
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCross-coupling of aryl aldehydes and benzyl chlorides enabled by dual N-heterocyclic carbene/cobalt catalysis
Hassan
Jomaa
,
David
Martin
 and
Eder
Tomás-Mendivil
*
and
Eder
Tomás-Mendivil
*
Univ. Grenoble Alpes, CNRS, DCM, UMR 5250, 38000 Grenoble, France. E-mail: eder.tomas-gonzalez-de-mendivil@univ-grenoble-alpes.fr
First published on 21st April 2026
Abstract
We have developed a dual N-heterocyclic carbene/cobalt-salen catalytic system for the cross-coupling between aryl aldehydes and benzyl chlorides via direct C–H activation of aldehydes. The catalysts employed are readily accessible and the transformation affords ketones in moderate to high yields.
N-heterocyclic carbene (NHC) catalysis is an attractive synthetic tool allowing for mild reaction conditions and chemo- and/or enantio-selective transformations.1 The most common substrates are aldehydes, which react with NHCs to form enaminols (so-called Breslow intermediates)2 able to react with a wide variety of electrophiles. Traditionally, ionic pathways dominated the field (Scheme 1a); however, in recent years, radical NHC catalysis has been developed for the coupling of unprecedented substrates.1c–f Among readily accessible electrophiles, organic halides have been coupled with aldehydes employing NHC catalysis.3 Aryl iodides4 and primary and secondary alkyl iodides and bromides5,6 can be activated employing NHCs as sole catalysts. Cooperative dual NHC/metal radical catalysis7 employing nickel8 or palladium9 has been developed for tertiary alkyl iodides and bromides, respectively.
 | ||
| Scheme 1 Representative examples on NHC-catalysed cross-coupling between aldehydes and benzyl halides and cooperative dual NHC/metal catalysis. | ||
Strikingly, benzyl halides, classic electrophiles, have been employed only occasionally and with mediocre outcome. Representative examples include the work from Du and Deng et al. with a stoichiometric amount of a thiazolium salt for the coupling between aromatic aldehydes and benzyl bromides (and a chloride) in moderate to low yields (Scheme 1b)10 and the competition control experiment from Glorius et al. employing diarylbromomethanes and benzyl bromide,11 among others.6,12 Finally, the most remarkable finding was reported by Zhang and Ye and co-workers in 2023, on a cooperative dual NHC/Pd radical strategy that is applicable to benzyl bromides in good to excellent yields.9 In this work, it was proposed that enolate A− would reduce the metal center and Pd0 would activate the benzyl bromide. Then, persistent radical A˙ and transient Bn˙ would couple releasing the free carbene and product.
Coupling of benzyl chlorides is the next obvious challenge in the field of NHC organocatalysis. We envisioned that applying a dual NHC/metal strategy would promote their use as electrophiles. Square planar [CoI(salen)]− complexes are known to activate benzyl chlorides.13 The reduction potential of [CoII(salen)] complexes stand ca. −1.5 V vs. SCE,13b,e–f,14 and they fall within the range of the reduction power of Breslow-type enolates (−1.9 to −1.4 V vs. SCE).15 Therefore, it is reasonable to propose a dual NHC/Co catalytic cycle where the enolate is able to reduce the cobalt species in order to activate benzyl chlorides.
We began our studies by testing the cross-coupling between 4-chlorobenzaldehyde (1a) and benzyl chloride (2a) under NHC/metal free conditions, obtaining alkylated benzoin 3aa in 22% NMR yield (Table 1, entry 1), probably via NHC-catalyzed benzoin condensation and alkylation under basic conditions.16 Then, we tested an NHC1/Co-salen combination and 4aa was formed only in 6% NMR yield (entry 2). Surprisingly most of the aldehyde was still present; i.e. the benzoin was not formed, suggesting the deactivation of the NHC. We considered the more bulky (1-chloroethyl)benzene (2b) and, to our delight, ketone 4ab was formed quantitatively (99% NMR yield, entry 4). In the absence of the cobalt catalyst, a moderate yield of 33% was achieved (entry 3).
