
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Palladium-catalyzed denitrative C–N coupling of amides with nitroarenes enabled by Al(OTf)3
Linjie
Yang
*abc,
Xuejie
Wang
c and
Wanzhi
Chen
 c
c
aCollege of Chemistry and Environmental Engineering, Shenzhen University, Shenzhen 518060, China
bGuangdong Key Laboratory for Biomedical Measurements and Ultrasound Imaging, National-Regional Key Technology Engineering Laboratory for Medical Ultrasound, School of Biomedical Engineering, Shenzhen University Medical School, Shenzhen 518060, China
cDepartment of Chemistry, Zhejiang University, Hangzhou 310058, China. E-mail: 21937063@zju.edu.cn; chenwzz@zju.edu.cn
First published on 19th March 2026
Abstract
We report a Pd-catalyzed denitrative C–N coupling of amide nucleophiles with nitroarenes, providing direct and atom-economical access to N-aryl amides from readily available nitroarene feedstocks. Al(OTf)3 is essential, both promoting the reaction and suppressing BrettPhos oxidation to maintain the active Pd–phosphine species. The method shows broad amide scope and delivers products in up to 95% yield.
The N-aryl amide scaffold represents a valuable structural motif widely found in biologically active molecules and functional materials.1 Consequently, the development of efficient and versatile methods for synthesizing such compounds continues to attract significant interest. In efforts to expand the synthetic toolbox beyond classical amide-bond–forming protocols,2 approaches involving transition-metal catalysis—particularly Pd-catalyzed C–N cross-coupling reactions—have drawn increasing attention (Scheme 1a). This appeal stems from the ready availability of amides as nucleophiles and the broad range of (hetero)aryl electrophiles available for coupling.3 Despite challenges associated with their inherently low nucleophilicity4 and potential catalytic inhibition through κ2-N,O coordination to the metal center,5 Pd-catalyzed C–N cross-coupling has nevertheless emerged as an efficient and reliable strategy for constructing N-aryl amides.6
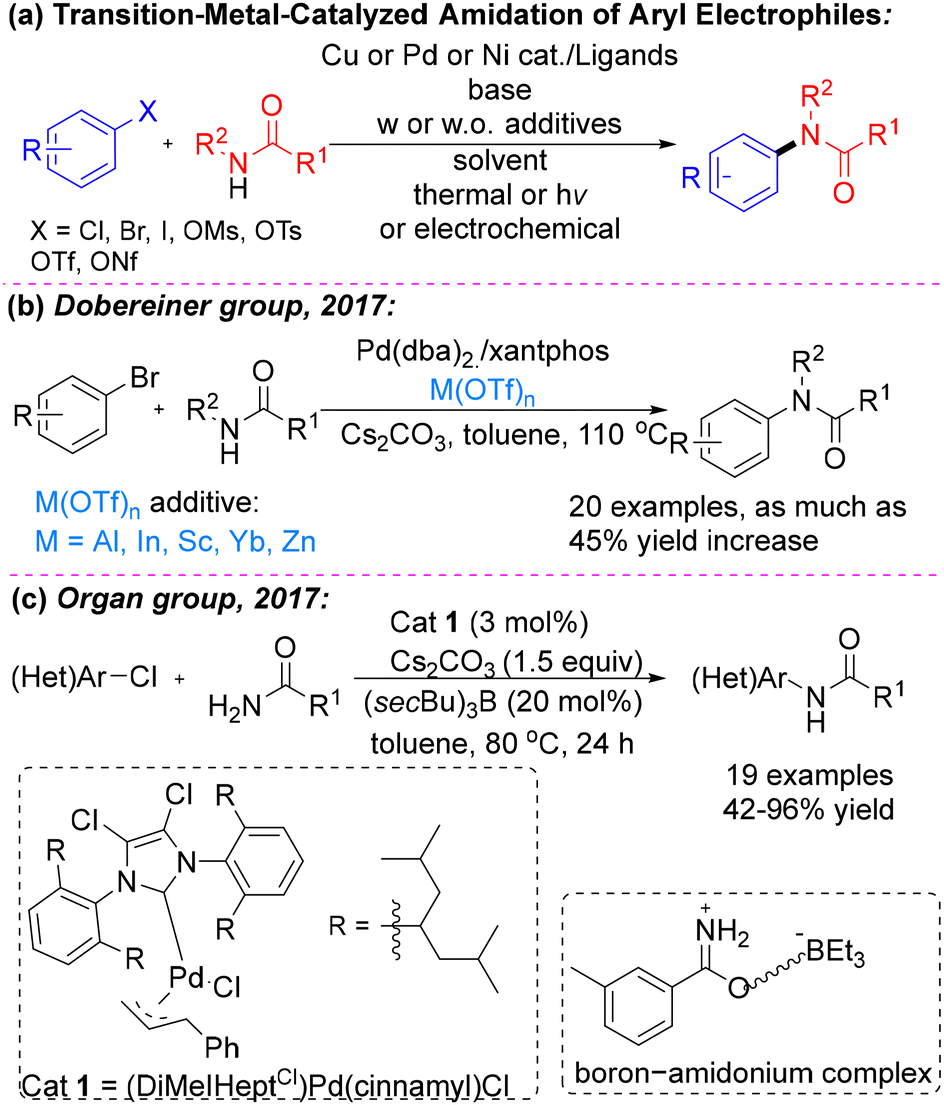 | ||
| Scheme 1 (a) Transition‑metal‑catalyzed amidation of aryl electrophiles, (b) Dobereiner group's work and (c) Organ group's work. | ||
Significant progress in amide N-arylation has been enabled by the development of phosphine7 and N-heterocyclic carbene (NHC) ligands8 with distinctive structural and electronic properties. Although ligand design has played a crucial role in advancing these transformations, persistent reactivity challenges continue to motivate exploration of alternative catalytic strategies and refinement of existing methodologies. One particularly effective approach for accelerating the turnover-limiting transmetalation step in Pd-catalyzed amide coupling involves the use of Lewis acid additives, as independently demonstrated by the research groups of Dobereiner (Scheme 1b)9 and Organ (Scheme 1c)8 in 2017.
In modern synthetic chemistry, achieving high atom economy and step efficiency is central to sustainable methodology design.10 In this context, employing nitroarenes as aryl donors in amide N-arylation offers a conceptually simple yet atom-economical strategy.11 By enabling direct amide bond formation from readily available nitro compounds, such reactions streamline synthetic routes, minimize waste generation, and provide a more environmentally responsible alternative to traditional multistep protocols.
Building on prior studies by our group,12 we hypothesized that relatively inert nitroarenes could serve as electrophilic partners in the Pd-catalyzed C–N coupling of amides. This approach would further broaden the scope of denitrative cross-coupling reactions (Scheme 2a). Guided by literature precedents and preliminary experiments, we speculated that under palladium catalysis, the use of electron-rich and sterically demanding phosphine or NHC ligands could facilitate cleavage of the C(sp2)–NO2 bond in nitroarenes,13 thereby enabling amides to participate as nucleophiles. The proposed mechanism is illustrated in Scheme 2b. To ensure efficient reaction progress, suppression of the formation of the catalytically inactive κ2-amidate complex E was pursued in our current work by carefully optimizing the reaction conditions.
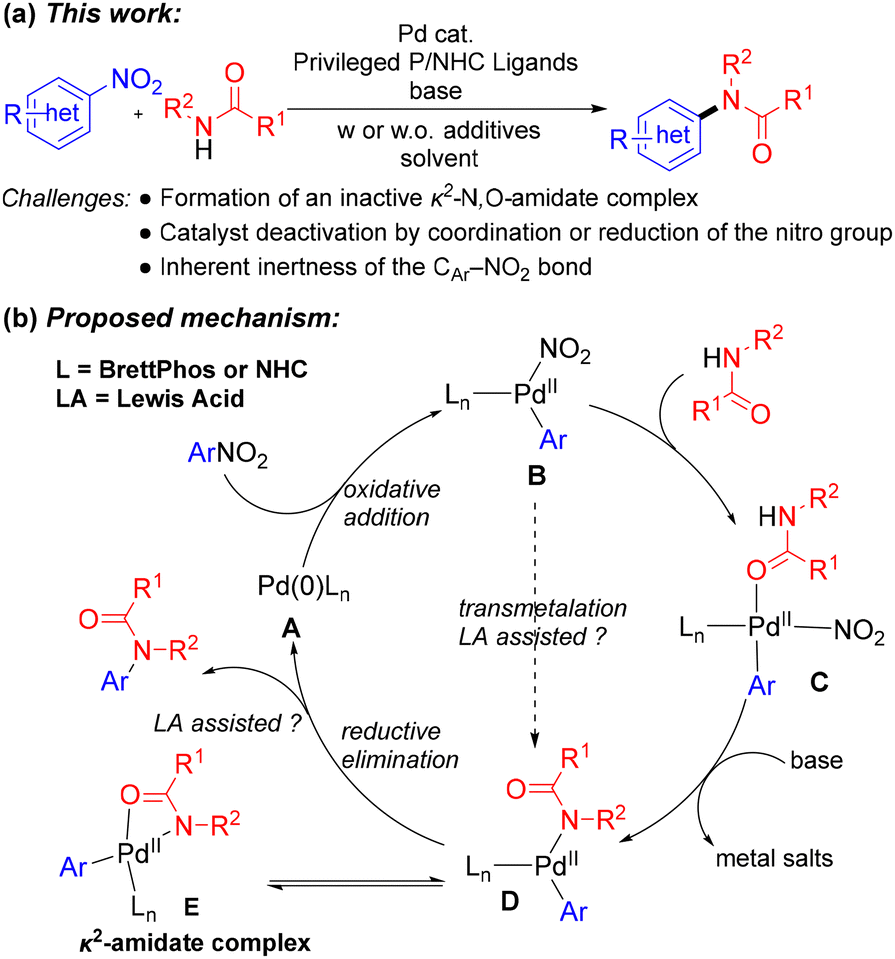 | ||
| Scheme 2 (a) This work and (b) the proposed mechanism. | ||
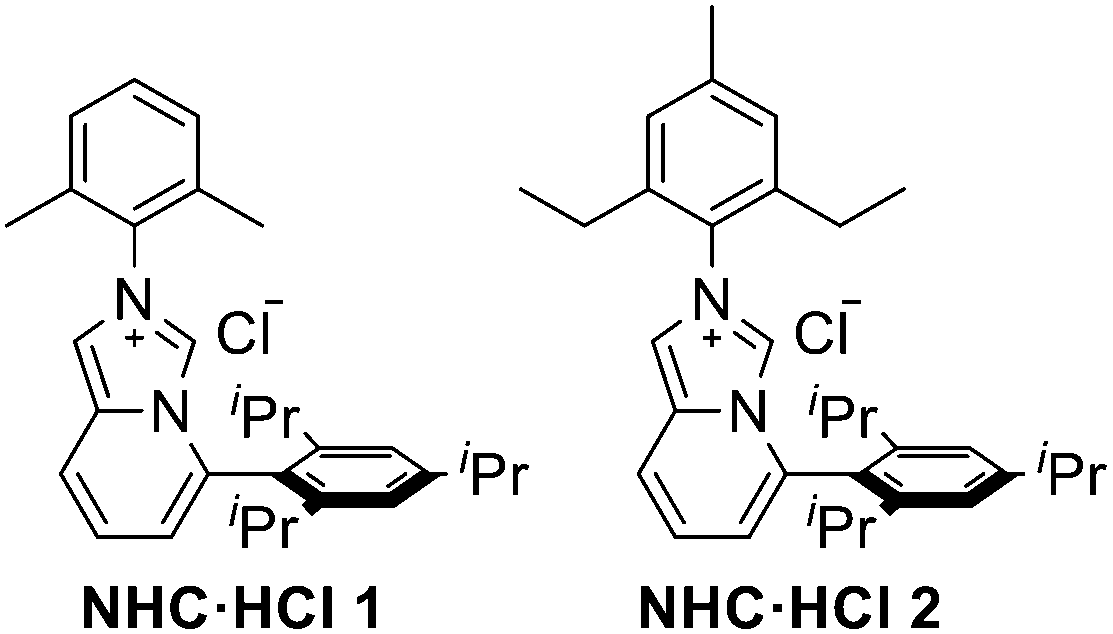
