
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
1,4-Bromoboration of nitroalkenes
Kanika
Vashisth
and
Caleb D.
Martin
 *
*
Baylor University, Department of Chemistry and Biochemistry, One Bear Place #97348, Waco, TX 76798, USA. E-mail: caleb_d_martin@baylor.edu
First published on 17th March 2026
Abstract
The selective 1,4-bromoboration of nitroalkenes occurs under mild conditions with a bromoborane featuring two 1-methyl-ortho-carborane substituents, BrBMeoCb2. The bromide is introduced on the β-carbon and the boryl moiety on the nitro group that is bound to boron in a κ2 manner. Screening a variety of other haloboranes gave either no reaction or indiscernible mixtures indicating the necessity for the BrBMeoCb2 reagent. The haloboration reaction tolerates a variety of aryl functional groups on the β-carbon as well as a hydrogen or methyl group on the α-carbon that all proceed with excellent regioselectivity and high isolated yields.
Boron addition reactions are powerful synthetic tools to introduce a boryl unit and additional functional group on an unsaturated substrate.1,2 The most prevalent boron addition reactions are hydroboration and carboboration that introduce a B–H and B–C bond as the second functional group, respectively.3–11 Despite the first haloboration reaction being reported over 80 years ago, examples of haloboration reactions are considerably more scarce than hydroboration and carboboration.12,13 The haloboration reaction installs a C–halogen bond that can be an effective synthetic handle to diversify the products.14
The most common haloboration reactions have been with alkynes to furnish boryl-vinyl-halides (I) with terminal alkynes being more reactive than internal alkynes (Fig. 1a).14 Pioneering work by Lappert and Eisch revealed that bromoboranes are much more effective than chloroboranes.12,15–18 This is due to the substrate interacting more favorably with electrophilic boron reagents as well as the increased lability of the boron-halogen bond with the heavier halogens.19,20 Recently, Ingleson, Pei, and Yuan revisited this historical reaction on alkyne substrates cleverly designed for cascade cyclizations to furnish polycyclic frameworks.21–23 From these efforts, biologically active compounds as well as B,N and B,O-containing analogues of polycyclic aromatic hydrocarbons have been accessed.23–26
 | ||
| Fig. 1 Representative haloboration reactions of unsaturated organic molecules. | ||
The haloboration of alkenes has been investigated but advancements have been hindered by the saturated products bearing a vicinal halide and boryl group being thermodynamically uphill thus reverting to starting materials, other products, or polyolefins.27–30 The first attempt was reported by Lappert on the haloboration of cycloalkenes with BBr3 that did not give any haloboration product.16 For example, the reaction with cyclohexene only gave CyBr, CyBBr2, and cyclohexenyl-BBr2.
In 1966, Lappert reported a haloboration with BCl3 on the unconjugated diene, norbornadiene, which furnished a 1,4-choroboration product with a C–C bond formed to give the cyclopropane containing product II (Fig. 1b).15,16 Allene haloboration has been more successful, however, the vinyl-BBr2/BCl2 products are susceptible to degradation, thus alkoxy groups have been used to isolate the haloboration products as boronic esters (III, Fig. 1c).31,32 In 2019, Chang reported the chloroboration of allenylsilanes with BCl3 and quenched with pinacol to furnish the 1,3-chloroboration product through an isomerization that was isolated as the Bpin species IV (Fig. 1d).33 In 2005, Bubov reported the reaction of allyl(dichloro)borane with alkynes to give the cis-allylboration intermediate V that underwent isomerization by an intramolecular chloroboration to give the cyclized product VI (Fig. 1e).34 Like alkenes, heteroatom multiple bonds are also rare with the minimal examples being with selected isocyanates, isothiocyanates, nitriles, isonitriles, and imines.35–42
In nitroalkenes, the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond is activated by the NO2 group that enhances electrophilicity at the β-carbon. We believed that the nitro group could also modulate the reactivity by coordination to boron to enable a haloboration reaction. In this work, we investigate if haloboranes react with nitroalkenes to furnish 1,4-haloboration products.
C bond is activated by the NO2 group that enhances electrophilicity at the β-carbon. We believed that the nitro group could also modulate the reactivity by coordination to boron to enable a haloboration reaction. In this work, we investigate if haloboranes react with nitroalkenes to furnish 1,4-haloboration products.
 | ||
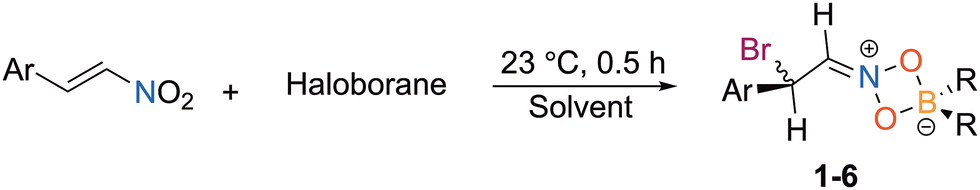
| Fig. 2 Solid state structures of 1–3. Ellipsoids depicted at the 50% probability level and hydrogen atoms omitted for clarity except the two on the formerly alkenyl carbons. | ||
We first screened the reactions of haloboranes that had been successful in literature reports for alkyne haloboration, specifically BCl3, BBr3, PhBCl2, PhBBr2, and Ph2BBr. The reactions with trans-β-nitrostyrene in benzene at 23 °C for 30 minutes were assessed by 1H NMR spectroscopy that revealed all five gave indiscernible mixtures (Table 1, entries 1–5). The reaction with the more Lewis acidic bromoborane featuring electron withdrawing perfluorophenyl groups, BrB(C6F5)2,43 also gave multiple products that we were unable to isolate (entry 6).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ar | Haloborane | R | Solvent | Isolated yield |
| 1 | Ph | BCl3 | Cl | C6D6 | Complex mixture |
| 2 | Ph | BBr3 | Br | C6D6 | Complex mixture |
| 3 | Ph | PhBCl2 | Ph/Cl | C6D6 | Complex mixture |
| 4 | Ph | PhBBr2 | Ph/Br | C6D6 | Complex mixture |
| 5 | Ph | Ph2BBr | Ph | C6D6 | Complex mixture |
| 6 | Ph | (C6F5)2BBr | C6F5 | C6D6 | Complex mixture |
| 7 | Ph | ClBMeoCb2 | Me oCb | C6D6 | NR |
| 8 | Ph | ClBTMSoCb2 | TMS oCb | C6D6 | NR |
| 9 | Ph | BrBTMSoCb2 | TMS oCb | C6D6 | NR |
| 10 | Ph | BrBMeoCb2 | Me oCb | C6H6 | 1 (83%) |
| 11 | Ph | BrBMeoCb2 | Me oCb | C7H8 | 1 (68%) |
| 12 | Ph | BrBMeoCb2 | Me oCb | CH2Cl2 | 1 (61%) |
| 13 | Ph | BrBMeoCb2 | Me oCb | C6H5F | 1 (81%) |
| 14 | 4-F-Ph | BrBMeoCb2 | Me oCb | C6H6 | 2 (74%) |
| 15 | 4-MeO-Ph | BrBMeoCb2 | Me oCb | C6H6 | 3 (79%) |
| 16 | 3,4-OCH2O-Ph | BrBMeoCb2 | Me oCb | C6H6 | 4 (98%) |
| 17 | 1-Np | BrBMeoCb2 | Me oCb | C6H6 | 5 (95%) |
| 18 | 2-thiophene | BrBMeoCb2 | Me oCb | C6H6 | 6 (94%) |
Recently, it has been demonstrated that ortho-carboranes are highly effective substituents to enhance the Lewis acidity at boron.44–55 Regarding boron addition reactions, the ortho-carborane substituted secondary borane, HBMeoCb2 (MeoCb = 1-methyl-ortho-carborane) and related carborane borenium compounds are extremely potent hydroboration reagents.45,54,55 We surmised the tremendous bulk and Lewis acidity of ortho-carborane substituted haloboranes could enable selective haloboration. Bis(ortho-carboranyl)boranes with a halogen on boron that feature methyl and trimethylsilyl (TMS) groups on the ortho-carbons have been reported, specifically ClBMeoCb2, BrBMeoCb2, and BrBTMSoCb2 (MeoCb = 1-methyl-ortho-carborane, TMSoCb = 1-trimethylsilyl-ortho-carborane).45,56 The chloro-borane with TMSoCb substituents, ClBTMSoCb2, was accessed by deprotonation of two equivalents of TMSoCb and reaction with BCl3 to complete the series. Reactions of both chloro species (ClBMeoCb2 and ClBTMSoCb2) and BrBTMSoCb2 did not result in any reaction, only unreacted starting material was detected by 1H NMR spectroscopy (entries 7–9).
The reaction of trans-β-nitrostyrene with BrBMeoCb2 led to a light-yellow solution that showed a dominant new species by 1H NMR spectroscopy with consumption of both starting materials (entry 10). The 11B{1H} NMR spectrum features a new peak in the tetracoordinate region at 8.8 ppm along with the disappearance of the peak corresponding to BrBMeoCb2 at 64.9 ppm. The 1H NMR spectrum has doublets at 5.57 and 5.12 ppm with matching coupling constants (J = 8 Hz). The product was isolated in 83% yield and the identity was determined by a single X-ray diffraction study as the 1,4-bromoboration product with the nitro group bound to boron in a κ2 fashion with an iminium center and the bromine introduced on the α-carbon (1, Fig. 2). The product bears a chiral center but crystallizes in the P21/c centrosymmetric space group indicating it is a racemate. The solid-state structure matches the solution NMR data with the tetracoordinate signal in the 11B{1H} NMR spectrum and the two doublets in the 1H NMR spectrum at 5.57 and 5.12 ppm are assigned to the protons on the iminium carbon (Ha) and halogen substituted tertiary carbon (Hb). To determine if there is a solvent effect in the haloboration of trans-β-nitrostyrene with BrBMeoCb2, we screened CH2Cl2, toluene, and fluorobenzene (entries 11–13). Dichloromethane and toluene resulted in a decrease in isolated yield of 1 compared to benzene (63% and 68% c.f. 83%) while fluorobenzene gave a comparable yield (81%). Hence, we moved forward with BrBMeoCb2 in benzene to investigate the breadth of the bromoboration reactions as the yield is the highest and enables monitoring reactions in situ in benzene-d6.
The reaction between (E)-1-fluoro-4-(2-nitrovinyl)benzene and BrBMeoCb2 led to a dark yellow solution which became colorless after stirring for 30 minutes at 23 °C (entry 14). The product (2) was isolated in 75% yield. Growing single crystals enabled determination of the solid-state structure as the iminium haloboration product akin to 1. The reaction tolerated alkoxy substitution with a methoxy group in the 4-position as well as a 3,4-methylenedioxy group that gave the iminium products in 79% (3) and 98% (4) isolated yields, respectively (entries 15 and 16). The structure of 3 was confirmed by single crystal X-ray diffraction. A naphthyl group was investigated to determine the impact of aryl fusion to the styrene. The reaction of BrBMeoCb2 with 1-(E)-[2-nitroethenyl]naphthalene afforded iminium product 5 in 95% isolated yield (entry 17). The reaction is tolerant to a heteroarene with a thiophene as the aryl group with (E)-2-(2-nitroethenyl)thiophene generating iminium 6 in 94% yield (entry 18). To determine if substitution on the β-carbon could be tolerated, trans-β-methyl-β-nitrostyrene was reacted with BrBMeoCb2 that smoothly generated the haloboration product 7 in 78% yield (Scheme 1).
 | ||
| Scheme 1 Reaction of trans-β-methyl-β-nitrostyrene with BrBMeoCb2. | ||
The 11B NMR signals in 1–7 for the tetracoordinate boron all fit in the narrow region of 8.2–9.2 ppm (Table 2). The di-substituted alkene products (1–6) feature doublets from 5.02–5.92 ppm with coupling constants of 8 Hz. The proton on the iminium carbon of 7 at 5.56 ppm lies in this chemical shift window. In 2, the 19F{1H} NMR spectrum features an upfield shift for the para-fluorine from the starting material (−109.0 c.f. −105.7 ppm).
|
|
|||
|---|---|---|---|
| Compound | Ha | Hb | 11B |
| 1 | 5.57 | 5.12 | 8.9 |
| 2 | 5.48 | 5.02 | 9.2 |
| 3 | 5.70 | 5.22 | 9.0 |
| 4 | 5.92 | 5.46 | 8.9 |
| 5 | 5.67 | 5.11 | 9.0 |
| 6 | 5.55 | 5.40 | 9.1 |
| 7 | N/A | 5.56 | 8.2 |
Examining the solid-state structures of 1–3 reveal short C(1)–N(1) bond distances consistent with the iminium structures with 2 and 3 having shorter bonds attributed to the electron withdrawing fluoro and methoxy substituents [Tables 3, 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.323(5) Å, 2:1.291(3) Å, 3:1.278(3) Å]. The C(1)–C(2) bonds in the three compounds range 1.472(3) to 1.493(6) Å that validate the elongation of the alkenes to single bonds. The N–O bond distances all range 1.331(2) to 1.347(4) Å revealing that the two nitrogen–oxygen bonds are within the error of measurement in the three compounds. The B–O bond lengths are consistent with both oxygen atoms binding boron in an equivalent manner [range = 1.557(6) to 1.573(5) Å]. The geometry at nitrogen is trigonal planar [Σ∠N range = 360.0(4)° to 360.1(3)°] but with a compressed O(1)–N(1)–O(2) bond angle due to the four membered ring [102.9(3) to 103.55(16)°].
:1.323(5) Å, 2:1.291(3) Å, 3:1.278(3) Å]. The C(1)–C(2) bonds in the three compounds range 1.472(3) to 1.493(6) Å that validate the elongation of the alkenes to single bonds. The N–O bond distances all range 1.331(2) to 1.347(4) Å revealing that the two nitrogen–oxygen bonds are within the error of measurement in the three compounds. The B–O bond lengths are consistent with both oxygen atoms binding boron in an equivalent manner [range = 1.557(6) to 1.573(5) Å]. The geometry at nitrogen is trigonal planar [Σ∠N range = 360.0(4)° to 360.1(3)°] but with a compressed O(1)–N(1)–O(2) bond angle due to the four membered ring [102.9(3) to 103.55(16)°].
| 1 | 2 | 3 | |
|---|---|---|---|
| C(1)–N(1) | 1.285(5) | 1.291(3) | 1.279(6) |
| C(1)–C(2) | 1.493(6) | 1.472(3) | 1.489(7) |
| C(2)–C(1)–N(1) | 121.0(3) | 121.3(2) | 119.5(4) |
| B(1)–O(1) | 1.569(5) | 1.570(3) | 1.562(6) |
| B(1)–O(2) | 1.573(5) | 1.562(3) | 1.557(6) |
| N(1)–O(1) | 1.334(4) | 1.331(2) | 1.345(5) |
| N(1)–O(2) | 1.347(4) | 1.339(2) | 1.343(5) |
| O(1)–N(1)–O(2) | 103.0(3) | 103.55(16) | 102.9(3) |
| Σ ∠N | 360.1(3) | 360.0(2) | 360.0(4) |
To verify that nitro groups interact with BrBMeoCb2, the reaction of BrBMeoCb2 with nitrobenzene was carried out in C6D6 at 23 °C. After 20 min, slight shifts were observed in the 1H NMR spectrum for the aryl peaks of nitrobenzene (up to 0.13 ppm) that indicates a coordinative interaction. The reaction of 1 with pyridine did not show any evidence of a retro-1,4-bromoboration, but rather a complex reaction mixture after stirring for 24 h at 23 °C in C6D6. To further determine if a retro-1,4-bromoboration occurs, the product with the electron withdrawing fluoride, 2, was reacted with trans-β-nitrostyrene in C6D6 at 23 °C that did not result in any change based on in situ1H and 11B NMR spectroscopy.
A proposed mechanism for the haloboration reaction is described in Scheme 2. The first step is coordination of the nitro oxygen to the haloborane to generate Int1 where the molecule is neutral with positive charge on nitrogen and negative on the boron center. Next, the oxygen delocalizes electron density to make a π bond with boron to facilitate the displacement of the bromide to furnish Int2. Finally, the bromide attacks the Michael acceptor carbon to access the 1,4-bromoboration iminium product.
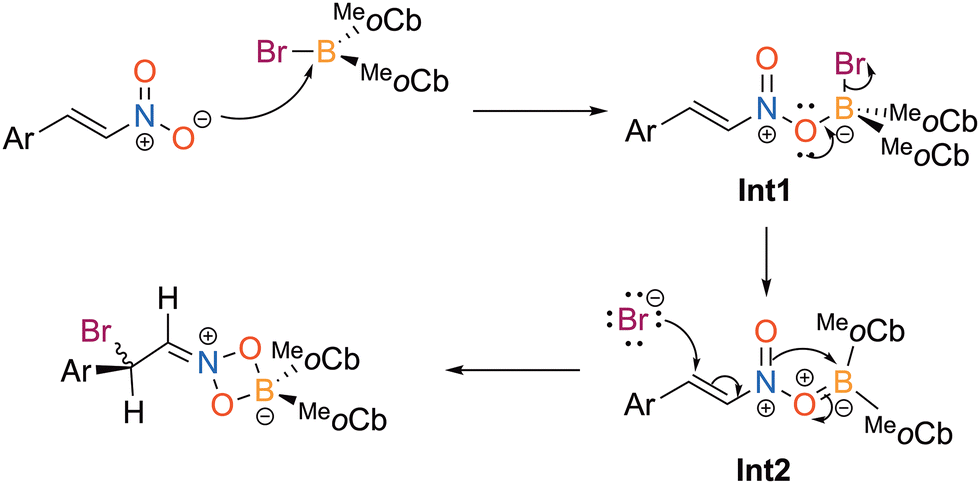 | ||
| Scheme 2 Proposed mechanism for the bromoboration of trans-β-nitrostyrenes. | ||
In this work, we shed light on rare haloboration reactions by demonstrating the 1,4-bromoboration of nitroalkenes in high isolated yields under mild reaction conditions. In haloborane screening, only BrBMeoCb2 was effective indicating that carboranyl substituted boranes can be leveraged to access unique reactivity. The products feature a chelated borate with an iminium nitrogen and bromide on the β-carbon as confirmed by single crystal X-ray diffraction and NMR spectroscopy. The reaction tolerates a variety of aryl groups on the β-carbon and a methyl group or hydrogen on the α-carbon. These findings greatly advance the miniscule amount of haloboration reactions in the literature that are potentially valuable tools for the synthetic chemist's arsenal.
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.Data availability
Supplementary information (SI): contains the experimental details, NMR spectra and the X-ray crystallographic details. See DOI: https://doi.org/10.1039/d6cc01239f.CCDC 2523539 (1) 2523540 (2) and 2523541 (3) contain the supplementary crystallographic data for this paper.57a–c
Acknowledgements
We are grateful to the National Science Foundation (Award No. 2349851) and the Welch Foundation (AA-2203-20240404) for their generous support of this work. We thank Sameera Ranasinghe for assistance with X-ray crystallography.References
- H. C. Brown and G. Zweifel, J. Am. Chem. Soc., 1959, 81, 4106–4107 CrossRef CAS.
- K. Burgess and M. J. Ohlmeyer, Chem. Rev., 1991, 91, 1179–1191 CrossRef CAS.
- Y. Su, K. Shen, P. Shi, C. Liu, S. Song, H. Yi, A. Lei, Y. Liu, H. Wang and X. Li, J. Am. Chem. Soc., 2025, 147, 39561–39571 CrossRef CAS PubMed.
- P. Huninik, P. Sharma, V. B. Saptal, M. Slaby, R. Langer, P. Kumar, A. Shayesteh Zeraati, X. Wang, M. Petr and M. Otyepka, ACS Catal., 2025, 15, 17347–17360 CrossRef CAS PubMed.
- G. Kehr and G. Erker, Chem. Commun., 2012, 48, 1839–1850 Search PubMed.
- G. Kehr and G. Erker, Chem. Sci., 2016, 7, 56–65 RSC.
- J. R. Lawson, V. Fasano, J. Cid, I. Vitorica-Yrezabal and M. J. Ingleson, Dalton Trans., 2016, 45, 6060–6070 RSC.
- L. C. Wilkins, Y. Soltani, J. R. Lawson, B. Slater and R. L. Melen, Chem. – Eur. J., 2018, 24, 7364–7368 CrossRef CAS PubMed.
- T. A. Gazis, B. A. M. Thaker, D. Willcox, D. M. Ould, J. Wenz, J. M. Rawson, M. S. Hill, T. Wirth and R. L. Melen, Chem. Commun., 2020, 56, 3345–3348 RSC.
- R. L. Melen, M. M. Hansmann, A. J. Lough, A. S. K. Hashmi and D. W. Stephan, Chem. – Eur. J., 2013, 19, 11928–11938 CrossRef CAS PubMed.
- M. M. Hansmann, R. L. Melen, M. Rudolph, F. Rominger, H. Wadepohl, D. W. Stephan and A. S. K. Hashmi, J. Am. Chem. Soc., 2015, 137, 15469–15477 Search PubMed.
- M. Lappert and B. Prokai, J. Organomet. Chem., 1964, 1, 384–400 CrossRef CAS.
- H. Arnold, US Pat, 2402589, 1946 Search PubMed.
- S. Kirschner, K. Yuan and M. J. Ingleson, New J. Chem., 2021, 45, 14855–14868 RSC.
- F. Joy and M. Lappert, Proc. Chem. Soc., 1960, 1, 353–354 Search PubMed.
- F. Joy, M. Lappert and B. Prokai, J. Organomet. Chem., 1966, 5, 506–519 CrossRef CAS.
- J. J. Eisch and H. P. Becker, J. Organomet. Chem., 1979, 171, 141–153 CrossRef CAS.
- J. J. Eisch and L. J. Gonsior, J. Organomet. Chem., 1967, 8, 53–64 CrossRef CAS.
- S. Yang, A. Alix, C. Bour and V. Gandon, Inorg. Chem., 2021, 60, 5507–5522 CrossRef CAS PubMed.
- J. Stosek, H. Semrád, C. Mazal and M. Munzarová, J. Phys. Chem. A, 2023, 127, 6135–6146 CrossRef CAS PubMed.
- A. J. Warner, K. M. Enright, J. M. Cole, K. Yuan, J. S. McGough and M. J. Ingleson, Org. Biomol. Chem., 2019, 17, 5520–5525 RSC.
- A. J. Warner, J. R. Lawson, V. Fasano and M. J. Ingleson, Angew. Chem., Int. Ed., 2015, 127, 11397–11401 CrossRef.
- K. Yuan and M. J. Ingleson, Angew. Chem., Int. Ed., 2023, 62, e202301463 CrossRef CAS PubMed.
- N. Okada, S. Nakatsuka, R. Kawasumi, H. Gotoh, N. Yasuda and T. Hatakeyama, Chem. – Eur. J., 2023, 29, e202202627 CrossRef CAS PubMed.
- F.-D. Zhuang, J.-M. Han, S. Tang, J.-H. Yang, Q.-R. Chen, J.-Y. Wang and J. Pei, Organometallics, 2017, 36, 2479–2482 Search PubMed.
- X. Feng, Z. Liu, Q.-Y. Ni, B. Wang, M. J. Ingleson and K. Yuan, Org. Lett., 2024, 26, 10339–10344 CrossRef CAS PubMed.
- C. Wang and M. Uchiyama, Eur. J. Org. Chem., 2012, 6548–6554 CrossRef CAS.
- M. F. Hawthorne and J. A. Dupont, J. Am. Chem. Soc., 1958, 80, 5830–5832 CrossRef CAS.
- H. C. Brown and K. A. Keblys, J. Am. Chem. Soc., 1964, 86, 1795–1801 CrossRef CAS.
- R. Köster, G. Griasnow, W. Larbig and P. Binger, Justus Liebigs Ann. Chem., 1964, 672, 1–34 CrossRef.
- S. Hara and A. Suzuki, Tetrahedron Lett., 1991, 32, 6749–6752 CrossRef CAS.
- S. Hara, S. Takinami, S. Hyuga and A. Suzuki, Chem. Lett., 1984, 345–348 Search PubMed.
- Z. Yang, T. Liu, X. Chen, R. Wan, Y. Li, X. Wang, C.-H. Yang and J. Chang, Org. Lett., 2019, 21, 9541–9544 CrossRef CAS PubMed.
- Y. N. Bubnov, N. Y. Kuznetsov, F. V. Pastukhov and V. V. Kublitsky, Eur. J. Chem., 2005, 4633 Search PubMed.
- A. Meller and W. Maringgele, Monatsh. Chem., 1968, 99, 2504–2513 Search PubMed.
- A. Meller and A. Ossko, Monatsh. Chem., 1969, 100, 1187–1194 Search PubMed.
- A. Meller, W. Maringgele and G. Maresch, Monatsh. Chem., 1974, 105, 637–647 Search PubMed.
- B. Bonnetot, B. Frange, F. Guilhon, H. Mongeot and W. Einholz, Polyhedron, 1994, 13, 2211–2216 Search PubMed.
- A. Meller and H. Batka, Monatsh. Chem., 1969, 100, 1823–1828 Search PubMed.
- F. S. Mair, R. Manning, R. G. Pritchard and J. E. Warren, Chem. Commun., 2001, 1136–1137 RSC.
- Y. Segawa, Y. Suzuki, M. Yamashita and K. Nozaki, J. Am. Chem. Soc., 2008, 130, 16069–16079 Search PubMed.
- M. Lappert and B. Prokai, J. Chem. Soc., 1963, 4223–4229 Search PubMed.
- A. Wong, G. Alcántara, M. Avalos, G. Wu and G. Ménard, Z. Anorg. Allg. Chem., 2023, 649, e202300007 CrossRef CAS.
- M. O. Akram, C. D. Martin and J. L. Dutton, Inorg. Chem., 2023, 62, 13495–13504 CrossRef CAS PubMed.
- M. O. Akram, J. R. Tidwell, J. L. Dutton and C. D. Martin, Angew. Chem., Int. Ed., 2023, 62, e202307040 Search PubMed.
- S. Ranasinghe, Y. Li, M. E. Andrews, M. O. Akram, R. A. Thornton and C. D. Martin, Chem. Commun., 2025, 61, 10182–10185 RSC.
- L. Xiang, A. Matler, L. Tan and Q. Ye, Dalton Trans., 2024, 53, 11655–11658 RSC.
- L. Xiang, J. Wang, A. Matler and Q. Ye, Chem. Sci., 2024, 15, 17944–17949 RSC.
- C. Zhang, J. Wang, Z. Lin and Q. Ye, Inorg. Chem., 2022, 61, 18275–18284 CrossRef CAS PubMed.
- M. O. Akram, K. A. French, K. L. Shuford and C. D. Martin, J. Am. Chem. Soc., 2025, 147, 33120–33126 CrossRef CAS PubMed.
- V. Rubio, M. Durham, M. Scurria, T. Kerr, O. Gutierrez and A. Spokoyny, ChemRxiv, 2025, preprint, chemrxiv-2025-n9bqp DOI:10.26434/chemrxiv-2025-n9bqp.
- K. Vashisth and C. D. Martin, Inorg. Chem., 2025, 64, 24192–24200 Search PubMed.
- Y. Liu, W. Dong, Z. H. Li and H. Wang, Chem, 2021, 7, 1843–1851 CAS.
- X. Tan and H. Wang, ChemCatChem, 2023, 15, e202300734 CrossRef CAS.
- X. Tan, X. Wang, Z. H. Li and H. Wang, J. Am. Chem. Soc., 2022, 144, 23286–23291 CrossRef CAS PubMed.
- K. Vashisth, R. Thornton, B. Yoon and C. Martin, ChemRxiv, 2025, preprint, chemrxiv-2025-04xgt DOI:10.26434/chemrxiv-2025-04xgt.
- (a) CCDC 2523539: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qpyhw; (b) CCDC 2523540: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qpyjx; (c) CCDC 2523541: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qpyky.
| This journal is © The Royal Society of Chemistry 2026 |