|
|
|||
|---|---|---|---|
| Entry | Benzyl chloride | [Co] (x mol%) | Yield (%)b |
| a Standard reaction conditions: 1a (0.3 mmol), 2a-b (0.25 mmol), NHC1 (0.038 mmol; 15 mol%), [Co(salen)] (when added), and Cs2CO3 (0.3 mmol) in 1 mL of dry THF under argon atmosphere for 16 h at 60 °C. b NMR yields for products are given employing 1,3,5- trimethoxybenzene (TMB) as internal standard. | |||
| 1 | 2a | 0 mol% | 22% (3aa) |
| 2 | 2a | 10 mol% | 6% (4aa) |
| 3 | 2b | 0 mol% | 33% (4ab) |
| 4 | 2b | 10 mol% | 99% (4ab) |

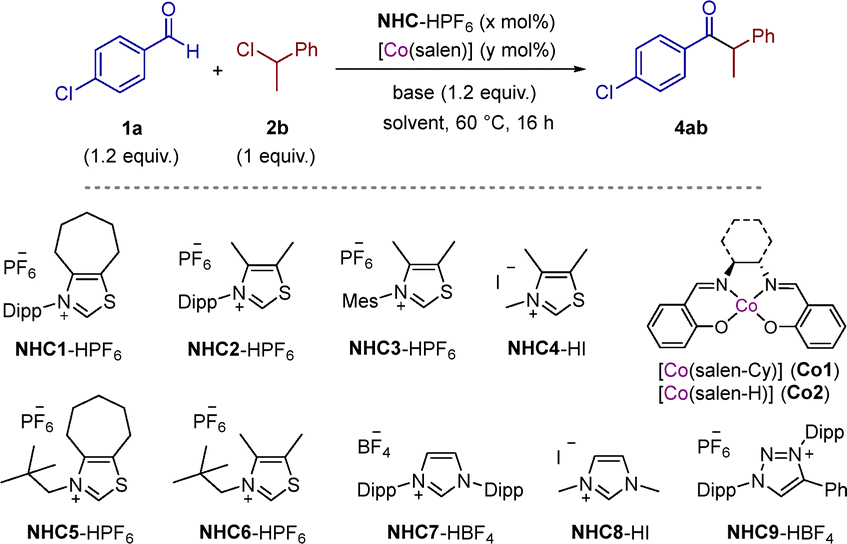
We chose (1-chloroethyl)benzene 2b as model substrate to find optimal conditions for the reaction. Initial reaction conditions for the cross-coupling between 1a (1.2 equiv.) and 2b (1 equiv.) employing 15 mol% of NHC1, 10 mol% of [Co(salen-Cy)] (Co1) and Cs2CO3 as base in THF gave 4ab in 99% NMR yield (Table 2, entry 1). We found that in the absence of salen ligand (entry 2) and cobalt complex (entry 3), the yield dropped to ca. 30%. This suggests that the carbene is able to promote by itself the cross-coupling albeit with low efficiency. In the absence of NHC1 the starting materials were recovered (entries 4 and 5). The use of K2CO3 instead of Cs2CO3 was detrimental (entry 6), probably due to the stabilization of the enolate, lowering its reducing power.15 Then, we decreased both catalysts’ loadings to 10 mol% of NHC1 and 5 mol% of Co1 and the yield maintained quantitative (entry 7). The use of the simplest cobalt complex Co2, bearing the ethylene-bridged (salen-H) ligand, gave comparable outcome (96%; entry 8). The use of other solvents like acetonitrile, toluene or dimethylsulfoxide instead of tetrahydrofuran gave lower yields (entries 9–11). Finally, we screened different NHCs. Simplification of the backbone of thiazol-2-ylidene from cycloheptyl ring (NHC1) to readily accessible dimethyl backbone (NHC2) resulted in full conversion and 75% isolated yield (entry 12). Decreasing the steric factors of the carbene by replacing the N-(2,6-diisopropylphenyl) (Dipp) group by 2,4,6-trimethylphenyl (Mes), methyl or neopentyl gave lower yields (entries 13–16). This is probably due to the higher pyramidalization around the N-atom which lowers the reducing power of the enolate.15 The use of Arduengo's imidazolydenes (NHC7-8; entries 17 and 18) or Bertrand's mesoionic carbenes (NHC9; entry 19) did not lead to product 4ab.
|
|
||
|---|---|---|
| Entry | Reaction conditions | Yield (%)b |
| a Standard reaction conditions: 1a (0.3 mmol), 2b (0.25 mmol), NHC-HX, [Co(salen)], and base (0.3 mmol) in 1 mL of dry solvent under argon atmosphere for 16 h at 60 °C. b NMR yields for 4ab are given employing 1,3,5- trimethoxybenzene (TMB) as internal standard. c Isolated yield. | ||
| 1 | NHC1 (15 mol%), Co1 (10 mol%), Cs2CO3, THF | 99 |
| 2 | NHC1 (15 mol%), CoBr2 (10 mol%), Cs2CO3, THF | 28 |
| 3 | NHC1 (15 mol%), Cs2CO3, THF | 33 |
| 4 | Cs2CO3, THF | 0 |
| 5 | Co1 (10 mol%), Cs2CO3, THF | 0 |
| 6 | NHC1 (15 mol%), Co1 (10 mol%), K2CO3, THF | 54 |
| 7 | NHC1 (10 mol%), Co1 (5 mol%), Cs2CO3, THF | 99 |
| 8 | NHC1 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 96 |
| 9 | NHC1 (10 mol%), Co2 (5 mol%), Cs2CO3, Acetonitrile | 91 |
| 10 | NHC1 (10 mol%), Co2 (5 mol%), Cs2CO3, Toluene | 73 |
| 11 | NHC1 (10 mol%), Co2 (5 mol%), Cs2CO3, DMSO | 70 |
| 12 | NHC2 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 99 (75)c |
| 13 | NHC3 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 56 |
| 14 | NHC4 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 20 |
| 15 | NHC5 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 35 |
| 16 | NHC6 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 36 |
| 17 | NHC7 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 0 |
| 18 | NHC8 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 0 |
| 19 | NHC9 (10 mol%), Co2 (5 mol%), Cs2CO3, THF | 0 |
Then, we studied the scope of the reaction employing the simplest catalytic system: NHC2 and Co2 (Scheme 2). Regarding aldehydes, para- and meta-chloro substitution gave similar yields (61% for 4bb). Replacing the halogen atom in para position with bromine (4cb) or fluorine (4db) led to lower yields of 27% and 48%, respectively. Electron withdrawing trifluoromethyl (4eb) and cyano (4fb) groups led to lower yields (25% and 30%, respectively). An electron donating group such as methoxy (4gb) gave an isolated yield of 41%. N-Dipp substituted thiazol-2-ylidenes may find ortho substitution problematic.8 Our system made no exception and ortho-methoxy substituted product (4mb) was not observed. On the contrary, the use of less hindered N-neopentyl NHC517 led to a poor yield of 11%. 2-naphthyl (4hb) gave the highest yield reaching 96%. Heteroaryl functionalities were also compatible: furyl (4ib), thiofuryl (4jb) and pyridyl (4kb and 4lb) gave moderated isolated yields. Aliphatic aldehydes are not compatible and the use of carbene NHC5 gave only traces of the desired product 4nb. Next, benzyl chloride derivatives were screened. Lengthening the aliphatic chain (4ac) or incorporating it into a cyclic structure (4ad) gave moderate yields. The use of a tertiary chloride gave 4ae in 42% yield. This is an interesting result from a mechanistic point of view, since the substrate is very unlikely to be activated via a SN2 mechanism; instead XAT is probably preferred (vide infra). Diphenylchloromethane and 9-chlorofluorene gave 4af and 4ag in modest isolated yields, respectively. para-Cyano (4ah) and para-methoxy (4ai) substituted benzyl chlorides are also compatible with higher yield for the electron deficient substrate. The pyridine group in the benzylic fragment gave good yield (4aj). As previously stated, unsubstituted benzyl chloride 2a gives very low yield employing NHC2. Employing NHC5, 4aa was obtained in 5% yield. Highly challenging unactivated tert-butyl chloride and cyclohexyl chloride did not lead to the desired ketones 4ak and 4al, respectively.