At the outset of this study, p-toluamide (1a) and α-nitronaphthalene (2a) were selected as model substrates to test the reaction parameters. The initial denitrative amidation conducted in toluene (150 °C) with Pd(acac)2 (5 mol%), BrettPhos (10 mol%), and K3PO4 (3.0 equiv.) afforded no detectable product (Table 1, entry 1). Inspired by the work of Dobereiner9 and Stradiotto,14 Al(OTf)3 (10 mol%) was introduced as a Lewis acid additive, and it furnished the coupling product 3a in 37% isolated yield (entry 2). Subsequent optimization revealed K3PO4 to be the optimal base: K2CO3 or Cs2CO3 led to slightly lower yields, while strong bases, namely t-BuOK or DBU, completely suppressed the reaction (entries 3–6). Solvent screening identified PhCF3 as superior to 1,4-dioxane, anisole, and p-xylene (entries 7–10). Ligand evaluation showed that BrettPhos outperformed other biarylphosphines (RuPhos, XPhos, tBuBrettPhos), while imidazo[1,5-a]pyridine-derived NHC ligands (NHC·HCl 1, NHC·HCl 2) also promoted the reaction effectively (entries 11–15). Ultimately, BrettPhos was identified as the optimal ligand. Increasing the loadings of both the catalyst and ligand improved the yield of 3a to 85%, whereas deviations in temperature reduced efficiency (entries 16–18). Under optimized conditions—Pd(acac)2 (10 mol%), BrettPhos (20 mol%), Al(OTf)3 (10 mol%), and K3PO4 (3.0 equiv.) in PhCF3 at 150 °C and a reaction time of 12 h—the coupling product 3a was obtained in 85% isolated yield (entry 16).
|
|
||||
|---|---|---|---|---|
| Entry | Catalyst | Base | Solvent | Yielda (%) |
| Reaction conditions: p-toluamide 1a (0.4 mmol), α-nitronaphthalene 2a (0.6 mmol), Pd(acac)2 (5 mol%), ligand (10 mol%), base (3.0 equiv.), Al(OTf)3 (10 mol%) and solvent (2.0 mL, 0.2 M) at 150 °C under N2 for 12 h.a Isolated yields.b No additive.c Pd(acac)2 (10 mol%), ligand (20 mol%).d 140 °C.e 160 °C. N.D. = not detected. | ||||
| 1b | BrettPhos | K3PO4 | Toluene | N.D. |
| 2 | BrettPhos | K3PO4 | Toluene | 37 |
| 3 | BrettPhos | K2CO3 | Toluene | 20 |
| 4 | BrettPhos | Cs2CO3 | Toluene | 34 |
| 5 | BrettPhos | t-BuOK | Toluene | N.D. |
| 6 | BrettPhos | DBU | Toluene | N.D. |
| 7 | BrettPhos | K3PO4 | Dioxane | 39 |
| 8 | BrettPhos | K3PO4 | Anisole | 32 |
| 9 | BrettPhos | K3PO4 | p-Xylene | 30 |
| 10 | BrettPhos | K3PO4 | PhCF3 | 40 |
| 11 | RuPhos | K3PO4 | PhCF3 | 28 |
| 12 | XPhos | K3PO4 | PhCF3 | 24 |
| 13 | t BuBrettPhos | K3PO4 | PhCF3 | 31 |
| 14 | NHC·HCl 1 | K3PO4 | PhCF3 | 35 |
| 15 | NHC·HCl 2 | K3PO4 | PhCF3 | 36 |
| 16c | BrettPhos | K3PO4 | PhCF3 | 85 |
| 17cd | BrettPhos | K3PO4 | PhCF3 | 82 |
| 18ce | BrettPhos | K3PO4 | PhCF3 | 79 |
|
||||
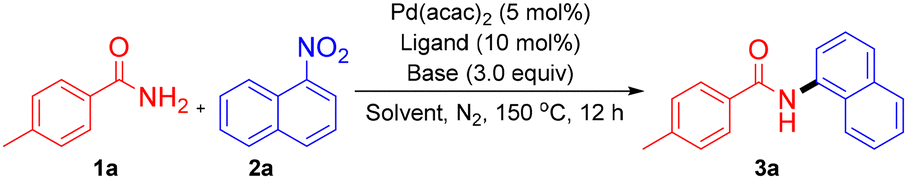
With the optimized conditions in hand, the nitroarene substrate scope was next examined (Scheme 3). Both 1-nitronaphthalene and nitrobenzene underwent smooth coupling to afford 3a and 3b in 85% and 86% yields, respectively. Scaling up the reaction of 2a to 5.0 mmol afforded 3a in 76% yield. The polycyclic aromatic substrate 1-nitropyrene exhibited moderate reactivity, yielding 3r (52%). Alkyl-substituted nitrobenzenes afforded 3d–f in 88–92% yields, irrespective of substitution pattern. Electron-donating substituents, namely methoxy (3c, 3j) and tert-butyl (3p), were well tolerated, as were ester-functionalized substrates (3n). In contrast, nitroarenes bearing strongly electron-withdrawing groups (e.g., cyano) failed to undergo coupling. A substrate containing a base-sensitive functionality, namely benzoyl (3h), gave a significantly reduced yield (15%). Fluorinated substrates, including those bearing trifluoromethoxy (3g), fluoro (3i), and trifluoromethyl (3k) groups, proved compatible. Furthermore, heteroaromatic nitro compounds, namely oxygen-containing heterocycles (3l–m), carbazole (3o), and dibenzothiophene (3q), afforded the corresponding products in 30–91% yields.
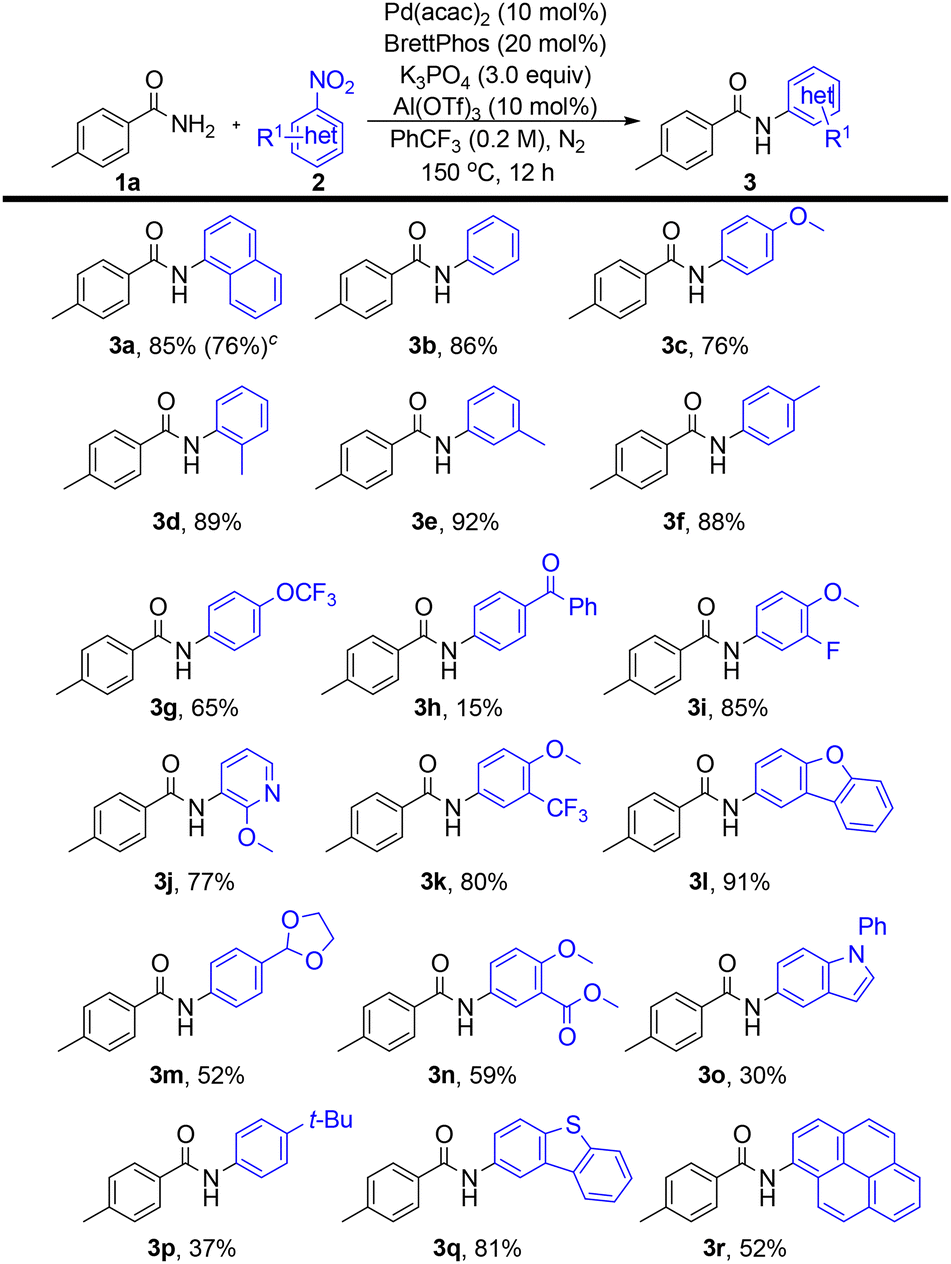 | ||
| Scheme 3 N-arylation of p-toluamide 1a with nitroarenes under palladium catalysis. | ||
Investigation of the amide substrate scope (Scheme 4) revealed similarly broad applicability. Benzamide afforded 4a in 78% yield, while meta- and ortho-methyl benzamides (4b, 4c) were obtained in 88% and 84% yields, respectively, indicating negligible steric effects. The electron-donating groups methoxy (4d) and tert-butyl (4e) were well tolerated; however, the strongly electron-withdrawing substituents cyano and trifluoromethyl completely suppressed the reaction. The heteroaromatic amide furan-2-carboxamide delivered 4f in 77% yield, and the fluorinated amide afforded 4g in 53% yield. Additional aryl amides, including 1-naphthamide (4h) and 2-naphthamide (4i), provided excellent yields (90–93%). Aliphatic amides were also tested. For example, pivalamide (lacking any α-C–H bond) afforded 4j in 95% yield. In contrast, cyclohexanecarboxamide gave 4k in 61% yield—with this efficiency being only moderate likely due to deprotonation at the acidic α–C–H site, leading to competitive deprotonation/enolization and reduced chemoselectivity under the reaction conditions, which can interfere with the coupling process.15 Secondary amides and lactams were not compatible with this protocol. Overall, taking our results together, the protocol provides a new and relatively general approach for accessing N-aryl amide substrates.
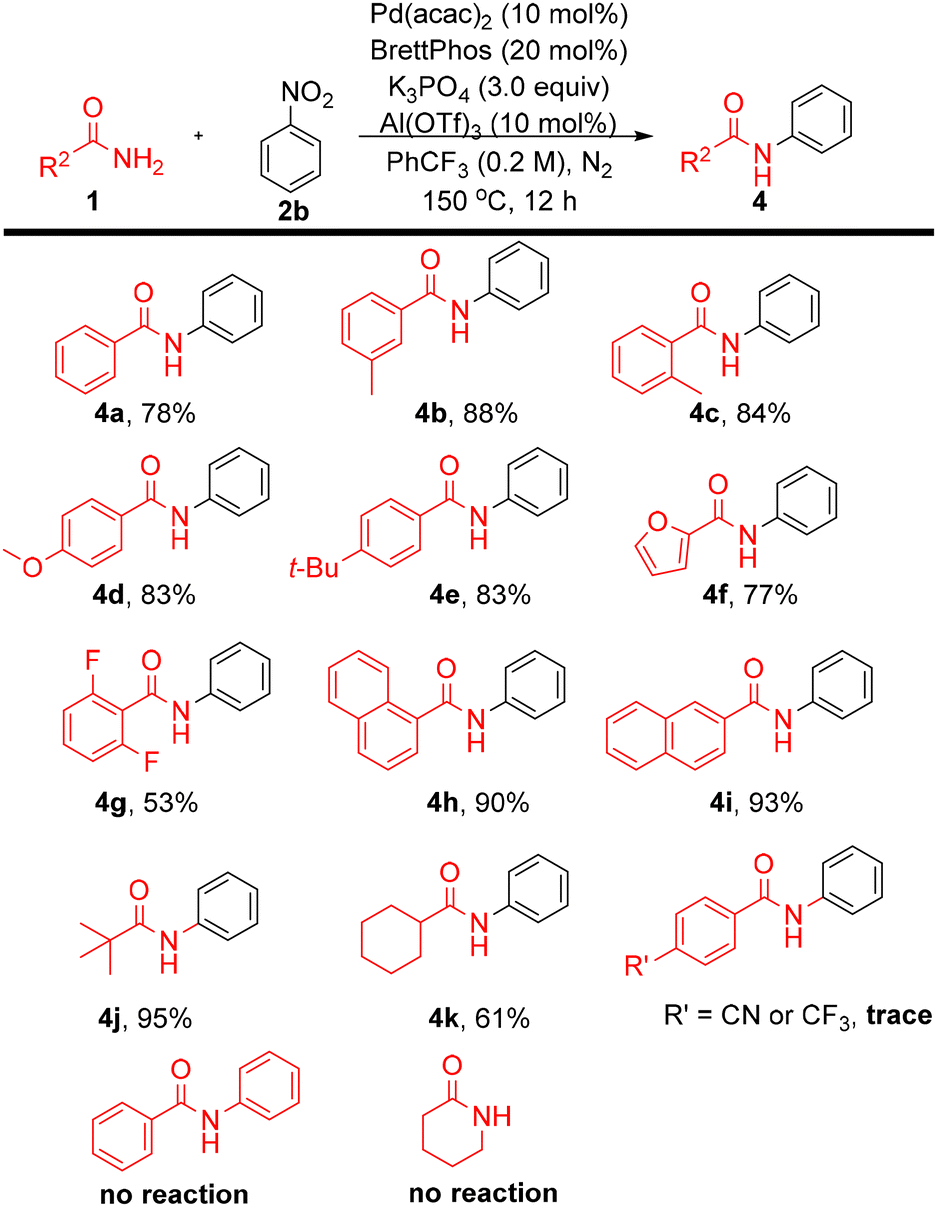 | ||
| Scheme 4 Scope of amide substrates. | ||
We also examined the influence of various Lewis acid additives on the reaction outcome (Scheme 5a). A series of air-stable metal triflates was tested, and the identity of the metal cation significantly affected the reactivity. Notably, the Group 13 metal triflates indium(III) triflate [In(OTf)3] and Al(OTf)3 exhibited the most pronounced promoting effects. Increasing the loading of Al(OTf)3 to 20 mol% did not lead to any appreciable improvement in yield. In contrast, the borane-type Lewis acid B(C6F5)3, previously employed by Organ and co-workers,8 proved ineffective in promoting the transformation in the current work.
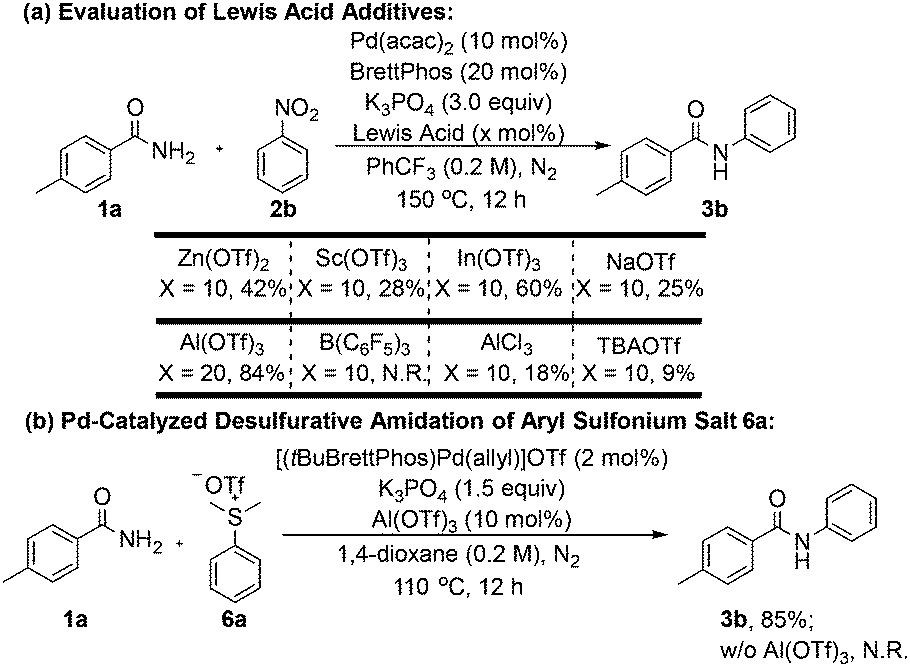 | ||
Scheme 5 Effects of Lewis acid additives. (a) a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 1a (0.4 mmol), 2b (0.6 mmol), Pd(acac)2 (10 mol%), BrettPhos (20 mol%), K3PO4 (3.0 equiv.), Lewis acid (10 mol%) in PhCF3 (2.0 mL, 0.2 M) at 150 °C for 12 h under an N2 atmosphere. bIsolated yields. (b) aReaction conditions: 1a (0.4 mmol), 6a (0.5 mmol), [(tBuBrettPhos)Pd(allyl)]OTf (2 mol%), K3PO4 (1.5 equiv.), Al(OTf)3 (10 mol%) in 1,4-dioxane (2.0 mL, 0.2 M) at 110 °C for 12 h under an N2 atmosphere. bIsolated yields. Reaction conditions: 1a (0.4 mmol), 2b (0.6 mmol), Pd(acac)2 (10 mol%), BrettPhos (20 mol%), K3PO4 (3.0 equiv.), Lewis acid (10 mol%) in PhCF3 (2.0 mL, 0.2 M) at 150 °C for 12 h under an N2 atmosphere. bIsolated yields. (b) aReaction conditions: 1a (0.4 mmol), 6a (0.5 mmol), [(tBuBrettPhos)Pd(allyl)]OTf (2 mol%), K3PO4 (1.5 equiv.), Al(OTf)3 (10 mol%) in 1,4-dioxane (2.0 mL, 0.2 M) at 110 °C for 12 h under an N2 atmosphere. bIsolated yields. | ||
We further investigated the C–N coupling reactions of amide nucleophiles with other electrophilic coupling partners, exemplified by dimethyl(phenyl)sulfonium triflate (6a) (Scheme 5b). In the absence of Al(OTf)3, the desired N-phenyl amide product was not detected; instead, thioanisole (i.e., the demethylated byproduct) was formed as the major product. Upon addition of 10 mol% Al(OTf)3, the desired coupling product 3b was isolated in 85% yield. Moreover, Al(OTf)3 effectively inhibited the oxidation of BrettPhos (Section II.5 of the Supporting Information), thereby maintaining a higher proportion of the active Pd–phosphine complex in the catalytic system and consequently enhancing the overall efficiency of the cross-coupling reaction.
In conclusion, we have developed a Pd-catalyzed denitrative C–N coupling between amide nucleophiles and nitroarenes using a catalytic amount of Lewis acid additives. This transformation employs nitroarenes as electrophilic partners. The Pd–Al cooperative catalytic system effectively inhibits the oxidation of the phosphine ligand. Importantly, the Lewis acid not only functions as an anion abstractor but also likely interacts with the amide substrate to form a catalytically active complex, accelerating the transmetalation step and preventing the formation of the inactive κ2-amidate species. Moreover, metal triflate additives may exert additional beneficial effects; further studies are required to elucidate their mechanistic role in Pd-catalyzed denitrative cross-coupling reactions.
Linjie Yang: conceived and performed the experiments, analyzed the data, and wrote the manuscript. Xuejie Wang: assisted with data analysis. Wanzhi Chen: administered the project, acquired funding, supervised the work, analyzed the data, and wrote the manuscript.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: methods, experimental procedures, product characterization data, and NMR spectra. See DOI: https://doi.org/10.1039/d6cc00697c.Acknowledgements
We thank the National Natural Science Foundation of China (No. 22071214) for financial support.References
- (a) A. L. Simplício, J. M. Clancy and J. F. Gilmer, Molecules, 2008, 13, 519–547 CrossRef PubMed; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471–479 CrossRef CAS PubMed; (c) R. M. de Figueiredo, J. S. Suppo and J. M. Campagne, Chem. Rev., 2016, 116, 12029–12122 CrossRef CAS PubMed; (d) A. R. Abduelkarem, H. S. Anbar, S. O. Zaraei, A. A. Alfar, O. S. Al-Zoubi, E. G. Abdelkarem and M. I. El-Gamal, Eur. J. Med. Chem., 2020, 188, 112029 CrossRef CAS PubMed.
- (a) J. R. Dunetz, J. Magano and G. A. Weisenburger, Org. Process Res. Dev., 2016, 20, 140–177 CrossRef CAS; (b) E. Massolo, M. Pirola and M. Benaglia, Eur. J. Org. Chem., 2020, 4641–4651 CrossRef CAS; (c) P. Acosta-Guzmán, A. Ojeda-Porras and D. Gamba-Sánchez, Adv. Synth. Catal., 2023, 365, 4359–4391 CrossRef.
- (a) C. L. Allen and J. M. J. Williams, Chem. Soc. Rev., 2011, 40, 3405 RSC; (b) S. Roy, S. Roy and G. W. Gribble, Tetrahedron, 2012, 68, 9867–9923 CrossRef CAS.
- (a) H. Sigel and R. B. Martin, Chem. Rev., 1982, 82, 385–426 CrossRef CAS; (b) F. Milletti, L. Storchi, L. Goracci, S. Bendels, B. Wagner, M. Kansy and G. Cruciani, Eur. J. Med. Chem., 2010, 45, 4270–4279 CrossRef CAS PubMed.
- K. I. Fujita, M. Yamashita, F. Puschmann, M. M. Alvarez-Falcon, C. D. Incarvito and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9044–9045 CrossRef CAS PubMed.
- P. Ruiz-Castillo and S. L. Buchwald, Chem. Rev., 2016, 116, 12564–12649 CrossRef CAS PubMed.
- (a) T. Ikawa, T. E. Barder, M. R. Biscoe and S. L. Buchwald, J. Am. Chem. Soc., 2007, 129, 13001–13007 CrossRef CAS PubMed; (b) J. D. Hicks, A. M. Hyde, A. M. Cuezva and S. L. Buchwald, J. Am. Chem. Soc., 2009, 131, 16720–16734 CrossRef CAS PubMed.
- S. Sharif, J. Day, H. N. Hunter, Y. Lu, D. Mitchell, M. J. Rodriguez and M. G. Organ, J. Am. Chem. Soc., 2017, 139, 18436–18439 CrossRef CAS PubMed.
- J. Becica and G. E. Dobereiner, ACS Catal., 2017, 7, 5862–5870 CrossRef CAS.
- P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 RSC.
- (a) K. Muto, T. Okita and J. Yamaguchi, ACS Catal., 2020, 10, 9856–9871 CrossRef CAS; (b) M. Kashihara and Y. Nakao, Acc. Chem. Res., 2021, 54, 2928–2935 CrossRef CAS PubMed; (c) K. Iizumi and J. Yamaguchi, Org. Biomol. Chem., 2025, 23, 1746–1772 RSC.
- (a) K. Chen, W. Chen, X. Yi, W. Chen, M. Liu and H. Wu, Chem. Commun., 2019, 55, 9287–9290 RSC; (b) W. Chen, K. Chen, W. Chen, M. Liu and H. Wu, ACS Catal., 2019, 9, 8110–8115 CrossRef CAS; (c) W. Chen, W. Chen, M. Liu and H. Wu, Org. Lett., 2022, 24, 6983–6987 CrossRef CAS PubMed; (d) W. Chen, K. Chen, X. Wang, L. Yang and W. Chen, RSC Adv., 2024, 14, 16624–16628 RSC; (e) L. Yang and W. Chen, Org. Lett., 2025, 27, 9721–9726 CrossRef CAS PubMed.
- (a) M. R. Yadav, M. Nagaoka, M. Kashihara, R. L. Zhong, T. Miyazaki, S. Sakaki and Y. Nakao, J. Am. Chem. Soc., 2017, 139, 9423–9426 CrossRef CAS PubMed; (b) F. Inoue, M. Kashihara, M. R. Yadav and Y. Nakao, Angew. Chem., Int. Ed., 2017, 56, 13307–13309 CrossRef CAS PubMed; (c) J. Schleinitz, C. Chinchilla-Garzon, A. Perfetto, E. Escoudé, A. Makhloutah, G. Gontard, M. R. Vitale, A. Goujon, F. X. Felpin, L. Binet, I. Ciofini, P. Hudhomme and L. Grimaud, Organometallics, 2024, 44, 29–35 CrossRef.
- M. J. Cotnam, N. Martinek and M. Stradiotto, Eur. J. Org. Chem., 2023, e202301004 CrossRef CAS.
- Z. Li, Y. Peng and T. Wu, Org. Lett., 2021, 23, 881–885 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |