
Sustainable steps forward in vitamin B12-catalysis
Shantanu
Nandi
 ,
Kitti Franciska
Szabó
and
Dorota
Gryko
*
,
Kitti Franciska
Szabó
and
Dorota
Gryko
*
Institute of Organic Chemistry, Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dorota.gryko@icho.edu.pl
First published on 2nd April 2026
Abstract
Vitamin B12, a naturally occurring cobalt complex, plays a vital role as a cofactor in biological processes for adenosylcobalamin- and methylcobalamin-dependent enzymes, where it is essential in DNA synthesis, neurological function, or red blood cell formation. Beyond its biological significance, vitamin B12 exhibits unique reactivity: in the presence of suitable reagents, it forms Co–alkyl complexes that, under light irradiation or thermal activation, generate C-centered radicals that engage in reactions with various SOMOphiles and electrophiles. The feature review highlights catalytic properties of this native cobalt-complex, with particular emphasis on emerging strategies that address the growing demand for more sustainable chemical processes. Remaining challenges and potential future directions are also discussed.
 Shantanu Nandi | Shantanu Nandi received his BSc and MSc degrees in Chemistry from Jadavpur University, India. Subsequently, he completed PhD in 2023 at CSIR-Indian Institute of Chemical Biology under the supervision of Dr Ranjan Jana. Following a brief postdoctoral stay at Osaka University, Japan, he joined Prof. Dorota Gryko's group at the IOC-PAS, Poland, in 2023 as a postdoctoral researcher. His research interests focus on photoredox and transition-metal catalysis, with an emphasis on the fixation and valorization of benign C1 feedstocks. |
 Kitti Franciska Szabó | Kitti Franciska Szabó obtained her BSc degree in Chemical Engineering from the University of Debrecen, Hungary. In 2019, she completed her MSc degree in Synthesis and Medicinal Chemistry at the Technical University of Denmark. In 2020, she joined Prof. Dorota Gryko's group at the Institute of Organic Chemistry of the Polish Academy of Sciences, as a PhD student. Her research interests include photoredox catalysis and cobalt catalysis in organic synthesis. |
 Dorota Gryko | Dorota Gryko obtained her PhD from the Institute of Organic Chemistry at the Polish Academy of Sciences in 1997 under the supervision of Prof. J. Jurczak. Following a post-doctoral stay with Prof. J. Lindsey at North Carolina State University (1998–2000), she started her independent career in Poland. In 2009 and 2018, she received the prestigious TEAM grants from the Foundation for Polish Science. Since 2015 she is full professor at the same institution. Her current research interest focuses on sustainable chemistry, with particular emphasis on solar-driven transformations, vitamin B12 catalysis, carbene chemistry, and micellar catalysis. |
1 Introduction
The chemical industry plays a pivotal role in modern society, and to mitigate the environmental impact of conventional chemical production, which often relies on harmful processes and toxic substances, there is an increasing demand for sustainable technologies.1,2 By integrating the principles of green chemistry,3–5 the replacement of stoichiometric reagents with catalytic amounts can significantly reduce waste and enhance the overall efficiency of chemical production.6 However, approximately 90% of catalysts used often rely on precious metals that are not only expensive but also toxic, limiting their widespread use in medicinal chemistry.7,8 To overcome these challenges, first-row transition metals, with cobalt leading the way, have been successfully employed in recent decades.9,10 Cobalt-based catalysts have facilitated a broad spectrum of transformations, including cycloaddition,11,12 hydroboration,13 C–H activation,8,14,15 and others.16 Among them, vitamin B12 (1, Fig. 1),17 a natural cobalt complex offers numerous advantages, including stability and non-toxicity, representing a significant advancement towards more sustainable cobalt catalysis. This fascinating molecule, recognized by Folkers and Smith in 1948,17–19 has inspired our interest in sustainable catalysis for more than a decade. | ||
| Fig. 1 (A) The structure of native vitamin B12, (B) general mechanism of alkyl radical formation using native or HME Co-catalysis, (C) structures of vitamin B12 derivatives. | ||
Vitamin B12 (1) is the first identified naturally occurring organometallic compound.20 As a cofactor, it plays pivotal roles in numerous biological processes.18,21,22 These include rearrangements involving adenosylcobalamin (1a), methylation with methylcobalamin (1b), isomerisation, dehalogenation,23–25 and more recently discovered light-dependent gene regulation.26,27 Building on these fundamental processes, organic chemists have leveraged the catalytic properties of this vitamin to forge both C–C and C–X bonds, as detailed in several comprehensive reviews.28–33
In this feature article, by no means comprehensive, we highlight newer trends in vitamin B12-catalysis. Following a brief introduction to the structure and fundamental properties of vitamin B12 and its analogues, we compare the principal modes of catalyst activation, including thermal, photochemical, and electrochemical approaches. We then show the evolution of reaction media, spanning from traditional organic solvents to more recent developments in aqueous systems. Finally, we highlight advances in dual catalytic strategies, where vitamin B12 catalysis is integrated with complementary systems such as transition-metal catalysis or hydrogen atom transfer (HAT) processes. In this course, we focus on methodologies that not only gave the desired products in high yields but also put emphasis on the sustainability and allow expansion of the chemical space. We included neither enzymatic transformations involving cobalamin nor thermal transformations, as there are numerous excellent reviews available on these topics.22,28–36
2 Vitamin B12 – structure and catalytic properties
It was in 1954, when Hodgkin reported the X-ray analysis of vitamin B12 (1c).37 It turned out to be a very complex molecule composed of the corrin ligand with the Co ion in the +3-oxidation state in the center and the ribonucleotide loop appended.30,35,38–40 The metal center is coordinated by four equatorial pyrroles through their nitrogen atoms, and by benzimidazole from the bottom α-side which is linked via a ribonucleoside to the macrocyclic ring (the nucleotide loop, Fig. 1A). In nature, the octahedral geometry of the vitamin is completed on the β-side of the corrin ring by one of four different ligands, giving rise to its naturally occurring forms: adenosylcobalamin (1a), methylcobalamin (1b), cyanocobalamin (1c), and aquacobalamin (1d).41 The central cobalt atom can exist in three oxidation states, each exhibiting distinct reactivity.42 This reversible redox characteristic predisposes vitamin B12 (1) and its derivatives for use in catalytic processes. The Co(III) oxidation state is the most stable and, in catalysis, it is regarded as a resting state (Fig. 1B). A single electron reduction leads to Co(II) complex IV, which is radical in nature, and adenosyl cobalamin (1a) operates via this mode of action. The Co(I) oxidation state is achievable via a two-electron reduction and is involved in processes, in which, methyl cobalamin (1b) is involved. This Co(I) complex II is highly nucleophilic, as such that is often referred to as ‘supernucleophile’.43,44The redox flexibility of vitamin B12 (1) governs its diverse catalytic activity, with most reported transformations operating through a radical mechanism. Typically, reduction of the Co(III) complex I generates the highly nucleophilic Co(I) species II which reacts with various electrophiles, usually via SN2 mechanism, to give an alkyl or acyl cobalamins III (Fig. 1B, path I). As an example, chemically generated Co(I) gives rise to alkyl radical from long-chain alkyl iodide.45 Cleavage of the cobalt–carbon bond can occur either homolytically and heterolytically; however, vitamin B12-catalysed processes most often follow the homolytic pathway triggered by heat or light to generate C-centered radicals that subsequently engage with a broad range of SOMOphiles or electrophiles. A key photochemical feature of alkylcobalt(III) complexes is ligand-to-metal charge transfer (LMCT)46 excitation which, upon visible-light irradiation, induces homolytic Co–C bond cleavage to generate Co(II) IV and an alkyl radical (Fig. 1B, inset). The resulting radical can either recombine within the solvent cage or escape to engage in free-radical pathways. The resulting Co(II) intermediate IV can then be reduced back to the active Co(I) form II, thereby completing the catalytic cycle. An alternative mechanism involves Co(II) metalloradical IV, which undergoes HAT to form Co(III)–H intermediate VI and alkene Vvia a formal β-hydride elimination process (Fig. 1B, path II, Co(III)–H). Notably, West and co-workers estimated this HAT process to be fast, occurring at rates of approximately 105 s−1.47 Overall, the operative pathway depends strongly on the choice of a reductant and an additive which can bias the system toward either the homolytic radical cycle or the metalloradiocal HAT pathway.
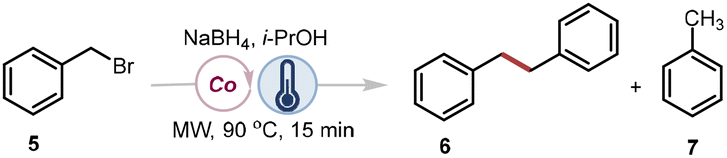
Naturally, vitamin B12 (1) is a hydrophilic molecule, soluble in water and some polar organic solvents only.30 For synthetic applications, however, compatibility with common nonpolar organic solvents is often required to address this limitation. Hydrophobic derivatives, such as cobyrinate 3 and cobalester 4 have been developed thereby broadening the applicability of vitamin B12 in organic synthesis (Fig. 1C). Cobyrinates lack the nucleotide loop, which influences the stability of alkyl cobalamins through the trans-effect.48 Through the trans-effect, an increase or decrease in the steric and electronic properties of the upper axial R group, respectively, leads to elongation or shortening of both Co–C and Co–N bond lengths. While an inverse trans influence is commonly observed for alkylcobalamins, theoretical studies suggest that strongly electron-withdrawing R groups may instead promote a normal trans-effect, indicating that the overall outcome depends on the interplay between steric and electronic factors48 Consequently, cobalester 4, in which the loop is preserved serves as a better mimic for the catalytic activity of vitamin B12 (1). All these derivatives catalyse the dimerisation of benzyl bromide – often considered the benchmark reaction in vitamin B12 catalysis, giving desired product 6 in varying amounts (Table 1).49 For both hydrophobic derivatives 3 and 4 (entries 2 and 3), full conversion was achieved; however, cobalester 4, retaining the nucleotide loop, gave the highest yield (entry 3).
Further experimental and theoretical studies on the dimerisation of 1,1-diphenylethylene proved that the base-on derivatives react with the olefin at the higher rate, however more controlled reaction with base-off derivatives led to the desired product with limited number of side products being formed.50
3. Activation modes: from chemical to electro- and photochemical conditions
The reactivity of vitamin B12 (1) is steered in three steps, (i) the reduction of Co(III) to its catalytically active Co(II) IV or Co(I) II species, (ii) Co(II) or Co(I) subsequently form alkylated Co(III) complex IIIvia interaction with radical intermediates or an electrophilic C-center and (iii) cleavage of the newly formed Co(III)–C bond to produce reactive C-centered radical species (Fig. 1B).3.1 Reduction of Co(III) to its catalytically active Co(II) IV or Co(I) II species
For cyanocobalamin (1c), cyclic voltammetry shows only one wave corresponding to a direct two electron reduction to Co(I) (Ep = −0.95 V versus Ag/AgCl) as a consequence of cyanide strong binding to the cobalt center. For aqua cobalamin (1d) two reduction waves occur at −0.00 and − 0.81 V versus Ag/AgCl. Hydrophobic cobyrinate 3 with two cyanide ligands that undergo exchange with solvent molecules exhibit two waves: E0[Co(III)/Co(II)] = −1.30 V, E0[Co(II)/Co(I)] = −1.50 V vs. Ag/AgCl. The reduction is executed predominantly through chemical methods, but several examples of efficient photo- or electrochemical conditions have been also reported.3.2 Cleavage of the newly formed Co(III)–C bond to produce reactive C-centered radical species
After the successful generation of supernucleophilic Co(I) form II, its attack on an electrophile is generally facile to produce Co(III)–C bond. The bond dissociation energy of the Co(III)–C bond is relatively low (BDE ∼31 kcal mol−1),51 allowing for its cleavage under mild conditions. This process can be triggered by thermal, electrochemical, and photochemical stimuli, in the context of sustainability, the latter two approaches are increasingly favoured as environmentally friendly alternatives in organic synthesis.The inherent reactivity of Co(I) species II remains largely unchanged, but the choice of reducing agent or activation method whether thermal, photochemical, or electrochemical governs the efficacy of the reaction and hence substrate scopes.
Achieving good yield in a specific reaction often requires a proper choice of the catalyst and careful adjustment of both stoichiometry of reagents, Zn/NH4Cl ratio, and the activation mode. Generally, with Zn/NH4Cl system, two electron reduction occurs but Stache and co-workers showed that simply changing the amount of the Zn/NH4Cl system can steer the reduction toward either Co(I) species II or Co(II) derivative IV (Scheme 1).52 Both active forms promote the atom transfer radical polymerisation (ATRA), but they do so in a different manner. The Co(I) catalyst II is highly reactive thus less selective while Co(II)-catalysed polymerisation enables efficient activation–deactivation steps, which in turn leads to polymers with lower dispersity.
 | ||
| Scheme 1 The influence of the amount of Zn on the vitamin B12 reduction. | ||
The activation mode is as important as the choice of a reducing system. For certain transformations, thermal and photochemical cleavage of the Co–C bond is equally effective, illustrating how light-based methods can substitute for conventional heating without compromising reactivity; however, in some cases, one pathway proves superior depending on the substrate's inherent reactivity. Along this line, the Co-catalysed polarity-reversal strain release functionalisation of donor–acceptor cyclopropanes (DAC) can be performed under both thermal and photochemical conditions (Fig. 2).53 But the reaction at ambient temperature (30 °C) proved the most efficient (Fig. 2), consistent with Scheffold's method for ring opening of achiral cyclopropanes under thermal condition.54
 | ||
| Fig. 2 Thermal versus light-induced functionalisation of DAC. | ||
Depending on the substrate, fine tuning of conditions were required as electrophilic olefins are prone to competing pathways such as dimerisation or reduction. Kinetic studies revealed that the reaction rate catalysed by HME 3 is higher than that observed in the presence of (CN)Cbl (1c). This difference proved crucial in cases where Michael acceptors react faster with reduced vitamin B12 than the cyclopropane itself, leading to undesired by-products. For example, the yield of desired product 10 from dimethyl fumarate, increased from 0% to 63% when the catalyst was changed to HME 3. Worth noting is the fact, that the dipolar nature of DACs directs nucleophiles to the donor (D)-substituted carbon and electrophiles to the acceptor (A)-substituted carbon. Consistent with this, Co(I)-mediated ring opening at the D-bearing carbon generates a nucleophilic radical that engages electrophilic alkenes, in sharp contrast to conventional reactivity patterns of these substrates.
On the other hand, vitamin B12-catalysed alkylation of vinyl azides with alkyl bromides in the presence of Zn/NH4Cl system is more efficient under blue light irradiation than when heated up to 60 °C (Scheme 2).55
 | ||
| Scheme 2 Vitamin B12-catalysed functionalisation of vinyl azides. | ||
Vitamin B12 strongly absorb light in the range 250–600 nm with characteristic absorption bands α,β-series at 450–600 and γ-350–370 nm that correspond to a number of excited states generated upon the π–π* transitions within the corrin ring.
For cobyrinate derivatives, the absorption maxima are red shifted by ∼50 nm. For the discussed alkylcobalamins and alkylcobyrrinates, UV/vis spectra are similar, just of lower intensity. Given the low BDE of the Co–C bond, a broad range of light (wavelengths) carry a sufficient energy for its homolysis as seen from Fig. 3 though the flux of photons matters. Gryko reported that the originally methyl pyrrolidine radical 14a generated follows different pathways depending on light used (Fig. 3).56 It can either undergo reduction to afford methylpyrrolidine 15 or recombine with the Co(II) species IV that after dehydrocobaltaion gives pyrrolidine 16. Under blue light irradiation (using LED tape) product 15a forms selectively in 35% yield, but when light intensity increased exocyclic olefin 16a predominates in the rection mixture (16a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :15a, 7:1).
:15a, 7:1).
 | ||
| Fig. 3 Influence of light intensity on the pyrrolidine synthesis. *10 W, 5a/5b 1/7. | ||
For reactions involving BCBs, the Giese type addition is driven by light irradiation, in this case, however, blue (87%) and white (84%) light overperform the green light (72%) induced reaction and the reason for that, at present, remains unknown (Scheme 3).57
 | ||
| Scheme 3 Vitamin B12-catalysed strain release functionalisation of bicyclobutanes. | ||
Similarly, for epoxides and oxetanes, the difference in the reaction outcome under irradiation with different light sources (blue, green, white) is less than 10 percentage points. The photon flux seems to have much stronger impact and it is not the case that the higher the better (Fig. 4).58,59 There is an optimum, which assumingly influences the reaction kinetics, particularly in the case of a reaction with electrophilic olefins that are prone to dimerisation and polymerisation.
 | ||
| Fig. 4 Influence of the photon flux on the reaction yield. | ||
In contrast, for the radical cyclopropanation of olefins with dichloromethane developed by Pitre and co-workers, the use of green (525 nm) instead of blue (456 nm) light led to a significant increase in the yield, which further improved to 78% after diminishing the photon flux by 25% (Scheme 4).60 It is assumed that the effect is caused by the greater spectral overlap of the absorption of the Co(III)–C ligand-to-metal charge transfer (LMCT) band as alkyl cobalamins absorb within the region 450–600 nm. Apparently, it is not the only factor, as some of the presented examples show superiority of blue light irradiation.
 | ||
| Scheme 4 The impact of blue and green light activation of the cyclopropane formation. | ||
In contrast, the analogous transformation of specifically engineered tetrachloro alkanols to cyclopropanes proceeds efficiently only with NaBH4, obviating the need for additional activation stimuli.61 Notably, the cyclisation occurs concomitantly with complete dechlorination. Reactions involving acyl radicals are stronger influenced by both activation mode and energy of the light used. This impact is well-observed in the reaction of electrophilic olefins with thiopyridine esters (24) as acylating reagents (Fig. 5).62 In this case, for acylcobalamin the homolytic cleavage is by far more efficient under light irradiation than under thermal conditions. While both white and blue light performed well, with less energetic green light, the yield of 25d diminished significantly (from 85%, 87% to 36%) contrasting the explanation proposed by Pitre and co-workers.60 Furthermore, a specifically designed 2-S-pyridyl thioesters 26 containing both alkyl bromide functionality helped to understand relative reactivity between alkyl bromides and thioester towards vitamin B12 (Fig. 6).63 Mechanistic studies revealed that alkyl bromides react with Co(I) complex instantly and at much higher rate as compared to thioesters to produce alkyl radical 26a. After the Giese addition from 26a the resulting thioester gives acyl radical 26b from reaction with Co(I) and therefore undergo Giese addition with another molecule of alkene 27. Again, irradiation with more energetic light allowed more efficient reaction, though UVA (black light) gave very complex mixture with almost no product being formed.
 | ||
| Fig. 5 Cobalester-catalysed acylation of olefins under light-irradiation. | ||
 | ||
| Fig. 6 Formation of alkyl and acyl radicals under light-irradiation. | ||
The Stache group has recently shown that even red light can induce vitamin B12-catalysed reactions, though these occur via a new activation mode, e.g. photothermal (Fig. 7).64 Here, the excitation event is followed by nonradiative relaxation pathways to generate heat that induce bond-forming and bond-cleavage processes. In particular, cobyrinate 3 is employed as a molecular photothermal agent that undergoes non-radiative decay upon light irradiation, generating localized heat gradients. In fact, this is thermal activation mode but under light irradiation. Given that cobyrinates absorb light from a broad range (300–600 nm), reactions can be activated by a broad range of light. Under blue, green, red, or even near infrared light irradiation, polymerisation of methyl acrylate with 2-bromoproprionate (29) in the presence of prereduced HME (Co(II)) enabled monomer conversion ∼50% in all cases.64 A thermal imaging showed a temperature increase in 8 °C after 2 minutes of irradiation, while without HME 3 added, the temperature increased by only 2 °C after 10 minutes of irradiation. The authors suggested that photoreduction of the cobalt center to Co(II) IV can be achieved via the excited state Co–S homolytic bond cleavage (Fig. 7).65 The excited state BDE for the Co–S is calculated to be −11.82 kcal mol−1. This activation mode proved useful in atom transfer radical polymerisation, and in the synthesis of photodegradable non-alternating acrylate–isocyanide copolymer.
 | ||
| Fig. 7 Red light induced vitamin B12-catalysed reaction polymerisation. | ||
An intriguing example of vitamin B12-catalysis is the reaction of olefins 31 with ethyl diazoacetate (EDA, 32) (Fig. 8). In 2004, Zhang showed that at the elevated temperature in trifluoroethanol,66 the reaction affords cyclopropanes;33 while under light irradiation, the Gryko group observed alkylation of styrenes.67 Under the thermal conditions, EDA (32) was assumed to reduce vitamin B12 to its active Co(II)-form and generate Co(III)–carbene intermediate 32a with concomitant release of nitrogen. This intermediate subsequently participate in a cyclopropanation reaction. In contrast, alkylation proved effective in the presence of CNCbl (1c) but it requires Zn/NH4Cl as a reducing system having strong impact on the reaction selectivity. In this light-induced process, a “supernucleophilic” Co(I) intercepts EDA (32) to form an alkyl–cobalester(III) intermediate which undergoes homolytic cleavage to generate a carbon-centered radical. This radical subsequently adds to the olefin, forming a new C–C bond. The resulting radical then recombines with the Co(II) species and undergoes dehydrocobalation, affording desired product 34. To add complexity to this picture, Zhang showed that under thermal conditions, diazo compounds react with secondary nitroalkanes to give alkylated derivatives.25 They proposed a radical mechanism though no evidences were given. Conducting the reaction under LED irradiation was found to be critical for achieving high yields and selectivity.
 | ||
| Fig. 8 Cobalester-catalysed functionalisation of sp2 C–H bonds with diazo reagent. | ||
In certain cases, NaBH4 is as efficient as the most often used Zn/NH4Cl system or even superior. This cannot, however, be predicted in advance. Petrovic used NaBH4 as the reducing agent for vitamin B12 (1) for producing Co(I) species II and made it react with tetrachloroalkanol 36.61 Complete dehalogenation led to formation of cyclopropane alcohols 37 (Scheme 5a). The most striking difference was observed in the ATRA (Scheme 5b).68 In the presence of Zn, product 40 does not form while NaBH4 enabled the synthesis of the desired product in a satisfactory yield. Intriguingly, the reaction occurs only if it is conducted in a microwave reactor. Further advancement came from the West group, who made a similar observation in the photochemical regioselective synthesis of subterminal olefins 42 from alkyl halides 41; only NaBH4 gave desired products in synthetically useful yields (Scheme 5c).47 The reaction was proposed to involve hydrogen atom transfer (HAT) as a key step.
 | ||
| Scheme 5 The impact of the reductant used on vitamin B12-photocatalysed reactions. | ||
There are isolated examples, where other reducing agents were employed. In 1998, Takai developed a vitamin B12 catalysed and CrCl2 mediated reaction between dienes and alkyl aldehydes to give homoallylic alcohols.69 Chromium catalyst plays dual role in this process, it reduces vitamin B12 (1) to the Co(I) species that forms cobalt hydride Co(III)–H and produce a chromium reagent from radicals generated in the Co-catalysed cycle.
The influence of light has been also observed in the vitamin B12-catalysed Giese addition of pseudohalides 43 to electrophilic olefins 44 when manganese was employed as a reducing agent (Fig. 9).70 In the dark, only traces (5%) of product 45d formed in this reaction. At ambient light-conditions (daylight) the yield increased to 15% while irradiation with blue light significantly pushed the reaction forward (61%). The optimised conditions enabled the synthesis of products from olefins of different electrophilicity. In this case, the replacement of Mn with Zn diminished the yield by 10%.
 | ||
| Fig. 9 Mn as a reductant in vitamin B12/photocatalysed addition to olefins. | ||
At present the choice of a reducing agent depends on the reaction – the presented examples, however, clearly show that Zn/NH4Cl system is the most versatile, particularly when merged with blue light irradiation. Seemingly, at present the arbitrary choice of the light wavelength for a certain reaction is not possible and the trial-and-error approach needs to be replaced by the rational choice. Apparently, the spectral overlap is not the only criteria. Considering that the mechanism of vitamin B12 catalysed reactions consists of several steps, the subtle balance of their rates seems crucial. For that reason, further research on this aspect is needed.
The photochemical reduction of vitamin B12 (1) and its derivatives has mainly been studied with TiO2, a widely used photocatalyst valued for its non-toxicity and photostability. Upon UV light irradiation of this heterogeneous catalyst, the electrons in the valence band (VB) are excited to the conduction band (CB) (Fig. 10). This excited state TiO2 exhibits reduction potential of approximately E° = −0.5 or −0.3 V (vs. NHE at pH = 7.0) for anatase and rutile, respectively. Since the Co(II)/Co(I) couple of vitamin B12 derivative lies around E° = −0.61 V vs. SCE (−0.37 V vs. NHE), the reduction of vitamin B12 derivatives to Co(I) species II is thermodynamically feasible with this system. In this case, Co(I) species II is generated by the reduction with CB electrons, while required amines act as a sacrificial hole-quencher in the VB.71,72
 | ||
| Fig. 10 Vitamin B12/TiO2 hybrid catalysis under UV irradiation. | ||
The Hisaeda group has extensively investigated strategies employing pre-reduced Co(II)-complex and made important contributions to the development of fully photochemical vitamin B12-catalysis.71,72 Nevertheless, knowing that in the presence of TiO2, HME 3 generates Co(I) species under UV-light irradiation from its +2 oxidation state, they reasoned that immobilisation of the complex on the TiO2 surface would enhance catalytic efficacy, due to the shorter distance between the cobalt center and the semiconductor (Fig. 10). Indeed, when cobyrinic acid was immobilized on TiO2, the resulting hybrid catalyst underwent photoexcitation under UV light irradiation and successfully reduced both Co(III) or Co(II) derivatives to the ‘supernucleophilic’ Co(I) form. The TiO2 hybrid catalyst proved effective in a wide variety of transformations. For example, it catalysed dehalogenation of diethyl 2-bromomethyl-2-phenylmalonate (47) with successive protonation/deuteration from the alcohol solvents or sacrificial reagents such as triethanolamine (entry a).73 The use of this system, was also extended to free-radical initiated ring expansion reaction of α-bromomethyl cyclo β-keto esters 49, thus eliminating the need for toxic n-Bu3SnH/AIBN (entry b).74 The formation of Co(III)–H upon proton capturing by the Co(I) species was proposed for the hydride transfer to the alkene double bond occurring in the Markovnikov manner (entry c).75 Furthermore, under nitrogen atmosphere, the transformation of α-trifluoromethylstyrene (53) showed marked solvent dependence: in MeOH the alkane product predominated, whereas in acetonitrile gem-difluoroolefins were favoured (entry d).76 Even more intriguingly, under aerobic conditions, trichloromethylbenzene (56) in MeOH yielded methyl benzoate (57) in 99% yield (entry e).77 The mechanism is believed to involve generation of dichloromethylbenzene radical that rapidly reacts with oxygen to form in turn benzoyl chloride, which subsequently reacts with methanol to give the desired ester, or with amines to afford amides are formed instead. These examples demonstrate how immobilisation of vitamin B12 on TiO2 provides one of the mildest known routes to esters and amides from trichlorinated compounds. Importantly, in all these processes the hybrid catalyst outperformed the separate use of vitamin B12 derivatives and TiO2, which were limited to dehalogenation of haloalkanes and for which diminished yields were observed. Notably, immobilisation also removed the need for preactivation of the cobyrinate to its Co(II) state IV.
Although, Co(II)/Co(I) potentials of different vitamin B12 derivatives fall within the redox window of many photocatalysts, the Co(III)/Co(II) transformation still represents a challenge. Consequently, many established methods rely on pre-reduced Co(II) complexes IV, especially hydrophobic cobyrinates. The Gryko group have shown that, under UV-light irradiation, TiO2 effectively reduced native vitamin B12 to its catalytically active state, which in turn promoted the deprotection of (allyloxy)arenes 58 (Scheme 6).78 In this transformation, the Co(I) species undergoes nucleophilic substitution (SN2′) at the allyl group to yield phenols and an allyl–Co(III) complex, which is subsequently cleaved photochemically to regenerate Co(I) complex II. This catalytic system is comparable in efficiency to chemical reductants; however, it also promotes double-bond isomerisation, necessitating a subsequent acid treatment to obtain the desired product.
 | ||
| Scheme 6 Vitamin B12/photocatalysed allyl deprotection of phenols. | ||
Despite significant advances, the main limitation of the semiconductor-vitamin B12 hybrid catalysis remains its reliance on high-energy UV light, which is generally unsuitable for widespread applications. As a result, research has increasingly focused on longer wavelengths, particularly visible light. With an appropriate photoredox catalyst, the reduction to the Co(I) species should be achieved in a photocatalytic manner. Building on their vit B12–TiO2 systems, Hisaeda, Shimakoshi and co-workers modified TiO2 with Rh(III) species and then immobilized vitamin B12 derivatives on its surface (Scheme 7).79 A new hybrid photocatalyst, (B12–Rh–TiO2) capable of absorbing visible light, enabled the synthesis of amides 64 under blue light irradiation (420 nm), using triethylamine as both a sacrificial electron donor and a coupling partner.
 | ||
| Scheme 7 Vitamin B12–Rh–TiO2 promoted synthesis of amides. | ||
Both heterogenous and homogeneous photocatalytic systems have their pros and cons. Heterogeneous systems allow easy recovery and reuse of catalysts but often face mass-transfer limits. Homogeneous systems give fast reactions and better contact with reactants, yet the catalyst recovery is difficult and stability can be lower. Following the similar rationale, the Hisaeda and Shimakoshi group also developed homogeneous, purely photocatalytic systems, mostly based on the Co(II) form IV, for the generation of Co(I) species II and the subsequent homolysis of the Co(III)–C bond (Fig. 11). Based on Walder and co-workers studies,80 dechlorination of dichlorodiphenyltrichloroethane (DDT) in the presence of reduced HME (Co(II)) and Ru(bpy)3Cl2 as a photocatalyst yielded mainly dichlorodiphenyldichloroethane (DDD, 77%).81 The excited Ru photocatalyst provided sufficient reducing power to generate Co(I) species II from the Co(II) complex, and the dual catalytic systems worked effectively even in ionic liquids, which facilitated catalysts recovery and reuse.82 Alternatively, polymeric catalysts can be more convenient. Along this line, both B12 complex and Ru(bpy)3(ClO4)2 were incorporated into a polymeric framework, and this catalyst outperformed an action of cobyrinate and the Ru-complex when being used separately (82 versus 59%, entries a and b).83 Although both Ru- and Ir-complexes possess sufficient reducing power to generate Co(I) from Co(II) species, reaction efficacy often depends on the substrate.84 For example, in the functionalisation of phenylacetylenes 70 with alkyl bromides 69 (entry c), the yield increased by 30 percentage points when Ir(ppy)2(dtbbpy)PF6 was used, while the analogous reaction with styrene was much more efficient with [Ru(bpy)3Cl]2 (81% versus 16%). For the stronger reducing Ir-catalysts, overreduction often occurs, which diminishes the yield substantially.85 From a green chemistry perspective, minimizing the use of transition metals is desirable due to their availability and toxicity. Recent discoveries show that organic photocatalysts can provide a viable alternative. Although many photocatalytic systems are available for the reduction of Co(II) to Co(I) state, the direct two electron reduction of native vitamin B12 (1) from Co(III) to Co(I) remains challenging. Rose Bengal sodium salt efficiently reduces cobyrinate 3, in its Co(II)-form, to catalytically active Co(I) species (entry d).86 In 2021, Barata-Vallejo and Postigo demonstrated that this photocatalyst, in its lactone form, can even reduce native vitamin B12 (both cyano- and hydroxy-complexes, 1c and 1d) directly from Co(III) to Co(I) state.87 The use of less energetic light is advantageous, as it limits side reactions and broadens the substrate scope. To address this issue, amino BODIPY with a maxima absorption at λ = 521 nm, was appended to cobyrinate derivative, thus enabling activation of the catalyst under green light irradiation with no interference from vitamin B12 (entry e).88 This catalyst showed excellent reactivity in dehalogenation reactions from trichloromethylbenzenes 74 and outperformed the two catalysts used separately.
 | ||
| Fig. 11 Hisaeda's works on B12/photoredox dual catalysis without metal reductants. | ||
For industrial applications, heterogenous catalysis is particularly attractive. Along this line, the Gryko group showed that the inexpensive and widely available MOF photocatalyst, MIL-125-NH2, enabled direct, visible light-induced reduction of native vitamin B12 to the supernucleophilic Co(I) form for the first time (Scheme 8).89 Remarkably, the amount of vitamin B12 required was as low as 0.5 mol%, and the MOF co-catalyst could be recovered with little loss of activity. The utility of this system was demonstrated in the synthesis of pyrrolidine derivatives and the dehalogenation of DDT. In 2024, the Chu group reported new modes of reactivity for vitamin B12 under visible-light irradiation.90 Acting as a bifunctional catalyst, vitamin B12 enabled meta-bromination/chlorination of phenol derivatives while vitamin B1 was used as a ligand.
 | ||
| Scheme 8 Visible light-induced radical cyclisation in the presence of MIL-125-NH2(Ti). | ||
The presented examples demonstrate that photoredox catalysis could be directly merged with Co(III)–Co(II)–Co(I) cycles, devoid of any classical Zn/NH4Cl system to drive chemical reactions. Nevertheless, obstacles remain, including efficient two-electron photoreduction, particularly under visible light irradiation.
The use of electrochemistry in vitamin B12-catalysis can be traced back to 1980s when Scheffold successfully generated Co(I) form II at Hg-cathode while using Pt as a sacrificial anode (Fig. 12, entry a).91,92 This species enabled generation of alkyl radicals from alkyl bromides 78 and subsequent 1,4-intramolecular addition. Rusling expanded this methodology for the elimination reaction of vicinal dibromides 80 (entry b).93 They showed that in the aerosol-OT/water/isooctane emulsion the Co(II)/Co(I) reduction occurs at about −0.9 to −1.0 V vs. SCE, whereas in aqueous acetonitrile at low pH it slightly shifts towards a more positive potential (≈−0.75 V vs. SCE).93 At glassy carbon cathode, the corrin complex was efficiently reduced to Co(I) at about −0.75 V in aq. acetonitrile. They showed that upon addition of vicinal dibromide the anodic peak for oxidation of Co(I) readily disappeared as it reacted with the substrate to recycle Co(II) at the electrode. To note, direct reduction of vicinal dibromides requires potentials near −1.6 V vs. SCE, whereas using electrogenerated Co(I) allows reduction to occur effectively at potentials ∼0.8 V more positive. Gryko and Hisaeda explored electrocatalysis for B12-catalysed homocoupling of benzyl bromide via benzyl radical generation (entry c).94
 | ||
| Fig. 12 Electrochemically driven vitamin B12 catalysed reactions. | ||
Extending beyond alkyl radicals, Scheffold and Orliński demonstrated that acyl radicals can also be generated electrochemically via vitamin B12-mediated redox catalysis, establishing an early paradigm for acyl radical chemistry under mild conditions.95 Generated Co(I) engages carboxylic anhydrides to form acyl–Co(III) intermediates that undergo electro- or photo-induced Co–C bond cleavage, releasing acyl radical at potentials substantially more positive than direct substrate reduction.
An intriguing contribution by Hisaeda and Shimakoshi demonstrated that electrolysis of trichloromethylated substrates 84 under aerobic conditions gives access to esters 85 (entry d).96 Under mild cathodic polarisation, the Co(I) species was generated in situ directly from either Co(III) or Co(II) complexes without external reductants, despite the presence of molecular oxygen, highlighting the unique ability of electrochemistry to transiently access highly reactive low-valent cobalt species under otherwise prohibitive conditions. Electroanalytical and mechanistic studies indicate that the electrode-driven Co(II)/Co(I) couple functioned as the central redox gate, mediating halogen abstraction and radical generation at potentials substantially more positive than those required for direct substrate reduction. Notably, competition between oxygen quenching of Co(I) and catalytic turnover did not suppress reactivity but instead redirected the catalytic pathway through oxygen-derived intermediates. The same group extended the scope in their following report by changing the nucleophile to amine which afforded amides 87 (entry e).97
Beyond radical-generation chemistry, recent studies have significantly broadened the electrocatalytic scope of vitamin B12 catalysis by exploiting its accessible cobalt redox manifold for fundamentally different transformations. Zhang and Verpoort demonstrated that cyanocobalamin (1c) can operate as a homogeneous molecular electrocatalyst for water oxidation (entry f),98 accessing high-valent cobalt states under neutral conditions with low overpotential and high stability, thereby extending B12 electrocatalysis into oxidative regimes. Complementarily, Ruccolo and co-workers reported a vitamin B12-catalysed, electrochemically driven reduction of disulfide bonds (88), in which Co(I) species II mediates selective S–S bond cleavage across small molecules, peptides, and proteins at potentials far milder than direct electroreduction, establishing vitamin B12 (1) as a versatile redox mediator for both oxidative and reductive electrocatalysis (entry g).99
Among various electrochemical protocols, constant potential electrolysis is often preferred due to its ability to deliver controlled and efficient generation of the Co(I) form II.
Electrocatalysis with vitamin B12 offers an attractive blueprint for sustainable redox chemistry; however, the practical costs of high current densities, sacrificial electrodes, and parasitic electrochemical reactions presently offset these advantages when benchmarked against photocatalysis.
4. Reaction media: from organic solvents to water
Most reported reactions catalysed by vitamin B12 (1) and its derivatives are considered non-toxic, however they are typically carried out in organic solvents, and the reaction conditions often deviate from the principles of green chemistry. Utilizing native vitamin B12 (1) under more environmentally benign conditions may enhance its applicability in the chemical industry, particularly as sustainability becomes a central concern. In this context, replacing conventional organic solvents, which contribute substantially to chemical waste, with greener alternatives, especially water, represents a promising strategy for the development of more sustainable synthetic methodologies.100,101In Postigo and Barata-Vallejo's report, they performed perfluoroalkylation of activated arenes 90 with the corresponding bromides in water (Fig. 13A).87 While the reaction follows the general mechanistic pathway, a noteworthy feature is the photochemical reduction of the Co(III) species to the catalytically active Co(I) form in the presence of Rose Bengal. However, the main limitation of this methodology lies in the solubility of solid substrates, which still necessitates the use of water–acetonitrile mixture. Using vitamin B12 catalysis, Sharghi and co-workers accomplished the synthesis of benzoxazoles 94 from catechol derivatives 92 and primary amines 93 in aqueous conditions (Fig. 13A).102 The proposed mechanism is unusual for vitamin B12 catalysis as it involves hydroxyl radical generated from hydroxylcobalamin that abstracts a hydrogen atom from catechol. An intermediate ketone formed reacts with benzylamine to give an imine, which after cyclisation furnishes benzoxazoline.
 | ||
| Fig. 13 Vitamin B12-catalysed reactions in (A) aqueous and (B) micellar media. | ||
To avoid the solubility mismatch (vitamin B12 hydrophilic, organic reagents mostly hydrophobic), the Gryko group merged vitamin B12 catalysis with micellar system (Fig. 13B).103 The native vitamin-catalysed tandem radical addition/1,2-aryl migration reaction, when performed in polar organic solvents, yielded desired product 97a in only 33% yield. In micellar solution, the choice of surfactant proved critical: sodium dodecyl trimethylammonium chloride (DTAC) not only improved the yield of the desired product 97a to 80% but also eliminated the need for NH4Cl, a common additive in B12-catalysed reactions.
NMR and theoretical studies provided insights into the localisation of the components within the micellar environment. Vitamin B12 is present in bulk water and only after it is reduced to the active Co(I) species, relocates to the Stern layer (micelle–water interface), where it reacts with alkyl bromides to generate radical species. The subsequent addition to the olefin occurs there as the open-shell species are short lived. The spatial arrangements of reactants in micellar solutions are crucial for efficient reaction. Consequently, both the chain length of the alkyl bromide and the nature of functional groups on the aromatic olefin strongly influence the reaction outcome. The same methodology enabled fully regioselective ring-opening of epoxides and aziridines 98 with electrophilic olefins 99 under blue or green LED irradiation respectively (Fig. 13B).59 Both alkyl- and aryl-substituted epoxides furnished desired products 100 in good yields though fine tuning of the reaction conditions was required for each group of substrates. For aziridines, under non-photocatalytic conditions, Scheffold successfully opened bicyclic aziridines to produce allylic amines in a buffered solution.104 Here in Gryko's method using micellar photocatalysis, products were obtained in satisfactory yields only from alkyl-substituted derivatives. The limitations assumingly stem from different partitioning of reactants within the micellar system under study.
These examples emphasize that water is indeed a suitable solvent for vitamin B12-catalysed reactions.
5. Cooperative catalysis – from monocatalytic system to dual catalysis
Although monocatalytic systems have enabled a wide range of valuable transformations, they often face limitations in reactivity, selectivity, or substrate scope when multiple activation steps are required. Cooperative catalysis overcomes these constrains by orchestrating two complementary catalysts that act synergistically to activate distinct reaction partners or elementary steps, thereby expanding the accessible chemical space beyond that of single catalytic systems. Such strategies have recently begun to influence vitamin B12 catalysis.33 We focus mainly on Co/Ni and Co/HAT cooperative catalysis to highlight their key characteristic and reactivity profiles.5.1 Vitamin B12/Ni
Merging vitamin B12 (1) and nickel catalysis enables the complementary activation of electrophiles within a single reaction manifold. This cooperative approach harnesses the strengths of both catalytic platforms to access electrophile–electrophile couplings that are inaccessible to either system alone. In 2020, the Gryko group utilized vitamin B12 catalysis to generate radicals from strained bicyclic molecules 101 (Scheme 9).57 The nucleophilic Co(I) species attacks the bridgehead C-center to produce alkyl–Co(III) complex that upon light irradiation releases the cycloalkyl radical. Then, it enters the Ni-catalytic cycle where it is captured by Ni(II)–aryl complex coming from oxidative addition of Ni(0) to aryl iodides 104 and forms Ni(III) complex. Then, a facile reductive elimination provides 3-aryl cyclobutanes 106. While previous methods for strained-release functionalisation's were typically restricted to functionalisation with nucleophiles, contrastingly this polarity reversal functionalisation has been attained with electrophiles. Here, Zn acted as reducing agent for both Co and Ni catalysts to continue the catalytic cycles. Due to the mild reaction condition, a broad range of functional groups are also compatible in this methodology. This dual vitamin B12/Ni/photocatalytic system facilitated also the synthesis of extremely challenging linear products from aryl epoxides 107, which have been unattainable with other methods (Fig. 14A).105 Scheffold showed earlier that the vitamin B12 could enable ring opening of symmetric alkyl epoxides to produce allylic alcohols in selective manner,106,107 which was later utilized in epoxide ring opening and successive intramolecular cyclization.108 In case of aryl epoxides, driving the functionalisation far from stabilized benzylic center to the terminal position had been the most challenging barrier for the linear-type product formation. In Gryko's strategy, it is the vitamin B12 which governs the regioselectivity of the reaction – this bulky Co-catalyst preferentially attacks the less-substituted C-center of the epoxide to give, after photohomolytic cleavage, a primary alkyl radical. The ring-opening approach was extended to 4-membered oxetane system (Fig. 14B).58 Although the strain of oxetanes 110 (106 kJ mol−1) is like that of epoxides (112 kJ mol−1), ring-opening of the former is kinetically unfavourable. Thus, to lower the activation energy, the use of one oxophilic Lewis acid (TMSBr) was essential. Mechanistic studies revealed that the first step involves ring-opening by the TMSBr which is, according to DFT calculations, barrierless. The halohydrin generated enters the vitamin B12-catalytic cycle when merged with Ni-catalysis, it enabled coupling with aryl halides. | ||
| Scheme 9 Merging vitamin B12 and Ni catalyses enable electrophile–electrophile coupling. | ||
 | ||
| Fig. 14 Light induced vitamin B12/Ni catalysed ring opening of oxo-heterocycles. (A) epoxides, (B) oxetanes. | ||
5.2 Vitamin B12/HAT
By merging B12 catalysis with HAT catalysis, the West group realized selective hydrogenative ring opening of epoxides 107 and synthesized otherwise hardly achievable Markovnikov alcohols 112 (Fig. 15A).109 Similarly to the Gryko's strategy,105 the terminal radical forms as a result of ring opening of the epoxide at the less hindered position by the Co(I) species and the subsequent light-induced homolysis of Co(III)–carbon center. Then, the open-shell species abstracts hydrogen from 2,4,6-triisopropylbenzenethiol serves as a hydrogen-atom transfer (HAT) agent. The solvent MeOH acted as H-atom donor to continue the HAT catalytic cycle. | ||
| Fig. 15 Light-induced vitamin B12/HAT transformations involving (A) epoxides, (B) oxetanes. | ||
Very recently, the same group extended this strategy to ring-opening of oxetanes 110 (Fig. 15B).110Via the similar protocol as their epoxide ring-opening, use of TRIP-disulphide as HAT reagent in protic solvent provided hydrogenative ring-opening of oxetanes yielding alkyl alcohols. The reaction goes through light-induced homolysis of Co(III)–C bond and thus generation of hydroxy alkyl radical and Co(II) metalloradical II. The alkyl radical undergoes HAT from the thiol or Co–H to generate alkyl alcohol. Alternatively, in absence of HAT-agent and in aprotic solvent the same alkyl radical generated from light-induced homolysis provided allylic alcohols via H-atom abstraction.
Vitamin B12/Co–salen The West group, using a dual vitamin B12/Co–salencatalytic system, successfully demonstrated a tandem olefination/isomerisation reaction of primary alkyl halides or pseudohalides 114 (Scheme 10).47 As in most vitamin B12-catalysed reactions, under visible light irradiation, a Co(III)–alkyl intermediate generates a Co(II)-metalloradical intermediate that after hydrogen atom transfer produces a terminal olefin. Subsequently, in the presence of the Co–salen complex, isomerisation of the terminal olefin occurs, yielding the subterminal olefin in a regioselective manner.
 | ||
| Scheme 10 Remote elimination of alkyl halides via dual cobalt catalysis. | ||
Merging vitamin B12 catalysis with other activation modes has proven highly useful by enabling previously inaccessible reactions or selectivities and opening new pathways in chemical synthesis.
6 Conclusions and future perspectives
As the chemical science increasingly align with the principles of sustainability, vitamin B12 stands at a strategic intersection of bioinspired design, radical chemistry, and green process engineering. It has evolved from a biological cofactor to a powerful cobalt catalyst for C–C bond forming reactions, including the addition to multiple bonds, electrophile–electrophile couplings, dimerisations, rearrangements, and functionalisations involving ring-opening.Vitamin B12 intrinsic characteristics – natural origin, low toxicity, water solubility, and well-defined redox chemistry – make it a uniquely attractive platform for the development of sustainable synthetic methodologies. In the context of greener chemistry, the shift from thermal activation toward photochemical strategies represent a significant advance. Light serves as an abundant and renewable energy source, enabling mild reaction conditions and eliminating the need for elevated temperature. Nevertheless, many current vitamin B12-catalytic systems still rely on stoichiometric metal reductants to access the catalytically active Co(I) state, which diminishes atom economy and generates waste. The development of effective fully photoredox-driven system would mark a significant advance. The isolated examples, do exist, but they are far from being general and an effective. In parallel, electrochemistry, as a traceless reducing platform, offers an equally compelling opportunity in this regard. Though the integration of vitamin B12-catalysis with electrochemistry dates back to 1980s, its full potential has not been recognized yet. Merging vitamin B12 catalysis with electrochemical or photochemical systems could therefore enable closed, reagent free redox cycles with improved sustainability metrics.
Another central parameter in sustainable chemistry which should be considered is solvent selection. Although vitamin B12 is naturally occurring molecule, many reported methods rely on hydrophobic derivatives to enable solubility in petroleum-derived solvents, commonly used by synthetic chemists, undermines its green potential. Future efforts should prioritize greener solvents, ideally water whenever possible. Micellar catalysis has indeed demonstrated that native vitamin B12 can operate efficiently in aqueous systems, and its integration with photocatalysis or electrochemistry opens promising avenue toward environmentally benign transformations.
Looking ahead, future research should focus on developing photo- and electrochemical platforms for efficient vitamin B12 reduction, improving catalyst efficiency, and enabling stereoselective transformations, most likely via cooperative catalysis. Such advances hold promises not only for greener vitamin B12-catalysed transformations but also for the broader transition toward more sustainable practices in synthetic chemistry.
Conflicts of interest
There are no conflicts to declare.Data availability
This review does not provide original data. All data can be found in the original literature.Acknowledgements
Financial support for this work was provided by the National Science Centre, Poland, grant OPUS UMO-2024/55/B/ST5/01006.Notes and references
- T. Collins, Science, 2001, 291, 48 CrossRef CAS PubMed.
- A. Cannon, S. Edwards, M. Jacobs, J. W. Moir, M. A. Roy and J. A. Tickner, RSC Sustainability, 2023, 1, 2092 RSC.
- J. H. Clark, Green Chem., 1999, 1, 1 RSC.
- H. C. Erythropel, J. B. Zimmerman, T. M. de Winter, L. Petitjean, F. Melnikov, C. H. Lam, A. W. Lounsbury, K. E. Mellor, N. Z. Janković, Q. Tu, L. N. Pincus, M. M. Falinski, W. Shi, P. Coish, D. L. Plata and P. T. Anastas, Green Chem., 2018, 20, 1929 RSC.
- K. N. Ganesh, D. Zhang, S. J. Miller, K. Rossen, P. J. Chirik, M. C. Kozlowski, J. B. Zimmerman, B. W. Brooks, P. E. Savage, D. T. Allen and A. M. Voutchkova-Kostal, Environ. Sci. Technol. Lett., 2021, 8, 487 CrossRef CAS.
- R. A. Sheldon, Pure Appl. Chem., 2000, 72, 1233 CrossRef CAS.
- J. N. Armor, Catal. Today, 2011, 163, 3 CrossRef CAS.
- P. Gandeepan and C.-H. Cheng, Acc. Chem. Res., 2015, 48, 1194 CrossRef CAS PubMed.
- G. Cahiez and A. Moyeux, Chem. Rev., 2010, 110, 1435 CrossRef CAS PubMed.
- K. Gao and N. Yoshikai, Acc. Chem. Res., 2014, 47, 1208 CrossRef CAS PubMed.
- P. R. Chopade and J. Louie, Adv. Synth. Catal., 2006, 348, 2307 CrossRef CAS.
- G. Domínguez and J. Pérez-Castells, Chem. Soc. Rev., 2011, 40, 3430 RSC.
- J. V. Obligacion and P. J. Chirik, J. Am. Chem. Soc., 2013, 135, 19107 CrossRef CAS PubMed.
- R. Santhoshkumar, S. Mannathan and C.-H. Cheng, Org. Lett., 2014, 16, 4208 CrossRef CAS PubMed.
- P. Gandeepan, K. Parthasarathy and C.-H. Cheng, J. Am. Chem. Soc., 2010, 132, 8569 CrossRef CAS PubMed.
- Y.-C. Wong, T. T. Jayanth and C.-H. Cheng, Org. Lett., 2006, 8, 5613 CrossRef CAS PubMed.
- E. L. Rickes, N. G. Brink, F. R. Koniuszy, T. R. Wood and K. Folkers, Science, 1948, 107, 396 CrossRef CAS PubMed.
- E. Kaczka, D. E. Wolf and K. Folkers, J. Am. Chem. Soc., 1949, 71, 1514 CrossRef CAS PubMed.
- R. West, Science, 1948, 107, 398 CrossRef CAS PubMed.
- F. Watanabe, Exp. Bio. Med., 2007, 232, 1266 CrossRef CAS PubMed.
- E. L. Smith, Br. J. Nutr., 1952, 6, 295 CrossRef CAS PubMed.
- K. Gruber, B. Puffer and B. Kräutler, Chem. Soc. Rev., 2011, 40, 4346 RSC.
- P. H. Degnan, M. E. Taga and A. L. Goodman, Cell Metab., 2014, 20, 769 CrossRef CAS PubMed.
- W. Wu, Z. Lin and H. Jiang, Org. Biomol. Chem., 2018, 16, 7315 RSC.
- Z. Zhang, M. Chen and G. Zheng, RSC Adv., 2024, 14, 29168 RSC.
- S. Zhang, L. N. Jeffreys, H. Poddar, Y. Yu, C. Liu, K. Patel, L. O. Johannissen, L. Zhu, M. J. Cliff, C. Yan, G. Schirò, M. Weik, M. Sakuma, C. W. Levy, D. Leys, D. J. Heyes and N. S. Scrutton, Nat. Commun., 2024, 15, 2740 CrossRef CAS PubMed.
- S. Padmanabhan, M. Jost, C. L. Drennan and M. Elías-Arnanz, Annu. Rev. Biochem., 2017, 86, 485 CrossRef CAS PubMed.
- R. Scheffold, G. Rytz, L. Walder, R. Orlinski and Z. Chilmonczyk, Pure Appl. Chem., 1983, 55, 1791 CrossRef CAS.
- R. Scheffold, S. Abrecht, R. Orlinski, H. R. Ruf, P. Stamouli, O. Tinembart, L. Walder and C. Weymuth, Pure Appl. Chem., 1987, 59, 363 CrossRef CAS.
- M. Giedyk, K. Goliszewska and D. Gryko, Chem. Soc. Rev., 2015, 44, 3391 RSC.
- M. Giedyk and D. Gryko, Chem. Catal., 2022, 2, 1534 CAS.
- T. Wdowik and D. Gryko, ACS Catal., 2022, 12, 6517 CrossRef CAS.
- A. J. Moser, B. E. Funk and J. G. West, ChemCatChem, 2024, 16, e202301231 CrossRef CAS PubMed.
- R. Banerjee and S. W. Ragsdale, Annu. Rev. Biochem., 2003, 72, 209 CrossRef CAS PubMed.
- K. L. Brown, Chem. Rev., 2005, 105, 2075 CrossRef CAS PubMed.
- K. Tahara, L. Pan, T. Ono and Y. Hisaeda, Beilstein J. Org. Chem., 2018, 14, 2553 CrossRef CAS PubMed.
- D. C. Hodgkin, J. Pickworth, J. H. Robertson, K. N. Trueblood, R. J. Prosen and J. G. White, Nature, 1955, 176, 325 CrossRef CAS PubMed.
- R. B. Woodward, Pure Appl. Chem., 1973, 33, 145 CAS.
- A. Eschenmoser and C. E. Wintner, Science, 1977, 196, 1410 CrossRef CAS PubMed.
- I. A. Dereven’kov, D. S. Salnikov, R. Silaghi-Dumitrescu, S. V. Makarov and O. I. Koifman, Coord. Chem. Rev., 2016, 309, 68 CrossRef.
- M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117 CrossRef CAS PubMed.
- H. E. Simmons and R. D. Smith, J. Am. Chem. Soc., 1959, 81, 4256 CrossRef CAS.
- G. N. Schrauzer, E. Deutsch and R. J. Windgassen, J. Am. Chem. Soc., 1968, 90, 2441 CrossRef CAS PubMed.
- G. N. Schrauzer and E. Deutsch, J. Am. Chem. Soc., 1969, 91, 3341 CrossRef CAS PubMed.
- L. Auer, C. Weymuth and R. Scheffold, Helv. Chim. Acta, 1993, 76, 810 CrossRef CAS.
- F. Juliá, ChemCatChem, 2022, 14, e202200916 CrossRef.
- R. Bam, A. S. Pollatos, A. J. Moser and J. G. West, Chem. Sci., 2021, 12, 1736 RSC.
- L. Randaccio, S. Geremia, G. Nardin and J. Wuerges, Coord. Chem. Rev., 2006, 250, 1332 CrossRef CAS.
- M. Giedyk, S. N. Fedosov and D. Gryko, Chem. Commun., 2014, 50, 4674 RSC.
- M. Karczewski, M. Ociepa and D. Gryko, Eur. J. Org. Chem., 2019, 469 CrossRef CAS.
- K. P. Jensen and U. Ryde, J. Am. Chem. Soc., 2005, 127, 9117 CrossRef CAS PubMed.
- S. Dadashi-Silab, C. Preston-Herrera and E. E. Stache, J. Am. Chem. Soc., 2023, 145, 19387 CrossRef CAS PubMed.
- J. Turkowska, J. Durka, M. Ociepa and D. Gryko, Chem. Commun., 2022, 58, 509 RSC.
- T. Troxler and R. Scheffold, Helv. Chim. Acta, 1994, 77, 1193 CrossRef CAS.
- K. R. Dworakowski, S. Pisarek, S. Hassan and D. Gryko, Org. Lett., 2021, 23, 9068 CrossRef CAS PubMed.
- S. Smoleń, A. Wincenciuk, O. Drapała and D. Gryko, Synthesis, 2020, 1645 Search PubMed.
- M. Ociepa, A. J. Wierzba, J. Turkowska and D. Gryko, J. Am. Chem. Soc., 2020, 142, 5355 CrossRef CAS PubMed.
- A. Potrząsaj, M. Ociepa, W. Chaładaj and D. Gryko, Org. Lett., 2022, 24, 2469 CrossRef PubMed.
- K. F. Szabó, T. Wdowik, A. Krzeszewska, K. Mazurek, M. P. Andersson and D. Gryko, Org. Lett., 2025, 27, 5642 CrossRef PubMed.
- J. H. G. Teye-Kau, M. J. Ayodele and S. P. Pitre, Angew. Chem., Int. Ed., 2024, 63, e202316064 CrossRef CAS PubMed.
- Z. Petrović, Z. Bugarčić, L. Marjanović and S. Konstantinović, J. Mol. Catal. A: Chem., 1999, 142, 393 CrossRef.
- M. Ociepa, O. Baka, J. Narodowiec and D. Gryko, Adv. Synth. Catal., 2017, 359, 3560 CrossRef CAS.
- A. Potrząsaj, M. Ociepa, O. Baka, G. Spólnik and D. Gryko, Eur. J. Org. Chem., 2020, 1567 CrossRef.
- C. Preston-Herrera, S. Dadashi-Silab, D. G. Oblinsky, G. D. Scholes and E. E. Stache, J. Am. Chem. Soc., 2024, 146, 8852 CrossRef CAS PubMed.
- S. Dadashi-Silab, C. Preston-Herrera, D. G. Oblinsky, G. D. Scholes and E. E. Stache, J. Am. Chem. Soc., 2024, 146, 34583 CrossRef CAS PubMed.
- Y. Chen and X. P. Zhang, J. Org. Chem., 2004, 69, 2431 CrossRef CAS PubMed.
- M. Giedyk, K. Goliszewska, K. Ó. Proinsias and D. Gryko, Chem. Commun., 2016, 52, 1389 RSC.
- K. ó Proinsias, A. Jackowska, K. Radzewicz, M. Giedyk and D. Gryko, Org. Lett., 2018, 20, 296 CrossRef CAS PubMed.
- K. Takai and C. Toratsu, J. Org. Chem., 1998, 63, 6450 CrossRef CAS.
- K. Komeyama, T. Michiyuki, Y. Teshima and I. Osaka, RSC Adv., 2021, 11, 3539 RSC.
- H. Shimakoshi and Y. Hisaeda, ChemPlusChem, 2017, 82, 18 CrossRef CAS PubMed.
- H. Shimakoshi, E. Sakumori, K. Kaneko and Y. Hisaeda, Chem. Lett., 2009, 38, 468 CrossRef CAS.
- H. Shimakoshi, M. Abiru, S.-I. Izumi and Y. Hisaeda, Chem. Commun., 2009, 6427 RSC.
- S.-i Izumi, H. Shimakoshi, M. Abe and Y. Hisaeda, Dalton Trans., 2010, 39, 3302 RSC.
- H. Shimakoshi and Y. Hisaeda, ChemPlusChem, 2014, 79, 1250 CrossRef CAS.
- H. Tian, H. Shimakoshi, K. Imamura, Y. Shiota, K. Yoshizawa and Y. Hisaeda, Chem. Commun., 2017, 53, 9478 RSC.
- H. Shimakoshi and Y. Hisaeda, Angew. Chem., Int. Ed., 2015, 54, 15439 CrossRef CAS PubMed.
- M. Giedyk, J. Turkowska, S. Lepak, M. Marculewicz, K. Ó. Proinsias and D. Gryko, Org. Lett., 2017, 19, 2670 CrossRef CAS PubMed.
- K. Shichijo, M. Fujitsuka, Y. Hisaeda and H. Shimakoshi, J. Organomet. Chem., 2020, 907, 121058 CrossRef CAS.
- B. Steiger, E. Eichenberger and L. Walder, Chimia, 1991, 45, 32 CrossRef CAS.
- H. Shimakoshi, M. Tokunaga, T. Baba and Y. Hisaeda, Chem. Commun., 2004, 1806 RSC.
- H. Shimakoshi, S. Kudo and Y. Hisaeda, Chem. Lett., 2005, 34, 1096 CrossRef CAS.
- H. Shimakoshi, M. Nishi, A. Tanaka, K. Chikama and Y. Hisaeda, Chem. Commun., 2011, 47, 6548 RSC.
- L. Chen, Y. Kametani, K. Imamura, T. Abe, Y. Shiota, K. Yoshizawa, Y. Hisaeda and H. Shimakoshi, Chem. Commun., 2019, 55, 13070 RSC.
- L. Chen, Y. Hisaeda and H. Shimakoshi, Adv. Synth. Catal., 2019, 361, 2877 CrossRef CAS.
- K. Tahara and Y. Hisaeda, Green Chem., 2011, 13, 558 RSC.
- D. E. Yerien, A. Postigo, M. Baroncini and S. Barata-Vallejo, Green Chem., 2021, 23, 8147 RSC.
- Y. Anai, K. Shichijo, M. Fujitsuka, Y. Hisaeda and H. Shimakoshi, Chem. Commun., 2020, 56, 11945 RSC.
- K. R. Dworakowski, A. Chołuj, M. J. Chmielewski and D. Gryko, Chem. Commun., 2023, 59, 11236 RSC.
- W. Zhang, X. Zhu, H. Tong, H. Zhao, Y. Gu and W. Chu, Org. Lett., 2024, 26, 9073 CrossRef CAS PubMed.
- R. Scheffold, M. Dike, S. Dike, T. Herold and L. Walder, J. Am. Chem. Soc., 1980, 102, 3642 CrossRef CAS.
- A. Ruhe, L. Walder and R. Scheffold, Helv. Chim. Acta, 1985, 68, 1301 CrossRef CAS.
- J. F. Rusling, T. F. Connors and A. Owlia, Anal. Chem., 1987, 59, 2123 CrossRef CAS PubMed.
- M. Giedyk, H. Shimakoshi, K. Goliszewska, D. Gryko and Y. Hisaeda, Dalton Trans., 2016, 45, 8340 RSC.
- R. Scheffold and R. Orlinski, J. Am. Chem. Soc., 1983, 105, 7200 CrossRef CAS.
- H. Shimakoshi, Z. Luo, T. Inaba and Y. Hisaeda, Dalton Trans., 2016, 45, 10173 RSC.
- M. Moniruzzaman, Y. Yano, T. Ono, K. Imamura, Y. Shiota, K. Yoshizawa, Y. Hisaeda and H. Shimakoshi, J. Org. Chem., 2021, 86, 16134 CrossRef CAS PubMed.
- H. M. Shahadat, H. A. Younus, N. Ahmad, S. Zhang, S. Zhuiykov and F. Verpoort, Chem. Commun., 2020, 56, 1968 RSC.
- S. Ruccolo, M. Emmert, C. Bottecchia, Y. Qin, R. Barrientos, K. Raymond and M. Haley, Org. Lett., 2024, 26, 6169 CrossRef CAS PubMed.
- F. Zhou, Z. Hearne and C.-J. Li, Curr. Opin. Green Sustainable Chem., 2019, 18, 118 CrossRef.
- V. Hessel, N. N. Tran, M. R. Asrami, Q. D. Tran, N. Van Duc Long, M. Escribà-Gelonch, J. O. Tejada, S. Linke and K. Sundmacher, Green Chem., 2022, 24, 410 RSC.
- H. Sharghi, M. Aali Hosseini, J. Aboonajmi and M. Aberi, ACS Sustainable Chem. Eng., 2021, 9, 11163 CrossRef CAS.
- A. Wincenciuk, P. Cmoch, M. Giedyk, M. P. Andersson and D. Gryko, J. Am. Chem. Soc., 2024, 146, 19828 CrossRef CAS PubMed.
- Z. D. Zhang and R. Scheffold, Helv. Chim. Acta, 1993, 76, 2602 CrossRef CAS.
- A. Potrząsaj, M. Musiejuk, W. Chaładaj, M. Giedyk and D. Gryko, J. Am. Chem. Soc., 2021, 143, 9368 CrossRef PubMed.
- H. Su, L. Walder, Z.-D. Zhang and R. Scheffold, Helv. Chim. Acta, 1988, 71, 1073 CrossRef CAS.
- P. Bonhôte and R. Scheffold, Helv. Chim. Acta, 1991, 74, 1425 CrossRef.
- G. Prina Cerai and B. Morandi, Chem. Commun., 2016, 52, 9769 RSC.
- B. E. Funk, M. Pauze, Y.-C. Lu, A. J. Moser, G. Wolf and J. G. West, Cell Rep. Phys. Sci., 2023, 4, 101372 CrossRef CAS PubMed.
- B. Funk, S. R. Buzsaki, G. P. Allen, J. C. Bravo and J. G. West, Chem. Catal., 2026, 6, 101607 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |