy is smaller compared to CO as a ligand) and excitations of Mo 2p electrons into this orbital appear as a pre-edge peak (in Mo(CO)6, the strong ligand σ-donation pushes the same transitions up in energy and into the main edge of the Mo L-edge spectrum). Upon R-OH to OH-R exchange, the increase in σ-donation (see Scheme 2) destabilizes the same orbital slightly, which is evident in the minor shift of the pre-edge intensity to higher incidence energy (see Fig. 2).
Besides its sensitivity to the degree of σ-donation, L-edge absorption additionally has been shown to exhibit sensitivity to backdonation interactions.18,32 In octahedral systems, these interactions are typically evaluated via the degree of hybridization between occupied metal 4d(t2g) orbitals and the unoccupied ligand π* orbitals of the same symmetry. Here, however, the close energetic proximity of the CO π* t2g manifold to the unoccupied 4d(eg)-derived orbitals leads to highly mixed transitions in the main edge for all three species Mo(CO)6, Mo(CO)5-R-OH and Mo(CO)5-OH-R that are consequently largely uninformative regarding the degree of π-backdonation (see transitions with strongest oscillator strength at 2524.8 eV in Fig. 2b). Instead of being sensitive to π-backdonation onto the (CO)5 fragment, the L-edge exhibits sensitivity to the minor backdonation from the Mo center onto the coordinated CH group in Mo(CO)5-R-OH species. This effect can be observed via the calculated transitions at 2529 eV (Fig. 2b), which correspond to Mo 2p excitations into the σ* orbital of the C-H group that has adapted minor Mo 4d character via backdonation from the metal.33,34 At a time delay of 0.3 ns, when Mo(CO)5-R-OH species are present, transient intensity is indeed experimentally observable at the same energy, albeit at limited signal-to-noise ratio.
In summary, we have used time-resolved IR and X-ray spectroscopy to study the photoinduced ligand-exchange dynamics in Mo(CO)6 in 1-pentanol solution. We show how CO specific IR transitions provide high sensitivity to π-backdonation from Mo to the carbonyl manifold, whereas the Mo-specific X-ray absorption spectra predominantly reflect changes in ligand-to-Mo σ-donation for the different intermediates with different ligands along the photochemical pathway. This complementary information content is not restricted to ligand-exchange dynamics, but enables detailed insight into the evolution of the valence electronic structure from the metal and the ligand perspective in all photocatalytic applications involving transition metal complexes and catalysts with ligands that provide strong IR marker modes.
R. M. J., E. T. J. N. and P. W. originated the project concept. R. M. J., T. D., M.-O. W., D. R., R.-P. W., T. L., R. S., C. N. B., G. S., N. H., T. H., E. T. J. N. and P. W. planned and executed the experiments. R. M. J and T. D. analyzed the experimental data. R. M. J, T. D., A. B. and M. O. performed and analyzed the theoretical calculations. R. M. J. and P. W. wrote the paper with input from all the authors.
Conflicts of interest
There are no conflicts to declare.Data availability
Additional data are available from the authors upon request.All data supporting the conclusions of this article are included in the main text and the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6cc01470d.
Acknowledgements
We acknowledge the Paul Scherrer Institut, Villigen, Switzerland, for provision of beamtime at the PHOENIX beamline of the Swiss Light Source (SLS). We like to thank R. Wetter and Ch. Frieh for important contributions to the experimental design and for setting up the experiment at the PHOENIX beamline. The computations were enabled by resources in project [UPPMAX 2025/2-10] provided by Uppsala University at UPPMAX. R. M. J. acknowledges funding from the Swedish Research Council under Grant Agreement No. 2024-03901. M.-O. W., D. R., and E. T. J. N. acknowledge funding by the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (ERC grant agreement no. 788704, E. T. J. N.). R.-P. W. acknowledges funding from the German Ministry of Education and Research (BMBF), project ID 05K19GU2. M. O. acknowledges funding from Swedish Research Council grant agreement no. 2021-04521. N. H. acknowledges funding from the Cluster of Excellence “CUI: Advanced Imaging of Matter” of the Deutsche Forschungsgemeinschaft (DFG), EXC 2056, project ID 390715994. P. W. acknowledges funding from the Swedish Research Council (grant agreement no. 2019-04796, 2019-06093, and 2024-05246).References
- J. Hartwig, Organotransition Metal Chemistry: From Bonding to Catalysis, University Science Books, 2010 Search PubMed.
- R. N. Perutz, O. Torres and A. Vlček, in Comprehensive Inorganic Chemistry II, Elsevier, 2013, pp. 229–253 Search PubMed.
- J. J. Turner, M. W. George, M. Poliakoff and R. N. Perutz, Chem. Soc. Rev., 2022, 51, 5300–5329 RSC.
- P. T. Snee, C. K. Payne, S. D. Mebane, K. T. Kotz and C. B. Harris, J. Am. Chem. Soc., 2001, 123, 6909–6915 CrossRef CAS.
- J. E. Shanoski, E. A. Glascoe and C. B. Harris, J. Phys. Chem. B, 2006, 110, 996–1005 CrossRef CAS PubMed.
- S. A. Bartlett, N. A. Besley, A. J. Dent, S. Diaz-Moreno, J. Evans, M. L. Hamilton, M. W. D. Hanson-Heine, R. Horvath, V. Manici, X.-Z. Sun, M. Towrie, L. Wu, X. Zhang and M. W. George, J. Am. Chem. Soc., 2019, 141, 11471–11480 CrossRef CAS PubMed.
- T. P. Dougherty and E. J. Heilweil, Chem. Phys. Lett., 1994, 227, 19–25 CrossRef CAS.
- I. J. S. Fairlamb and J. M. Lynam, Acc. Chem. Res., 2024, 57, 919–932 CrossRef CAS PubMed.
- A. J. Cowan, P. Portius, H. K. Kawanami, O. S. Jina, D. C. Grills, X.-Z. Sun, J. McMaster and M. W. George, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6933–6938 CrossRef CAS PubMed.
- M. W. George, M. B. Hall, O. S. Jina, P. Portius, X.-Z. Sun, M. Towrie, H. Wu, X. Yang and S. D. Zaric, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 20178–20183 CrossRef CAS PubMed.
- B. J. Aucott, A.-K. Duhme-Klair, B. E. Moulton, I. P. Clark, I. V. Sazanovich, M. Towrie, L. A. Hammarback, I. J. S. Fairlamb and J. M. Lynam, Organometallics, 2019, 38, 2391–2401 CrossRef CAS.
- P. Wernet, K. Kunnus, I. Josefsson, I. Rajkovic, W. Quevedo, M. Beye, S. Schreck, S. Grübel, M. Scholz, D. Nordlund, W. Zhang, R. W. Hartsock, W. F. Schlotter, J. J. Turner, B. Kennedy, F. Hennies, F. M. F. De Groot, K. J. Gaffney, S. Techert, M. Odelius and A. Föhlisch, Nature, 2015, 520, 78–81 CrossRef CAS PubMed.
- E. J. Mascarenhas, M. Fondell, R. Büchner, S. Eckert, V. Vaz da Cruz and A. Föhlisch, Phys. Chem. Chem. Phys., 2022, 24, 17979–17985 RSC.
- R. M. Jay, M. R. Coates, H. Zhao, M.-O. Winghart, P. Han, R.-P. Wang, J. Harich, A. Banerjee, H. Wikmark, M. Fondell, E. T. J. Nibbering, M. Odelius, N. Huse and P. Wernet, J. Am. Chem. Soc., 2024, 146, 14000–14011 CrossRef CAS PubMed.
- R. M. Jay, A. Banerjee, T. Leitner, R.-P. Wang, J. Harich, R. Stefanuik, H. Wikmark, M. R. Coates, E. V. Beale, V. Kabanova, A. Kahraman, A. Wach, D. Ozerov, C. Arrell, P. J. M. Johnson, C. N. Borca, C. Cirelli, C. Bacellar, C. Milne, N. Huse, G. Smolentsev, T. Huthwelker, M. Odelius and P. Wernet, Science, 2023, 380, 955–960 CrossRef CAS PubMed.
- V. Vaz da Cruz, E. J. Mascarenhas, R. Büchner, R. M. Jay, M. Fondell, S. Eckert and A. Föhlisch, Phys. Chem. Chem. Phys., 2022, 24, 27819–27826 RSC.
- B. E. Van Kuiken, N. Huse, H. Cho, M. L. Strader, M. S. Lynch, R. W. Schoenlein and M. Khalil, J. Phys. Chem. Lett., 2012, 3, 1695–1700 CrossRef CAS PubMed.
- R. K. Hocking, E. C. Wasinger, F. M. F. De Groot, K. O. Hodgson, B. Hedman and E. I. Solomon, J. Am. Chem. Soc., 2006, 128, 10442–10451 CrossRef CAS PubMed.
- A. A. Cordones, J. H. Lee, K. Hong, H. Cho, K. Garg, M. Boggio-Pasqua, J. J. Rack, N. Huse, R. W. Schoenlein and T. K. Kim, Nat. Commun., 2018, 9, 1989 CrossRef PubMed.
- R. M. Jay, J. Norell, S. Eckert, M. Hantschmann, M. Beye, B. Kennedy, W. Quevedo, W. F. Schlotter, G. L. Dakovski, M. P. Minitti, M. C. Hoffmann, A. Mitra, S. P. Moeller, D. Nordlund, W. Zhang, H. W. Liang, K. Kunnus, K. Kubiček, S. A. Techert, M. Lundberg, P. Wernet, K. Gaffney, M. Odelius and A. Föhlisch, J. Phys. Chem. Lett., 2018, 9, 3538–3543 CrossRef CAS PubMed.
- R. M. Jay, A. Banerjee, M. Reinhard, H. Zhao, N. Huse, K. J. Gaffney, T. Kroll, D. Sokaras and P. Wernet, Chem. Sci., 2026 10.1039/D5SC09924B.
- T. Lian, S. E. Bromberg, M. C. Asplund, H. Yang and C. B. Harris, J. Phys. Chem., 1996, 100, 11994–12001 CrossRef CAS.
- A. G. Joly and K. A. Nelson, Chem. Phys., 1991, 152, 69–82 CrossRef CAS.
- W. Gawelda, M. Johnson, F. M. F. De Groot, R. Abela, C. Bressler and M. Chergui, J. Am. Chem. Soc., 2006, 128, 5001–5009 CrossRef CAS PubMed.
- J. K. McCusker, Science, 2019, 363, 484–488 CrossRef CAS PubMed.
- C. Kayran, M. Richards and P. C. Ford, Inorg. Chim. Acta, 2004, 357, 143–148 CrossRef CAS.
- H. Hermann, F. W. Grevels, A. Henne and K. Schaffner, J. Phys. Chem., 1982, 86, 5151–5154 CrossRef CAS.
- M. Tronc and C. Dezarnaud-Dandine, Chem. Phys. Lett., 1991, 184, 267–270 CrossRef CAS.
- M. Guo, T. Dederichs, L. Enzmann, A. Banerjee, V. Zamudio-Bayer, M. Lundberg, P. Wernet and R. M. Jay, J. Chem. Phys., 2025, 163, 465–502 Search PubMed.
- T. A. Albright, J. K. Burdett and M.-H. Whangbo, in Orbital Interactions in Chemistry, Wiley, 2013, pp. 465–502 Search PubMed.
- C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- R. M. Jay, S. Eckert, M. Fondell, P. S. Miedema, J. Norell, A. Pietzsch, W. Quevedo, J. Niskanen, K. Kunnus and A. Föhlisch, Phys. Chem. Chem. Phys., 2018, 20, 27745–27751 RSC.
- E. A. Cobar, R. Z. Khaliullin, R. G. Bergman and M. Head-Gordon, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6963–6968 CrossRef CAS PubMed.
- J. Y. Saillard and R. Hoffmann, J. Am. Chem. Soc., 1984, 106, 2006–2026 CrossRef CAS.
Footnotes |
| † Deutsches Elektronen-Synchrotron DESY, 22607 Hamburg, Germany. |
| ‡ Carl Zeiss MultiSEM GmbH, 73447 Oberkochen, Germany. |
| This journal is © The Royal Society of Chemistry 2026 |