 | ||
| Scheme 2 Scope of the reaction at 1 mmol scale. Isolated yields given are unless otherwise noted. | ||
Intrigued by the differing reactivity of benzyl chlorides 2a and 2b, we postulated three hypotheses (Scheme 3):
 | ||
| Scheme 3 Different hypotheses to understand the origin of different reactivity of benzyl chlorides 2a-b. | ||
Hypothesis 1: regioselectivity issues in the radical cross-coupling step. Experimental18 and theoretical19 studies had proposed that steric factors may rule the cross-coupling between radicals leading to productive or unproductive pathways, which may also be the case herein (Scheme 3a). Note also that, only a few carbenes are known to be compatible in NHC radical catalysis with unhindered Bn˙ radicals: (i) 1,2,4-triazol-5-ylidenes via oxidative (photo)redox catalysis,20,21 (ii) an imidazolydene,22 and (iii) inefficiently few thiazol-2-ylidenes,23 except for an efficient N-neopentyl thiazol-2-ylidene/Pd cooperative catalytic system.9 On the contrary, more sterically crowded substituted benzyl radicals (ArCHR)˙ generated through radical relay strategies employing olefins are widely applied.4a,b,1c–f,24 Note that (ArCHMe)˙ has successfully been employed in enantioselective approaches too.25
Hypothesis 2: the nucleophilic attack of NHCs to benzyl chlorides to form enamines (Scheme 3b).26 We reacted 2a-b with NHC2 under catalytic conditions for 15 minutes and then aldehyde 1a was added and reacted for another 4 hours. In both cases, starting materials were recovered, the corresponding benzoin was not observed and neither products 3 or 4. Since this experiment does not discriminate between substrates 2a-b, such hypothesis was ruled out.
Hypothesis 3: square-planar [CoI(salen)]− complexes are strong nucleophiles and react with ArCHRCl leading to [CoIII(ArCHR)(salen)] complexes through a SN2 mechanism. These species are in equilibrium with [CoII(salen)] and (ArCHR)˙.13,27 [CoIII(ArCHR)(salen)] can also be reduced to the unstable species [CoII(ArCHR)(salen)]− and its homolytic decomposition leads to [CoI(salen)]− and (ArCHR)˙. Kinetics control these events and an appropriate match under catalytic conditions may explain the efficient formation of 4ab in contrast to 4aa. To probe for the presence of transient benzyl radicals, we tested 2b under optimized catalytic conditions in the presence of ten equivalents of styrene, but the corresponding radical relay product 5ab was not formed (Scheme 4). On the contrary, tertiary benzyl chloride 2e gave ketone 5ae suggesting the efficient trapping of the transient radical by styrene. This suggests that in the first case a benzyl radical is not released to the medium while in the second case it is, probably via a XAT event. In other words, the mechanism, and thus the outcome of the reaction, are substrate dependent (2avs.2bvs.2e) and the role of the cobalt catalyst is crucial.
 | ||
| Scheme 4 Radical relay tests. NMR yields are given employing 1,3,5- trimethoxybenzene (TMB) as internal standard. | ||
In conclusion, we have developed an efficient NHC promoted cross-coupling reaction between aromatic aldehydes and benzyl chlorides aided by a readily and commercially available cobalt catalyst. In the literature, as far as we are aware, the direct coupling between such substrates yielding ketones has only been reported once employing nickel/photoredox catalysis.28 Our system is complementary to that one and the results described herein support that merging NHC catalysis with transition metal catalysis has the potential to activate substrates beyond known boundaries.
This work was funded by the French National Agency for Re-search (ANR-23-CE07–0004–01). H. J. is grateful for the funding of his PhD thesis by this program. We acknowledge the ICMG analytic platform (FR 2607), the CNRS, the University of Grenoble Alpes, the Labex Arcane and CBHEUR-GS (ANR-17-EURE-0003).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details. See DOI: https://doi.org/10.1039/d6cc01204c.References
- (a) D. Enders, O. Niemeier and A. Henseler, Chem. Rev., 2007, 107, 5606 CrossRef CAS PubMed; (b) D. M. Flanigan, F. Romanov-Michailidis, N. A. White and T. Rovis, Chem. Rev., 2015, 115, 9307 CrossRef CAS PubMed; (c) A. V. Bay and K. A. Scheidt, Trends Chem., 2022, 4, 277 CrossRef CAS PubMed; (d) K. Liu, M. Schwenzer and A. Studer, ACS Catal., 2022, 12, 11984 CrossRef CAS; (e) H. Cai, X. Yang, S.-C. Ren and Y. R. Chi, ACS Catal., 2024, 14, 8270 CrossRef CAS; (f) S. Chakraborty, S. Barik and A. T. Biju, Chem. Soc. Rev., 2025, 54, 1102 RSC.
- (a) T. Ukai, S. Tanaka and S. Dokawa, J. Pharm. Soc. Jpn., 1943, 63, 296 CrossRef CAS; (b) S. Mizuhara and P. Handler, J. Am. Chem. Soc., 1954, 76, 571 CrossRef CAS; (c) R. Breslow, J. Am. Chem. Soc., 1958, 80, 3719 CrossRef CAS.
- F. Gao, Z. Zhang and X. Yan, ChemCatChem, 2024, 16, e202301331 CrossRef CAS.
- (a) C. Liu, W. Liu, A. Vianna, Z. Zhang, S. Huang, L. Huang, M. Melaimi, G. Bertrand and X. Yan, Chem Catal., 2021, 1, 196 Search PubMed; (b) Y. Matsuki, N. Ohnishi, Y. Kakeno, S. Takemoto, T. Ishii, K. Nagao and H. Ohmiya, Nat. Commun., 2021, 12, 3848 CrossRef CAS PubMed; (c) N. Assani, L. Delfau, P. Smits, S. Redon, Y. Kabri, E. Tomás-Mendivil, P. Vanelle, D. Martin and J. Broggi, Chem. Sci., 2024, 15, 14699 RSC; (d) B. Huang, Z. Zhang, J. Jiao, W. Liu and X. Yan, Org. Lett., 2024, 26, 7419 CrossRef CAS PubMed; (e) S. Liu, S. Zheng, Y. Zhang, K. Lin, W. Li, Z. Li and T. Zhu, Org. Lett., 2026, 28, 896 CrossRef CAS PubMed.
- (a) C. Liu, Z. Zhang, L.-L. Zhao, G. Bertrand and X. Yan, Angew. Chem., Int. Ed., 2023, 62, e202303478 CrossRef CAS PubMed; (b) Q.-Z. Li, R. Zeng, P.-S. Xu, X.-H. Jin, C. Xie, Q.-C. Yang, X. Zhang and J.-L. Li, Angew. Chem., Int. Ed., 2023, 62, e202309572 CrossRef CAS PubMed; (c) J. Jiao, Z. Zhang, G. Lu, S. Huang, Y. Bian, F. Gao, G. Bertrand and X. Yan, Chem. Sci., 2025, 16, 9163 RSC.
- Q.-Z. Li, R. Zeng, P.-S. Xu, X.-H. Jin, C. Xie, Q.-C. Yang, X. Zhang and J.-L. Li, Angew. Chem., Int. Ed., 2024, 62, e202309572 CrossRef PubMed.
- Z.-F. Zhang, C.-L. Zhang and S. Ye, Chem. – Eur. J., 2024, 30, e202402259 CrossRef CAS PubMed.
- L. Delfau, E. Mauro, J. Pecaut, D. Martin and E. Tomás-Mendivil, ACS Catal., 2024, 14, 7149 CrossRef CAS.
- (a) Y. Huang, Y.-F. Han, C.-L. Zhang and S. Ye, ACS Catal., 2023, 13, 11033 CrossRef CAS; (b) Y. Huang, X.-H. Wang, C.-L. Zhang and S. Ye, Org. Lett., 2024, 26, 3441 CrossRef CAS PubMed; (c) Y. Huang, Y.-F. Han, C.-L. Zhang and S. Ye, Org. Lett., 2025, 27, 415 CrossRef CAS PubMed.
- L. Lin, Y. Li, W. Du and W.-P. Deng, Tetrahedron Lett., 2010, 51, 3571 CrossRef CAS.
- M. Padmanaban, A. T. Biju and F. Glorius, Org. Lett., 2011, 13, 98 CrossRef CAS PubMed.
- V. Hahnvajanawong, B. Pungpis and P. Theramongkol, Der Pharma Chem., 2016, 8, 112 CAS.
- (a) J.-C. Folest, J.-M. Duprilot, J. Perichon, Y. Robin and J. Devynck, Tetrahedron Lett., 1985, 26, 2633 CrossRef CAS; (b) A. J. Fry and U. N. Sirisoma, J. Org. Chem., 1993, 58, 4919 CrossRef CAS; (c) A. J. Fry and A. H. Singh, J. Org. Chem., 1994, 59, 8172 CrossRef CAS; (d) C. A. Bessel and D. R. Rolison, J. Am. Chem. Soc., 1997, 119, 12673 CrossRef CAS; (e) B.-L. Chen, H.-W. Zhu, Y. Xiao, Q.-L. Sun, H. Wang and J.-X. Lu, Electrochem. Commun., 2014, 42, 55 CrossRef CAS; (f) C. A. Malapit, M. Tanwar, A. D. Pendergast, S. Udyavara, W. D. Beck, R. E. Smith, S. Kadic, T. Primo, D. B. Wu, T. Stone, H. S. White, M. Neurock, M. S. Sigman and S. D. Minteer, ChemRxiv, 2021 DOI:10.26434/chemrxiv-2021-thftp; (g) C. Sandford, L. R. Fries, T. E. Ball, S. D. Minteer and M. S. Sigman, J. Am. Chem. Soc., 2019, 141, 18877 CrossRef CAS PubMed.
- S. Al Zubaydi, I. O. Onuigbo, B. L. Truesdell and C. S. Sevov, Angew. Chem., Int. Ed., 2024, 63, e202313830 CrossRef CAS PubMed.
- L. Delfau, S. Nichilo, F. Molton, J. Broggi, E. Tomás-Mendivil and D. Martin, Angew. Chem., Int. Ed., 2021, 60, 26783 CrossRef CAS PubMed.
- (a) H.-G. Heine, Liebigs Ann. Chem., 1970, 735, 56 CrossRef CAS; (b) Y. Ueno and M. Okawara, Synthesis, 1975, 268 CrossRef CAS; (c) Z. Wang, L. Gan, Z. Song, Y. Liu and J.-P. Wan, Chin. J. Chem., 2024, 42, 3041 CrossRef CAS.
- Y. Kakeno, M. Kusakabe, K. Nagao and H. Ohmiya, ACS Catal., 2020, 10, 8524 CrossRef CAS.
- M. N. Alam, S. R. Dash, A. Mukherjee, S. Pandole, U. K. Marelli, K. Vanka and P. Maity, Org. Lett., 2021, 23, 890 CrossRef CAS PubMed.
- A. V. Bay, K. P. Fitzpatrick, G. A. González-Montiel, A. O. Farah, P. H.-Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2021, 60, 17925 CrossRef CAS PubMed.
- A. V. Bay, K. P. Fitzpatrick, G. A. González-Montiel, A. O. Farah, P. H.-Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2021, 60, 17925 CrossRef CAS PubMed.
- (a) A. V. Davies, K. P. Fitzpatrick, R. C. Betori and K. A. Scheidt, Angew. Chem., Int. Ed., 2020, 59, 9143 CrossRef PubMed; (b) Q.-Y. Meng, L. Lezius and A. Studer, Nat. Commun., 2021, 12, 2068 CrossRef CAS PubMed; (c) H. Huang, Q.-S. Dai, H.-J. Leng, Q.-Z. Li, S.-L. Yang, Y.-M. Tao, X. Zhang, T. Qia and J.-L. Li, Chem. Sci., 2022, 13, 2584 RSC; (d) C.-Y. Tan, M. Kim and S. Hong, Angew. Chem., Int. Ed., 2023, 62, e202306191 CrossRef CAS PubMed; (e) C. R. Schull, J. Cao, S. R. Mitton-Fry, M. Mrksich and K. A. Scheidt, ACS Catal., 2025, 15, 1287 CrossRef CAS.
- M. Jakob, L. Steiner, M. Göbel, J. P. Götze and M. N. Hopkinson, ACS Catal., 2024, 14, 17642 CrossRef CAS.
- (a) S.-C. Ren, W.-X. Lv, X. Yang, J.-L. Yan, J. Xu, F.-X. Wang, L. Hao, H. Chai, Z. Jin and Y. R. Chi, ACS Catal., 2021, 11, 2925 CrossRef CAS; (b) Y. Man, S. Liu, B. Xu and X. Zeng, Org. Lett., 2022, 24, 944 CrossRef CAS PubMed.
- For selected examples, see: (a) T. Ishii, K. Ota, K. Nagao and H. Ohmiya, J. Am. Chem. Soc., 2019, 141, 14073 CrossRef CAS PubMed; (b) J.-L. Li, Y.-Q. Liu, W.-L. Zou, R. Zeng, X. Zhang, Y. Liu, B. Han, Y. He, H.-J. Leng and Q.-Z. Li, Angew. Chem., Int. Ed., 2020, 59, 1863 CrossRef CAS PubMed; (c) B. Zhang, Q. Peng, D. Guo and J. Wang, Org. Lett., 2020, 22, 443 CrossRef CAS PubMed; (d) I. Kim, H. Im, H. Lee and S. Hong, Chem. Sci., 2020, 11, 3192 RSC; (e) S. Jin, X. Sui, G. C. Haug, V. D. Nguyen, H. T. Dang, H. D. Arman and O. V. Larionov, ACS Catal., 2022, 12, 285 CrossRef CAS; (f) F. F. Mulks, M. Melaimi, X. Yan, M.-H. Baik and G. Bertrand, J. Org. Chem., 2023, 88, 2535 CrossRef CAS PubMed.
- (a) S. Byun, M. U. Hwang, H. R. Wise, A. V. Bay, P. H.-Y. Cheong and K. A. Scheidt, Angew. Chem., Int. Ed., 2023, 62, e202312829 CrossRef CAS PubMed; (b) X. Liu, S. Xu, H. Chen and Y. Yang, ACS Catal., 2024, 14, 9144 CrossRef CAS; (c) Y. Xu, H. Chen, L. Yu, X. Peng, J. Zhang, Z. Xing, Y. Bao, A. Liu, Y. Zhao, C. Tian, Y. Liang and X. Huang, Nature, 2024, 625, 74 CrossRef CAS PubMed; (d) C.-B. Li, X.-N. Li, Z.-C. Li, J. Li, Z.-X. Wang, Z.-H. Gao and S. Ye, Angew. Chem., Int. Ed., 2025, e202421151 CAS.
- C. E. I. Knappke, A. J. Arduengo III, H. Jiao, J.-M. Neudörfl and A. J. von Wangelin, Synthesis, 2011, 3784 CAS.
- (a) D. G. Boucher, A. D. Pendergast, X. Wu, Z. A. Nguyen, R. G. Jadhav, S. Lin, H. S. White and S. D. Minteer, J. Am. Chem. Soc., 2023, 145, 17665 CrossRef CAS PubMed; (b) J. Demarteau, A. Debuigne and C. Detrembleur, Chem. Rev., 2019, 119, 6906 CrossRef CAS PubMed; (c) F. T. T. Ng, G. L. Rempel, C. Mancuso and J. Halpern, Organometallics, 1990, 9, 2762 CrossRef CAS.
- X. Li, Y. Mao, P. Fan and C. Wang, Eur. J. Org. Chem., 2022, e202200214 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |