
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A direct route to ynones: Pd-catalyzed Sonogashira carbonylative coupling of ethers with alkynes via C–O cleavage
Xiaowen
Qin
a,
Shuhui
Sun
a,
Weijie
Zhang
a,
Tiefeng
Xu
 b,
Xiao-Feng
Wu
*c and
Zhiping
Yin
*a
b,
Xiao-Feng
Wu
*c and
Zhiping
Yin
*a
aSchool of Pharmacy, Jiangsu University, Zhenjiang 212013, P. R. China. E-mail: zhiping_yin@ujs.edu.cn
bNational Engineering Lab for Textile Fiber Materials & Processing Technology (Zhejiang), Zhejiang Sci-Tech University, Hangzhou 310018, China
cDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023, Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
First published on 24th February 2026
Abstract
Herein, we report a catalytic strategy that directly utilizes benzylic ethers as coupling partners through a palladium/copper co-catalyzed pathway. The reaction exhibits broad substrate scope, accommodating a wide range of functionalized benzylic alcohols and terminal alkynes, and provides direct access to valuable ynones.
Ynones represent a privileged structural motif prevalent in pharmaceuticals, natural products, and functional materials, serving as versatile intermediates for heterocycle synthesis and conjugate addition (Scheme 1a).1 Among various synthetic approaches, the carbonylative Sonogashira reaction has emerged as one of the most reliable methods for ynone formation. This process capitalizes on the unique efficiency of carbon monoxide, a C1 building block that enables streamlined carbonyl incorporation.2 Over the past few decades, this transformation has been extensively optimized, typically relying on palladium catalysis, carbon monoxide, and two key coupling partners: terminal alkynes and prefunctionalized electrophiles such as aryl- or benzyl-halides (Scheme 1b).3 Despite its widespread utility, this classical paradigm remains intrinsically constrained by its dependence on halide-based electrophiles, which necessitate additional synthetic steps for their installation, generate stoichiometric halide waste, and limit overall atom economy.
 | ||
| Scheme 1 Investigations of Sonogashira carbonylation synthesis of ynones. | ||
Recent years have witnessed growing interest in C–O bond functionalization as a sustainable alternative to conventional cross-coupling protocols.4 Notably, catalytic systems enabling the carbonylative transformation of benzylic alcohols into esters and related derivatives have been established through in situ activation strategies.5 However, the development of a direct deoxygenative carbonylative coupling of alcohols with alkynes to access ynones presents a distinct set of challenges. The simultaneous requirements of C–O bond cleavage, CO insertion, and alkyne coupling – each with competing mechanistic demands – have rendered this transformation a persistent unmet goal. Consequently, a catalytic system capable of integrating these steps efficiently, while operating under practical and mild conditions, remains highly desirable. A classical approach to ynone synthesis involves the nucleophilic addition of metal acetylides to carbonyl compounds. However, this route often necessitates prefunctionalized substrates or subsequent oxidation steps.6 Thus, the development of a direct deoxygenative carbonylative coupling of ethers with alkynes would not only offer practical advantages but also establish a new conceptual framework for catalytic ynone synthesis (Scheme 1c).
Building upon our group's longstanding focus on carbonylation reactions,7 we report a novel palladium-catalyzed carbonylative Sonogashira reaction via benzylic C–O bond cleavage. Our strategy focuses on in situ activation using pentafluoropyridine, which smoothly converts versatile inert benzylic alcohols into reactive benzylpyridinium intermediates. These species readily engage in a subsequent catalytic cycle, enabling direct coupling with terminal alkynes. This strategy introduces a catalytic system that merges C–O activation, CO insertion, and alkyne coupling in a single operation, thereby replacing conventional benzyl halides with widely available benzylic ethers as coupling partners.
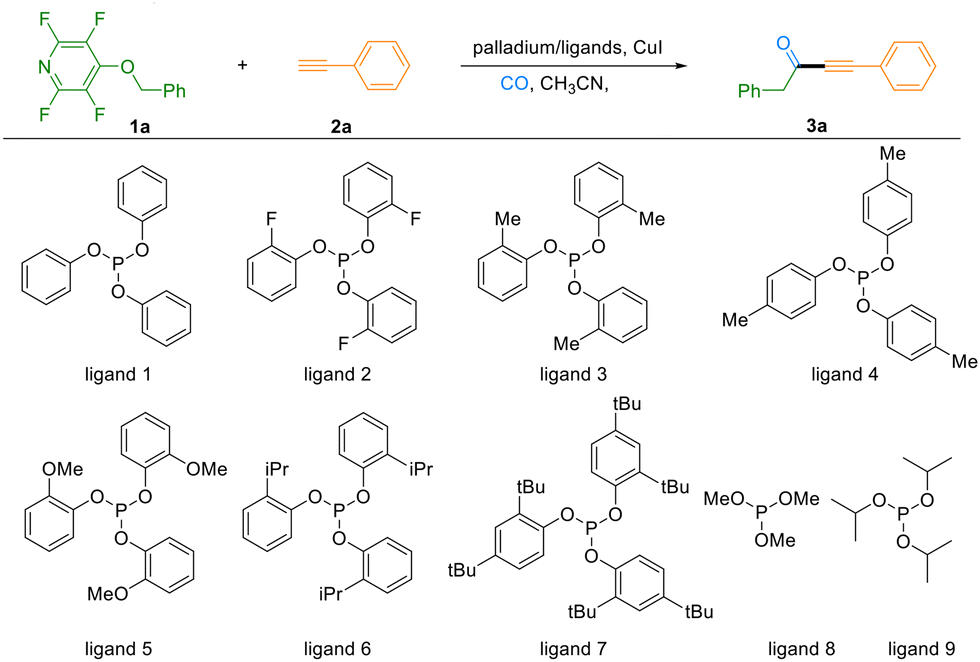
Our initial efforts focused on optimizing the reaction conditions using 4-(benzyloxy)-2,3,5,6-tetrafluoropyridine (1a) and phenylacetylene (2a) as model substrates in acetonitrile under a CO gas pressure (2 bar), with CuI and PdCl2(PPh3)2/(OPh)3P serving as the catalytic system. Gratifyingly, the desired product 3a was obtained in 37% GC yield (Table 1, entry 1). Preliminary solvent screening identified CH3CN as uniquely effective, as alternatives such as DMF, dioxane, and toluene proved unsuitable (see Section 3.1, SI). Further evaluation of palladium precursors revealed that Pd(OAc)2 significantly enhanced the reaction efficiency, elevating the yield to 66% (entries 2 and 3). Given the documented importance of phosphine ligands in suppressing alkyne homocoupling and enhancing catalytic performance in Sonogashira carbonylative processes, we systematically examined a range of phosphine ligands (entries 4–11).8 By introducing substituents with varying electronic and steric properties – including electron-withdrawing (F) and electron-donating (Me, OMe, tBu) groups at different positions on the phenyl rings of triphenyl phosphite derivatives – we identified tri-o-tolyl phosphite (ligand 3) as optimal, delivering 3a in 75% GC yield. A slight improvement was achieved by increasing the ligand loading to 15 mol% (entry 12). Ultimately, elevating the reaction temperature to 120 °C enabled a 90% GC yield, with an isolated yield of 84% for the target compound 3a (entry 13). Furthermore, control experiments underscored the essential roles of each catalytic component. Omission of CuI afforded only trace amounts of product, while reactions conducted in the absence of ligand or Pd(OAc)2 gave diminished yields of 31% and 54%, respectively (entries 15 and 16).
|
|
||
|---|---|---|
| Entry | Palladium/ligand | Yieldb |
| a Reaction conditions: 1a (103 mg, 0.4 mmol, 2.0 eq), 2a (22 µL, 0.2 mmol, 1.0 eq), palladium catalyst 5 mol%, ligand 10 mol%, CuI (7.6 mg, 20 mol%), CO 2 bar, CH3CN 2 mL, 110 °C, 24 h. b GC yields of mixture 3a were determined by using dodecane as the internal standard. c Ligand 3 15 mol%. d 120 °C. e Isolated yield is given in parentheses. f Without CuI. g Without Pd(OAc)2. h Without ligand. | ||
| 1 | PdCl2(PPh3)2/ligand 1 | 37 |
| 2 | Pd(OAc)2/ligand 1 | 66 |
| 3 | Pd(TFA)2/ligand 1 | 43 |
| 4 | Pd(OAc)2/ligand 2 | 40 |
| 5 | Pd(OAc)2/ligand 3 | 75 |
| 6 | Pd(OAc)2/ligand 4 | 68 |
| 7 | Pd(OAc)2/ligand 5 | 62 |
| 8 | Pd(OAc)2/ligand 6 | 66 |
| 9 | Pd(OAc)2/ligand 7 | 48 |
| 10 | Pd(OAc)2/ligand 8 | 51 |
| 11 | Pd(OAc)2/ligand 9 | 24 |
| 12c | Pd(OAc)2/ligand 3 | 83 |
| 13cd | Pd(OAc)2/ligand 3 | 90 (84e) |
| 14f | Pd(OAc)2/ligand 3, without CuI | Trace |
| 15g | without Pd(OAc)2/ligand 3 | 54 |
| 16h | Pd(OAc)2/without ligand 3 | 31 |
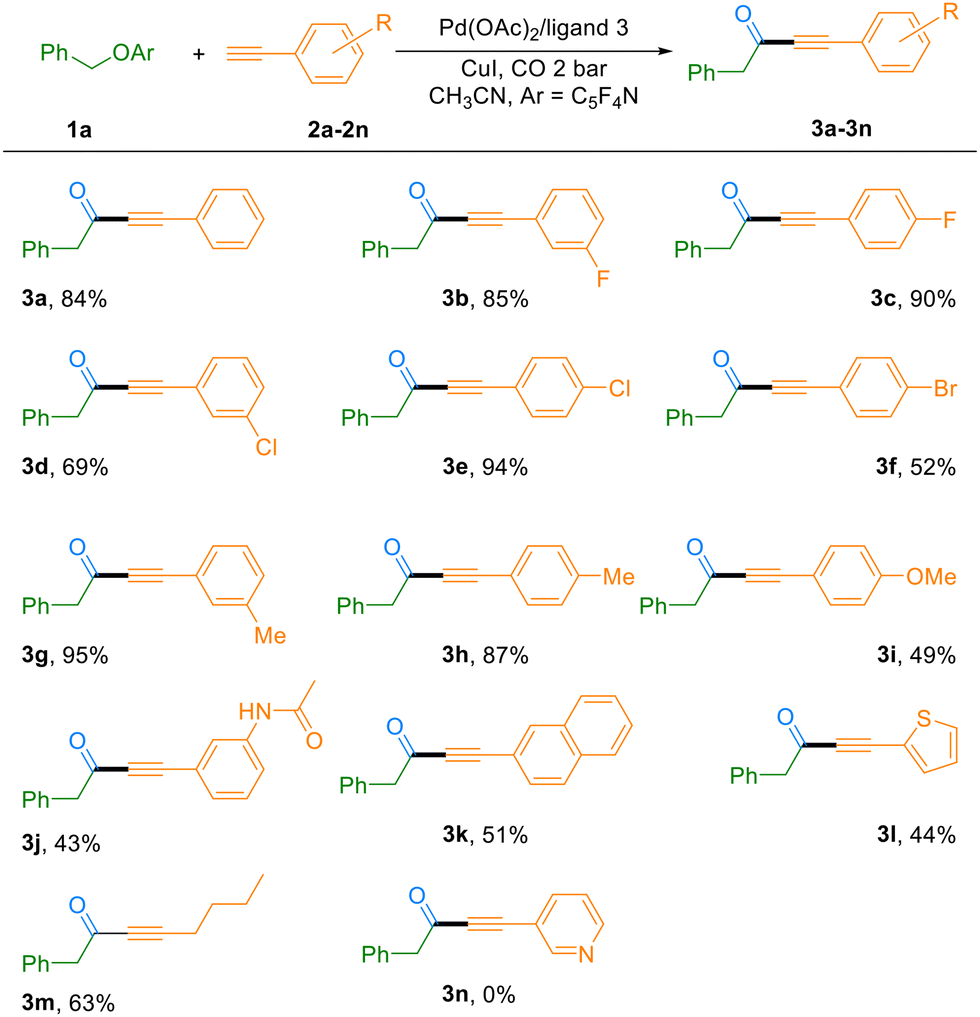
With the optimized reaction conditions in hand, we subsequently evaluated the substrate scope of this C–O bond carbonylation transformation. As summarized in Table 2, the reaction demonstrated excellent compatibility with a wide range of terminal alkynes. Electron-deficient alkynes featuring halogen substituents (F, Cl, and Br) at the meta- or para-positions of the phenyl ring reacted smoothly, affording the corresponding ynones in 52–94% yields (Table 2, 3b–3f). It is worth noting that bromo-substituted substrates not only participated efficiently in the reaction (52% yield, 3f), but also retained the synthetically versatile bromo group, allowing for further functionalization of the products. The catalytic system also proved highly effective for electron-rich alkynes. Substrates bearing methyl, methoxy, or amide groups were well tolerated, delivering the desired products in 43–95% yields (Table 2, 3g–j). Furthermore, naphthyl-substituted alkynes and heteroaromatic thiophene-based alkynes were also compatible reaction partners, yielding the corresponding ynones in 51% and 44% yields, respectively. However, a pyridine-containing alkyne failed to undergo the transformation, presumably due to coordination of the basic nitrogen atom to the catalytic metal center, which may inhibit the catalytic cycle (Table 2, 3n). Most notably, the methodology was successfully extended to an aliphatic terminal alkyne. Under the standard conditions, 1-hexyne participated effectively in the reaction, furnishing the ynone product 3m in 63% yield. This result highlights the broad substrate generality of the present catalytic system, including its applicability to non-aromatic alkynes.
| a Reaction conditions: 2a–2n (0.2 mmol), 1a (0.4 mmol, 2 equivalent), Pd(OAc)2 (5 mol%, 2.3 mg), ligand 3 (15 mol%), CuI (7.6 mg, 20 mol%), CO 2 bar, CH3CN (2 mL), 120 °C. 24 h. |
|---|
|

We next investigated the scope of benzyl alcohol-derived ether substrates in this C–O bond carbonylation system (Table 3). The transformation exhibited broad functional group tolerance, accommodating halogen (F, Cl) and trifluoromethyl substituents at various positions of the phenyl ring, with yields ranging from 62% to 77% depending on the substitution pattern (Table 3, 4a–4e). Electron-donating groups, including methyl, tert-butyl, phenyl, and naphthyl, were also compatible, affording the desired products in 47–71% yields (Table 3, 4f–4i). However, a methoxy-substituted ether afforded the target product in only 29% yield, presumably due to competing side reactions involving the ether moiety. In contrast, pyridine- and allylic ether-based substrates proved challenging under the standard conditions, yielding only trace amounts of the desired products. However, the reaction failed with aliphatic alcohols.
To demonstrate the practical utility of this method, a gram-scale reaction was conducted at a 4 mmol scale, delivering the corresponding ynone in 81% isolated yield (Scheme 2, I), thereby underscoring the potential of this protocol for industrial applications. Furthermore, as highlighted earlier, the resulting ynones serve as versatile synthetic intermediates for accessing diverse heterocyclic scaffolds. For instance, compound 3a underwent a copper-catalyzed cascade process involving addition/5-exo-dig cyclization/isomerization with isothiocyanate to furnish (Z)-2-ylidene-5-aminothiophen-3-one (Scheme 2, II).9 Additionally, the intramolecular ipso-iodocyclization of 1-(4-methoxyphenyl)-4-phenylbut-3-yn-2-one (4j) enabled efficient construction of spiro[4.5]trienone in 85% yield (Scheme 2, III).10 These post-functionalization examples collectively illustrate the synthetic versatility and robustness of the present carbonylation strategy.
 | ||
| Scheme 2 Large-scale synthesis and further synthetic application. | ||
To gain deeper mechanistic insight into the carbonylation process, a series of control experiments were conducted (Scheme 3). Firstly, the reaction pathway was probed using radical scavengers such as TEMPO and 1,1-diphenylethylene (Scheme 3, I). In the presence of 1,1-diphenylethylene, the transformation proceeded smoothly, affording product 3a in 93% yield (Scheme 3, II), thereby ruling out the involvement of radical intermediates. It is also noteworthy that alternative ether substrates, including (methoxymethyl)benzene and (benzyloxy)benzene, failed to produce 3a (Scheme 3, III, IV), highlighting the unique reactivity of the pentafluoropyridine-based system in facilitating this C–O activation process.
 | ||
| Scheme 3 Control experiments, GC yield. | ||
On the basis of these experimental results and relevant literature precedents,11 a plausible catalytic cycle is proposed (Fig. 1). The cycle begins with oxidative addition of the C–O bond in 1a to Pd(0), generating benzyl–palladium(II) species Int.-1. Carbon monoxide coordination and insertion then affords the acyl–palladium intermediate Int.-3. Meanwhile, the terminal alkyne is activated by CuI to form a copper–acetylide complex, which acts as a nucleophile attacking Int.-3 to give the palladium species Int.-4. Finally, reductive elimination from Int.-4 delivers the ynone product 3a and regenerates the active Pd(0) catalyst, closing the catalytic cycle.
 | ||
| Fig. 1 Proposed mechanism. | ||
In conclusion, we have developed a deoxygenative carbonylation reaction that successfully replaces classical benzyl halides with readily available benzylic ethers for ynone synthesis. The success of this transformation relies on efficient C–O activation mediated by pentafluoropyridine, enabling a sequential palladium-catalyzed process involving C–O cleavage, CO insertion, and terminal alkyne coupling. This operationally simple protocol features excellent functional group tolerance and provides a step-economical alternative to conventional approaches. Beyond its synthetic utility, this work establishes a new paradigm for employing inert C–O bonds as versatile handles in transition-metal-catalyzed carbonylation chemistry, offering a promising platform for the direct valorization of ether feedstocks.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d6cc00779a.Acknowledgements
We acknowledge the financial support from the Senior Talent Foundation of Jiangsu University (4111290008) and the Natural Science Foundation of Jiangsu Province (BK20250875). We also appreciate the general support of Professor Weiping Zheng at Jiangsu University.References
- (a) C. Nájera, L. K. Sydnes and M. Yus, Chem. Rev., 2019, 119, 11110–11244 CrossRef PubMed; (b) Y. Li, J. Yu, Y. Bi, G. Yan and D. Huang, Adv. Synth. Catal., 2019, 361, 4839–4881 CrossRef CAS; (c) L. Wang, H. Zhu, T. Peng and D. Yang, Org. Biomol. Chem., 2021, 19, 2110–2145 Search PubMed; (d) S. Bhagat, N. Goel, P. Kumari, Gunjan and A. Khullar, Org. Biomol. Chem., 2025, 23, 5273–5292 RSC; (e) C. Liu and M. Szostak, Org. Chem. Front., 2022, 9, 216–222 Search PubMed.
- (a) D.-M. Liu, B.-W. Sun, Z.-W. Sun, Q. Jin, Y. Sun and X. Chen, Acta Pharmacol. Sin., 2008, 29, 838–846 CrossRef CAS PubMed; (b) W.-C. Shen, X. Wang, W.-T. Qin, X.-F. Qiu and B.-W. Sun, Acta Pharmacol. Sin., 2014, 35, 1566–1576 CrossRef CAS PubMed; (c) X. Qiu, W. Shen, X. Wang, W. Qin and B. Sun, Pak. J. Pharm. Sci., 2015, 28, 281–292 Search PubMed; (d) X. F. Wu, P. J. Dyson and B. A. Arndtsen, J. Org. Chem., 2023, 88, 4891–4893 CrossRef CAS PubMed; (e) J. L. Wang, C. X. Miao, X. Y. Dou, J. A. Gao and L. N. He, Curr. Org. Chem., 2011, 15, 621–646 Search PubMed; (f) J. B. Peng, H. Q. Geng and X. F. Wu, Chem, 2019, 5, 526–552 CrossRef CAS; (g) C. S. Kuai, Y. Yuan and X. F. Wu, Chem, 2025, 11, 102503 Search PubMed; (h) X.-F. Wu, B. Han, K. Ding and Z. Liu, The chemical transformations of C1 compounds, John Wiley & Sons, 2022 CrossRef.
- (a) X.-F. Wu, H. Neumann and M. Beller, Org. Biomol. Chem., 2011, 9, 8003–8005 RSC; (b) X. Feng, J. Song, H. Liu, L. Wang, X. Yu and M. Bao, RSC Adv., 2013, 3, 18985–18991 RSC; (c) S. Perrone, F. Bona and L. Troisi, Tetrahedron, 2011, 67, 7386–7391 CrossRef CAS; (d) V. Sans, A. M. Trzeciak, S. Luis and J. J. Ziółkowski, Catal. Lett., 2006, 109, 37–41 CrossRef CAS.
- (a) G. Y. Song, F. Wang and X. W. Li, Chem. Soc. Rev., 2012, 41, 3651–3678 Search PubMed; (b) M. Tobisu and N. Chatani, Acc. Chem. Res., 2015, 48, 1717–1726 CrossRef CAS PubMed; (c) E. Bisz and M. Szostak, ChemSusChem, 2017, 10, 3964–3981 CrossRef CAS PubMed; (d) S. M. Pound and M. P. Watson, Chem. Commun., 2018, 54, 12286–12301 Search PubMed; (e) X. Zhang, F. Jordan and M. Szostak, Org. Chem. Front., 2018, 5, 2515–2521 RSC; (f) T. L. Zhou and M. Szostak, Catal. Sci. Technol., 2020, 10, 5702–5739 RSC; (g) P. Villo, A. Shatskiy, M. D. Kärkäs and H. Lundberg, Angew. Chem., Int. Ed., 2023, 62, e202211952 CrossRef CAS PubMed.
- (a) J. Duan, J. Jiang, J. Gong, Q. Fan and D. Jiang, J. Mol. Catal. A: Chem., 2000, 159, 89–96 CrossRef CAS; (b) S. Jayasree, A. Seayad and R. V. Chaudhari, Org. Lett., 2000, 2, 203–206 CrossRef CAS PubMed; (c) S. Sabater, M. Menche, T. Ghosh, S. Krieg, K. S. L. Rück, R. Paciello, A. Schäfer, P. Comba, A. S. K. Hashmi and T. Schaub, Organometallics, 2020, 39, 870–880 CrossRef CAS.
- (a) R. E. Whittaker, A. Dermenci and G. B. Dong, Synthesis, 2016, 161–183 Search PubMed; (b) A. Behera, Adv. Syn. Catal., 2024, 366, 1044–1058 CrossRef CAS; (c) S. Bhagat, N. Goel, P. Kumari and A. Khullar, Org. Biomol. Chem., 2025, 23, 5273–5292 RSC.
- (a) X. Liu, T. An, Z. Yin and W. Zhang, Chem. – Eur. J., 2023, 29, e202202880 CrossRef CAS PubMed; (b) W. Yuan, T. An, C. Liu, X. Qin and Z. Yin, Asian J. Org. Chem., 2024, 13, e202400078 CrossRef CAS; (c) T. An, C. Liu, W. Yuan, X. Qin and Z. Yin, Chem. Commun., 2024, 60, 3389–3392 RSC; (d) C. Liu, T. An, W. Yuan, H. Dai, X. Liang and Z. Yin, Chem. Commun., 2023, 59, 12891–12894 RSC; (e) Z. Yin, J. Xu and X.-F. Wu, ACS Catal., 2020, 10, 6510–6531 CrossRef CAS; (f) Z. Yin, W. Shi and X.-F. Wu, J. Org. Chem., 2023, 8, 4975–4994 CrossRef PubMed; (g) N. N. Zhang, Y. L. Zhao, D. M. Fan, J. B. Xiao, K. W. Cheng and M. F. Wang, Food Hydrocoll., 2020, 108, 106073 Search PubMed; (h) I. A. Khan, J. Luo, H. B. Shi, Y. Zou, A. Khan, Z. S. Zhu, W. M. Xu, D. Y. Wang and M. Huang, Food Chem., 2022, 368, 130845 Search PubMed; (i) N. N. Liang, B. Q. Shi, X. T. Hu, W. T. Li, X. W. Huang, Z. H. Li, X. N. Zhang, X. B. Zou and J. Y. Shi, Food Chem., 2024, 460, 140570 Search PubMed.
- (a) J. M. Brunel and B. Faure, J. Mol. Catal. A: Chem., 2004, 212, 61–64 Search PubMed; (b) A. A. Dabbawala, R. V. Jasra and H. C. Bajaj, Catal. Commun., 2010, 11, 616–619 CrossRef CAS; (c) E. Jung, K. Park, J. Kim, H. T. Jung, I. K. Oh and S. Lee, Inorg. Chem. Commun., 2010, 13, 1329–1331 CrossRef CAS; (d) P. A. Evans, E. A. Clizbe, M. J. Lawler and S. Oliver, Chem. Sci., 2012, 3, 1835–1838 RSC; (e) M. V. Sokolovskaya, S. E. Lyubimov, I. S. Mikhel, K. P. Birin and V. A. Davankov, J. Organomet. Chem., 2018, 861, 230–233 CrossRef CAS; (f) J. E. Smart, J. Emerson-King, R. J. Jeans, T. M. Hood, S. Lau, A. Bara-Estaún, U. Hintermair, P. G. Pringle and A. B. Chaplin, ACS Catal., 2024, 14, 11803–11807 CrossRef CAS PubMed; (g) P. Basnet, R. K. Dhungana, S. Thapa, B. Shrestha, S. Kc, J. M. Sears and R. Giri, J. Am. Chem. Soc., 2018, 140, 7782–7786 CrossRef CAS PubMed.
- Z. Jin, C. Li, G. Shen, H. Kang and X. Lv, Adv. Synth. Catal., 2024, 366, 5151–5159 CrossRef CAS.
- X. Zhang and R. C. Larock, J. Am. Chem. Soc., 2005, 127, 12230–12231 Search PubMed.
- (a) Y. Jiang, Y. Wei, Q.-Y. Zhou, G.-Q. Sun, X.-P. Fu, N. Levin, Y. Zhang, W.-Q. Liu, N. Song, S. Mohammed, B. G. Davis and M. J. Koh, Nature, 2024, 631, 319–327 Search PubMed; (b) X. Qin, H. Li, S. Sun, S. Shen, T. Xu, X.-F. Wu and Z. Yin, Org. Lett., 2025, 27, 12524–12529 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |