
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFe(0)-catalyzed alkyne carboxylation with CO2 involving spin crossover
Huili
Wang†
a,
Qiqi
Zhang†
b,
Haizhu
Yu
*b,
Kane
Jacob
c,
Azzedine
Bousseksou
*c,
Philippe
Hapiot
 d,
Jean-Luc
Pozzo
*a and
Didier
Astruc
*a
d,
Jean-Luc
Pozzo
*a and
Didier
Astruc
*a
aUniversité de Bordeaux, ISM, UMR CNRS N°5255, 33405, Talence Cedex, France. E-mail: didier.astruc@u-bordeaux.fr
bDepartment of Chemistry and Center for Atomic Engineering of Advanced Materials, Anhui Province Key Laboratory of Chemistry for Inorganic/Organic Hybrid Functionalized Materials, Anhui University, Hefei, Anhui 230601, People's Republic of China. E-mail: yuhaizhu@ahu.edu.cn
cUniversité de Toulouse, LCC, UPR CNRS N°8241, 31077, Toulouse Cedex, France. E-mail: azzedine.bousseksou@lcc-toulouse.fr
dUniv. Rennes CNRS, ISCR (Institut des Sciences Chimiques de Rennes), UMR 6226, F-35000, Rennes, France
First published on 13th April 2026
Abstract
The first Fe(0)-catalyzed alkyne carboxylation using CO2 is reported using Fe(0)–phenanthroline complexes, and DFT calculations suggest a spin crossover from a triplet to a quintet state during the reaction.
Carbon dioxide (CO2) fixation in various forms is useful in order to limit the amount of greenhouse gases1 and, besides, CO2 is a cheap and readily available carbon source that is currently utilized for the formation of C–C, C–N and C–O bonds.2–9 In particular, propionic acids, which can be obtained via catalysed alkyne carboxylation, find applications in the manufacture of herbicides, rubber chemicals, emulsions, and environmentally friendly solvents for coating formulations, artificial fruit flavours, pharmaceuticals, and modified synthetic cellulose fibers, including cellulose acetate propionate.8–10 Catalysts employed in alkyne carboxylation for the formation of propionic acids mostly involved Ag11 and Cu.6,7,11,12 Iron, the most stable atom in the Universe and one of the most abundant elements in the Earth's crust, is often used in catalysis,13–15 particularly in heterogeneous catalysis, but its utilization in alkyne carboxylation to propionic acids is so far unknown.
First-row, late transition metal complexes with strong field ligands such as cyclopentadienyl and CO mostly fulfill the 18-electron rule, and they are “closed-shell” systems.16,17 On the other hand, inorganic nitrogen and oxygen ligands exert weak fields, so this weak ligand field leaves a relatively lower energy (Δ) between the three lower-energy eg orbitals and the two higher-energy t2g d orbitals in the case of an octahedral or a pseudo-octahedral geometry. The low Δ value in inorganic complexes opens the possibility of different orbital occupations by electrons, leading to the possibilities of two spin states, low spin or high spin.18,19 Different reactivities, including catalytic behaviours, are expected from these two spin states, which has already been exploited as a key feature since the 2000s in bio-inorganic catalytic systems.20–23 More recently, various studies have demonstrated the effect of the spin state and spin crossover in catalysis,24–37 but not yet for carboxylation reactions. Complexes with the general formula Fe(II)(1,10-phenanthroline)Cl2 bearing bulky substituents at the 2,9 positions have been reduced by the Zhu group to substituted Fe(0)(phenanthroline) complexes showing catalytic activities for reactions of alkenes and alkynes.27,37–43 This family of Fe(0) complexes are used in this study as alkyne carboxylation catalysts. The Fe electronic structure therein is 4s24p63d8, i.e. the eight d electrons occupy the five d orbitals. The chloro ligands have now been replaced by solvent ligands in the reduction process, and, according to both Pauli and Hund's rules, the three lower d orbitals are doubly occupied with electrons of opposite spins, and the two higher orbitals are singly occupied with electrons of the same spin, corresponding to a triplet spin ground state (S = 1; 2S + 1 = 3). Zhu et al. have taken their catalytic results into account using a two-spin-state reactivity, the phenanthroline acting as a redox ligand that could possibly accept an electron from Fe(0) to yield a quintet-state Fe(I) complex of phenanthroline radical anion.38 Under these conditions, among the five d orbitals, three become singly occupied, whereas one electron occupies a ligand orbital (S = 2). Such spin delocalization was reported in 2010–2012 with other iron catalysts containing a redox-active ligand; however, the relationship with reactivity effects was not rationalized.38
Herein, we report, using this family of iron–phenanthroline complexes, the first Fe(0)-catalyzed alkyne carboxylation reaction utilizing CO2 (eqn (1)), and DFT calculations suggest spin crossover during the catalyzed carboxylation process.
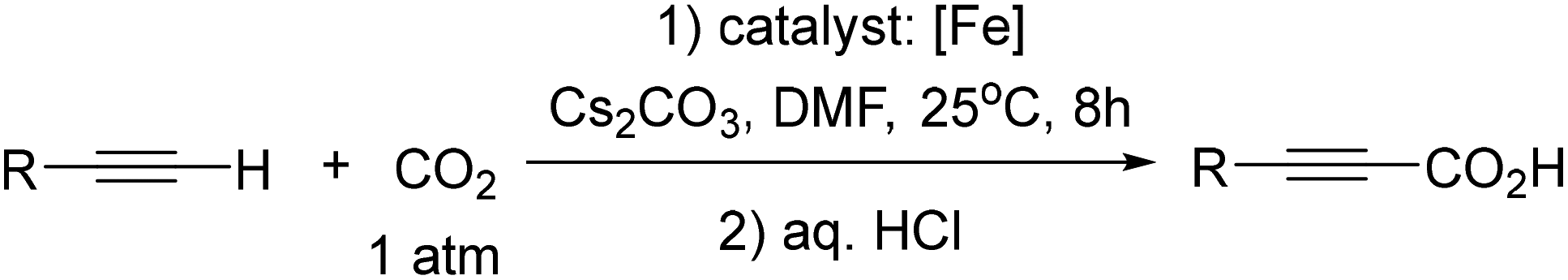 | (1) |
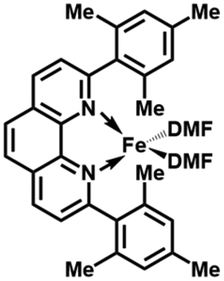
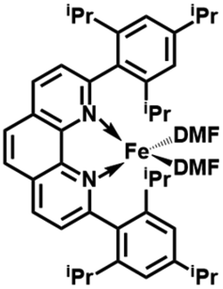
In order to obtain the Fe(0)(BTIPP) complex (BTIPP = 2,9-bis(2,4,6-triisopropylphenyl)-1,10-phenanthroline ligand), the Zhu group reduced the [Fe(II)(BTIPP)Cl2] complex using EtMgBr in THF and recorded the X-ray crystal structure of the Fe(II) precursor and a diolefin Fe(0) adduct.37,39
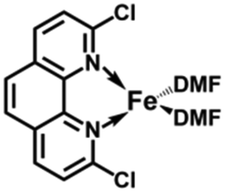
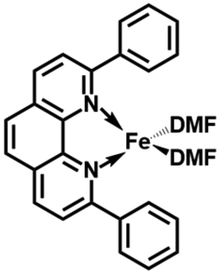
In the present work, the same and related Fe(II) precursors have been synthesized by stoichiometric reactions in THF between FeCl2 and the phenanthroline derivatives with substituents at the 2,9 positions (chloro, phenyl, mesityl and triisopropylphenyl), followed by precipitation from addition of excess hexane to a concentrated THF solution of the complexes and recrystallization from ether. The corresponding reduced black Fe(0) complexes have been generated in the same way as Zhu et al.37,39 by reduction of the Fe(II) precursor in DMF using EtMgBr in ether. Their cyclic voltammograms consistently show two successive irreversible cathodic waves at −1.2 and −2.1 V vs. Fc+/Fc on an Au disk electrode corresponding to the successive reductions of the first and second Cl ligands (SI). The resulting reactive Fe(0) catalysts that are very sensitive have not been isolated, but utilized in situ for carboxylation of various alkynes.
After reduction of these Fe(II) complexes to Fe(0) catalysts conducted using EtMgBr in ether/DMF, it is speculated that Fe is in the zero oxidation state with two or three solvent molecules (from the solvent used in the synthesis) in addition to the chelating L2 phenanthroline ligand, which would involve, respectively, 16-electron (2 weak solvent ligands) or 18-electron (3 weak solvent ligands) complexes. According to the DFT calculation results, in the presence of the sterically bulky BTIPP and DMF solvent, the coordination of three DMF molecules is unlikely. The two DMF molecules favorably adopt one η1- and one η2-coordination, and the Fe(0) complex is in a triplet spin state (Fig. 1).
 | ||
| Fig. 1 Relative energies (in kcal mol−1) of the BTIPP Fe(0) structures with coordination of two DMF ligands. The superscripts T, Q and S denote triplet, quintet and singlet states, respectively. | ||
Specifically, in the reduction step, the two chloro (X-type) ligands of the 14-electron Fe(II) precursors are replaced by the solvent:
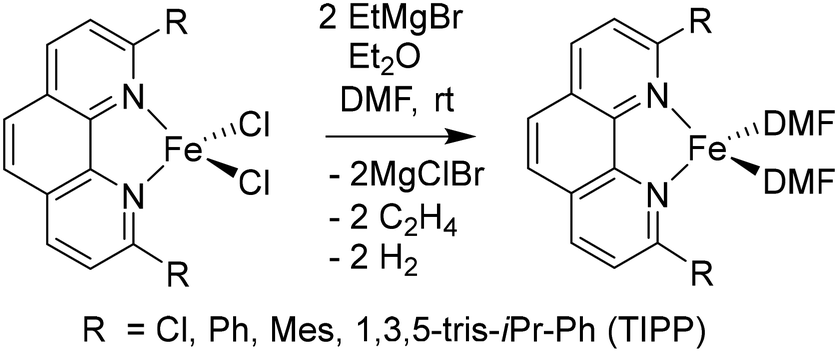 | (2) |
(L-type ligand, eqn (2)), yielding the Fe(N^N)L2 or Fe(N^N)L3 complexes.
During the catalytic process, the solvent ligands are readily displaced by alkyne and CO2 substrates. DFT calculations confirm that this ligand exchange is highly facile (Scheme 1).
 | ||
| Scheme 1 Gibbs free energy changes for the replacement of the DMF solvent by two alkynes (top) or an alkyne and CO2 (bottom). | ||
The alkyne carboxylation reactions catalyzed by the Fe(0) complexes were conducted using the four Fe(0) complexes at 25 °C and 80 °C, but it was found that the yields were approximately the same at both temperatures without any improvement at 80 °C, and therefore the yields obtained using different arylacetylene substrates at 25 °C are provided in Table 1. The best alkyne carboxylation yield (95%) was obtained using the bulkiest catalyst Fe(0)BTIPP with phenylacetylene, but, interestingly, all the other alkynes bulkier than phenylacetylene provided much lower yields, clearly demonstrating the predominance of steric effects over electronic substituent effects. With the other less bulky catalysts, the electron withdrawing substituents on the arene were beneficial to the reaction, in contrast to the arylacetylene bearing an electron-donating substituent. The catalysts bearing CF3 substituents led, due to steric bulk, to lower yields than those obtained with fluoroaryl substituents. The parent Fe(0)phenanthroline catalyst without substituents at the 1,9 positions gave very low yields (10% or less) with the various arylacetylenes, confirming the importance of the crucial steric effect in 2,9-disubstituted 1,10-phenanthroline positions observed with the Fe(0)BTIPP catalyst. With 1-butyne and 1-pentyne, the reaction provided modest yields; however, with the latter catalysts, mutual steric effects disfavored the reaction (Table 1). On the basis of the aforementioned experimental observations, DFT calculations were conducted on the modeling phenylacetylene carboxylation mechanism as well as the possible occurrence and effect of spin crossover during catalysis (see the SI for details).
|
|
||||
|---|---|---|---|---|
|
|
|
|
|
|
| Reaction conditions: 1 mmol substrate, 0.02 mmol ArPhenFe(0), 1 atm CO2 balloon, 1.5 mmol Cs2CO3 and 3 mL DMF. Sn = solvent ligand (n = 2 or 3). Isolated yield. | ||||
|
60% | 70% | 70% | 95% |
|
5% | 30% | 30% | 30% |
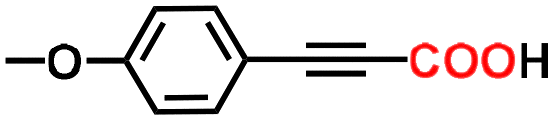
|
5% | 40% | 10% | 0% |

|
80% | 85% | 60% | 5% |

|
40% | 50% | 20% | 0% |

|
5% | 34% | 43% | 25% |

|
8% | 36% | 45% | 26% |

As shown in Fig. 2, the catalytic cycle begins with the triplet bis-alkyne adduct TIn1, whose energy lies below that of its quintet counterpart QIn1 and all other states. A spin crossover from a triplet to a quintet state could then occur via the MECP (minimum energy crossing point), whose energy is 31.0 kcal/mol higher than that of TIn1 to generate its quintet state QIn1. After that, the dissociation of one alkyne from QIn1 to generate the mono-alkyne coordinated quintet intermediate QIn2, and this step is slightly exergonic (ΔG0 = −4.5 kcal mol−1). From QIn2, the subsequent deprotonation and carboxylation proceed smoothly on the quintet potential energy surface, ultimately delivering the quintet carboxylate product QIn4. Ligand exchange between QIn4 and the alkyne substrates could regenerate the active catalyst TIn1, thereby finishing the catalytic cycle. After QIn2, the close energy of the triplet state intermediates and transition states to their quintet counterparts indicates that the deprotonation, carboxylation, and catalyst regeneration along the triplet potential energy surface could be competitive.
 | ||
| Fig. 2 The Gibbs free energy profile for the phenylacetylene carboxylation Fe(0) catalyst. The superscripts T, S, and Q denote triplet, singlet and quintet states, respectively. The relative electronic energy of MECP is given in grey color. | ||
Throughout this catalytic cycle (an illustrative diagram is provided in Fig. 3), the rate-determining step is the spin crossover step, i.e. the conversion from the triplet bis-alkyne adduct TIn1 to the quintet state. Compared to this mechanism, all the other mechanistic possibilities, such as the prior coordination of CO2 onto the Fe(0) center (i.e. the formation of T/Q/SIn5) or the involvement of the singlet mono-alkyne coordinated Fe(0) structure SIn2, and the CO2-coordinated structures (T/Q/SIn6 and QIn7– in supporting information) can be excluded due to their relatively high energy (Fig. 2). Specifically, similar to Zhu's observation,37,38 the spin distribution of the key species involved in the most feasible pathway indicate that the substituted phenanthroline acts as a redox ligand that accepts an electron from Fe(0) to provide a formal quintet-state Fe(I) complex of the phenanthroline radical anion (Table S1).
 | ||
| Fig. 3 Proposed catalytic cycle for the (phenanthrene)Fe(0)-catalyzed alkyne carboxylation. | ||
In summary, this study reports the first Fe(0)-phenanthroline catalyzed alkyne carboxylation using CO2 as a carbon source. Steric hindrance at the 2,9-positions of the ligand appears crucial for achieving high yields (up to 95%). DFT calculations reveal a unique catalytic pathway involving spin crossover from a triplet to a quintet state, which serves as the rate-determining step. Using earth-abundant iron, this work provides a sustainable and efficient alternative to precious or/and toxic metal catalysts for synthesizing propionic acid derivatives and a spin crossover phenomenon during the catalytic process.
H. Wang: investigation, data curation, methodology, software, validation, writing – original draft; Q. Zhang: DFT calculations; K. Jacob: investigation, data curation, software, validation; A. Bousseksou: methodology, validation, resources, funding acquisition, review and editing; Philippe Hapiot: methodology, validation, review and editing; H. Yu: methodology, software, data curation, validation, resources, funding acquisition, review and editing; J.-L. Pozzo: conceptualization, methodology, resources, project administration, review and editing; D. Astruc: conceptualization, resources, project administration, review and editing.
Conflicts of interest
There are no conflicts of interest to declare.Data availability
The data that support the findings of this study are available from the corresponding author upon reasonable request.Supplementary information (SI) is available. See DOI: https://doi.org/10.1039/d6cc00893c.
Acknowledgements
Financial support from the China Scholarship Council (CSC, PhD grants to H. W.), the Centre National de la Recherche Scientifique (CNRS), the National Natural Science Foundation of China (U23A2090), and the Universities of Bordeaux and Toulouse III, and technical support from the High-Performance Computing Platform of Anhui University are gratefully acknowledged.References
- A. S. Pottinger, R. Geyer, N. Biyani, C. C. Martinez, N. Nathan, M. R. Morse, C. Liu, S. Hu, M. de Bruyn, C. Boettiger, E. Baker and J. M. Douglas, Pathways to reduce global plastic waste mismanagement and greenhouse gas emissions by 2050, Science, 2024, 386, 1168–1173 CrossRef CAS PubMed.
- M. Aresta, A. Dibenedetto and A. Angelini, Catalysis for the valorization of exhaust carbon: from CO2 to chemicals, materials, and fuels. Technological use of CO2, Chem. Rev., 2014, 114, 1709–1742 CrossRef CAS PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Using carbon dioxide as a building block in organic synthesis, Nat. Commun., 2015, 6, 5933 CrossRef PubMed.
- A. Tortajada, F. Juliá-Hernández, M. Börjesson, T. Moragas and R. Martin, Transition–metal–catalyzed carboxylation reactions with carbon dioxide, Angew. Chem., Int. Ed., 2018, 57, 15948–15982 CrossRef CAS PubMed.
- A. R. Woldu, Z. Huang, P. Zhao, L. Hu and D. Astruc, Electrochemical CO2 reduction (CO2RR) to multi-carbon products over copper-based catalysts, Coord. Chem. Rev., 2022, 454, 214340 CrossRef CAS.
- A. L. Gu, Y. X. Zhang, Z. L. Wu, H. Y. Cui, T. D. Hu and B. Zhao, Highly Efficient Conversion of Propargylic Alcohols and Propargylic Amines with CO2 Activated by Noble-Metal-Free Catalyst Cu2O@ZIF-8, Angew. Chem., Int. Ed., 2022, 134, e202114817 CrossRef.
- J.-H. Ye, T. Ju, H. Huang, L.-L. Liao and D.-G. Yu, Radical carboxylative cyclizations and carboxylations with CO2, Acc. Chem. Res., 2021, 54, 2518–2531 CrossRef CAS PubMed.
- M. Miao, L. Zhu, H. Zhao, L. Song, S.-S. Yan, L.-L. Liao, J.-H. Ye, Y. Lan and D.-G. Yu, Visible-light-driven thio-carboxylation of alkynes with CO2: facile synthesis of thiochromones, Sci. China: Chem., 2023, 66, 1457–1466 CrossRef CAS.
- W. Wang, T. Wang, S. Chen, Y. Lv, L. Salmon, B. Espuche, S. Moya, O. Morozova, Y. Yun, D. Di Silvio, N. Daro, M. Berlande, P. Hapiot, J.-L. Pozzo, H. Yu, J.-R. Hamon and D. Astruc, Cu(I)-Glutathione Assembly Supported on ZIF-8 as Robust and Efficient Catalyst for Mild CO2 Conversions, Angew. Chem., Int. Ed., 2024, 63, e202407430 CrossRef CAS PubMed.
- K. L. Flannigan, K. M. Nieves, H. E. Szczepanski, A. Serra, J. W. Lee, L. A. Alston, H. Ramay, S. Mani and S. Hirota, The pregnane X receptor and indole-3-propionic acid shape the intestinal mesenchyme to restrain inflammation and fibrosis, Cell. Mol. Gastroenterol. Hepatol., 2023, 15, 765–795 CrossRef CAS PubMed.
- K. Sekine and T. Yamada, Silver-catalyzed carboxylation, Chem. Soc. Rev., 2016, 45, 4524–4532 RSC.
- L.-X. You, T.-C. Han, J.-R. Li, J. Guo, G. Xiong and Y.-G. Sun, An N-heterocyclic carbene–copper(I) functionalized metal-organic framework: synthesis and recoverable applications in the carboxylation of CO2 with terminal alkynes, J. Environ. Chem. Eng., 2025, 116785 CrossRef CAS.
- A. Fürstner, Iron catalysis in organic synthesis: a critical assessment of what it takes to make this base metal a multitasking champion, ACS Cent. Sci., 2016, 2, 778–789 CrossRef PubMed.
- I. Bauer and H.-J. Knölker, Iron catalysis in organic synthesis, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed.
- S. Rana, J. P. Biswas, S. Paul, A. Paik and D. Maiti, Organic synthesis with the most abundant transition metal–iron: from rust to multitasking catalysts, Chem. Soc. Rev., 2021, 50, 243–472 RSC.
- M. Elian, M. M. Chen, D. M. P. Mingos and R. Hoffmann, Comparative bonding study of conical fragments, Inorg. Chem., 1976, 15, 1148–1155 CrossRef CAS.
- R. Hoffmann, Building bridges between inorganic and organic chemistry (Nobel Lecture), Angew. Chem., Int. Ed. Engl., 1982, 21, 711–724 CrossRef.
- G. Molnár, S. Rat, L. Salmon, W. Nicolazzi and A. Bousseksou, Spin crossover nanomaterials: from fundamental concepts to devices, Adv. Mater., 2018, 30, 1703862 CrossRef PubMed.
- A. Bousseksou, G. Molnár, L. Salmon and W. Nicolazzi, Molecular spin crossover phenomenon: recent achievements and prospects, Chem. Soc. Rev., 2011, 40, 3313–3335 RSC.
- D. Schröder, S. Shaik and H. Schwarz, Two-state reactivity as a new concept in organometallic chemistry, Acc. Chem. Res., 2000, 33, 139–145 CrossRef PubMed.
- A. L. Buchachenko and V. L. Berdinsky, Electron spin catalysis, Chem. Rev., 2002, 102, 603–612 CrossRef CAS PubMed.
- J. N. Harvey, R. Poli and K. M. Smith, Understanding the reactivity of transition metal complexes involving multiple spin states, Coord. Chem. Rev., 2003, 238, 347–361 CrossRef.
- M. P. Shaver, L. E. Allan, H. S. Rzepa and V. C. Gibson, Correlation of metal spin state with catalytic reactivity: polymerizations mediated by alpha-diimine-iron complexes, Angew. Chem., Int. Ed., 2006, 45, 1241–1244 CrossRef CAS PubMed.
- S. Shaik, H. Hirao and D. Kumar, Reactivity of high-valent iron–oxo species in enzymes and synthetic reagents: a tale of many states, Acc. Chem. Res., 2007, 40, 532–542 CrossRef CAS PubMed.
- Y. Sun, H. Tang, K. Chen, L. Hu, J. Yao, S. Shaik and H. Chen, Two-state reactivity in low-valent iron-mediated C–H activation and the implications for other first-row transition metals, J. Am. Chem. Soc., 2016, 138, 3715–3730 CrossRef CAS PubMed.
- J. Liu, L. Hu, L. Wang, H. Chen and L. Deng, An iron(II) ylide complex as a masked open-shell iron alkylidene species in its alkylidene-transfer reactions with alkenes, J. Am. Chem. Soc., 2017, 139, 3876–3888 CrossRef CAS PubMed.
- P. He and S. F. Zhu, Spin crossover and its application in organometallic catalysis: concepts and recent progress, Chem. – Eur. J., 2024, 30, e202403437 CrossRef CAS PubMed.
- M. A. Halcrow, Manipulating metal spin states for biomimetic, catalytic and molecular materials chemistry, Dalton Trans., 2020, 49, 15560–15567 RSC.
- F. Wang, L. Hu and Y. Jing, Correlation between the spin effect and catalytic activity of two-dimensional metal organic frameworks for the oxygen evolution reaction, J. Mater. Chem. A, 2024, 12, 28764–28770 RSC.
- Z. Jiang, K. Tong, Z. Li, H. Tao and M. Zhu, Spin State Regulation for Peroxide Activation: Fundamental Insights and Regulation Mechanisms, Angew. Chem., 2025, 137, e202500791 CrossRef.
- Q. Huang, S. Xie, J. Hao, Z. Ding, C. Zhang, H. Sheng and J. Zhao, Spin-enhanced O−H cleavage in electrochemical water oxidation, Angew. Chem., Int. Ed., 2023, 62, e202300469 CrossRef CAS PubMed.
- Y. Li, D. Wang, Y. Guan, H. Liu, Y. Bao, N. Wu, X. Zhao, C. Sun, Z. Li and L. Lei, Theory-driven design of local spin-state modulation at atomically dispersed iron sites for enhanced CO2 electroreduction, Nano Energy, 2024, 132, 110370 CrossRef.
- J. Huang, C. N. Borca, T. Huthwelker, N. S. Yüzbasi, D. Baster, M. El Kazzi, C. W. Schneider, T. J. Schmidt and E. Fabbri, Surface oxidation/spin state determines oxygen evolution reaction activity of cobalt-based catalysts in acidic environment, Nat. Commun., 2024, 15, 3067 CrossRef CAS PubMed.
- G.-Z. Huang, Y.-S. Xia, F. Yang, W.-J. Long, J.-J. Liu, J.-P. Liao, M. Zhang, J. Liu and Y.-Q. Lan, On–off switching of a photocatalytic overall reaction through dynamic spin-state transition in a Hofmann clathrate system, J. Am. Chem. Soc., 2023, 145, 26863–26870 CrossRef CAS PubMed.
- A. Enríquez-Cabrera, Y. Lai, L. Salmon, L. Routaboul and A. Bousseksou, Spin-state effect on the efficiency of a post-synthetic modification reaction on a spin crossover complex, Commun. Chem., 2025, 8, 47 CrossRef PubMed.
- Y. Lai, L. Getzner, A. Enríquez-Cabrera, G. Molnár, L. Routaboul and A. Bousseksou, Spin State-Driven Modulation of Catalytic Activity of an Iron Spin-Crossover Complex: Toward Switchable Catalysis, ChemCatChem, 2025, e202500127 CrossRef CAS.
- M.-Y. Hu, Q. He, S.-J. Fan, Z.-C. Wang, L.-Y. Liu, Y.-J. Mu, Q. Peng and S.-F. Zhu, Ligands with 1, 10-phenanthroline scaffold for highly regioselective iron-catalyzed alkene hydrosilylation, Nat. Commun., 2018, 9, 221 CrossRef PubMed.
- P. He, M. H. Guan, M. Y. Hu, Y. J. Zhou, M. Y. Huang and S. F. Zhu, Iron-Catalyzed Allylic C(sp3)−H Silylation: Spin-Crossover-Efficiency-Determined Chemoselectivity, Angew. Chem., Int. Ed., 2024, 63, e202402044 CrossRef CAS PubMed.
- P. He, M.-Y. Hu, J.-H. Li, T.-Z. Qiao, Y.-L. Lu and S.-F. Zhu, Spin effect on redox acceleration and regioselectivity in Fe-catalyzed alkyne hydrosilylation, Natl. Sci. Rev., 2024, 11, nwad324 CrossRef CAS PubMed.
- M.-Y. Hu, P. He, T.-Z. Qiao, W. Sun, W.-T. Li, J. Lian, J.-H. Li and S.-F. Zhu, Iron-catalyzed regiodivergent alkyne hydrosilylation, J. Am. Chem. Soc., 2020, 142, 16894–16902 CrossRef CAS PubMed.
- M.-Y. Hu, J. Lian, W. Sun, T.-Z. Qiao and S.-F. Zhu, Iron-catalyzed dihydrosilylation of alkynes: efficient access to geminal bis (silanes), J. Am. Chem. Soc., 2019, 141, 4579–4583 CrossRef CAS PubMed.
- Q. Huang, Y.-X. Su, W. Sun, M.-Y. Hu, W.-N. Wang and S.-F. Zhu, Iron-catalyzed vinylzincation of terminal alkynes, J. Am. Chem. Soc., 2021, 144, 515–526 CrossRef PubMed.
- Q. Huang, W.-N. Wang and S.-F. Zhu, Iron-catalyzed alkylzincation of terminal alkynes, ACS Catal., 2022, 12, 2581–2588 CrossRef CAS.
Footnote |
| † Both authors (HW and QZ) contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2026 |