
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tetracationic phosphonium-bridged ladder stilbenes: redox states and electronic properties of highly charged P-heteropolycyclic materials
Sebastian Senna,
Nele Konradb,
Jens Müller b,
Joachim Ballmann†
a and
Lutz H. Gade*a
b,
Joachim Ballmann†
a and
Lutz H. Gade*a
aAnorganisch-Chemisches Institut Universität Heidelberg, Im Neuenheimer Feld 276, D-69120 Heidelberg, Germany. E-mail: lutz.gade@uni-heidelberg.de
bInstitut für Anorganische und Analytische Chemie, Universität Münster, Corrensstr. 28/30, 48149 Münster, Germany. E-mail: mueller.j@uni-muenster.de
First published on 3rd March 2026
Abstract
This work describes the development of an air-stable tetracationic P-heteropolycyclic material in which four phosphorus atoms are incorporated into an extended π-conjugated framework. The system, which may be viewed as a doubly phosphonium-bridged ladder stilbene, was found to display a rich redox chemistry and charge-dependent interaction with DNA.
Redox-active organic molecules with a range of specifically accessible oxidation states are of fundamental interest in the development of new and readily processible functional materials.1–4 In this context, polycyclic π-conjugated frameworks are capable of accommodating multiple charges while maintaining structural integrity and provide access to materials which are relevant for organic electronic and optoelectronic applications.1,5–15
From a practical point of view, the materials employed in such applications ideally require chemical robustness under ambient conditions.2,8,11,16 Air stability and water solubility13,17 are among the critical properties, especially for emerging biological and biomedical applications where oxidative stress and aqueous media impose stringent stability requirements.18,19
Beyond the extensive chemistry of N-heteropolycycles,20–24 the integration of other main-group elements into π-conjugated systems has emerged as a powerful strategy to access unusual electronic configurations and a rich redox chemical behaviour.1,7–9,11,16,25–29 Notable examples include the isolation of neutral, radical, and biradical states in diboraindeno[1,2-b]fluorene derivatives, demonstrating that heteroatom frameworks can stabilize multiple oxidation states within a molecular scaffold.30 Related concepts were introduced earlier by Yoshifuji, who reported the first synthesis and comprehensive characterization of diphosphaquinones and diphosphathienoquinones, compounds that exhibit pronounced redox activity associated with their quinoidal π-systems.31
Despite these advances, highly charged phosphorus-containing π-systems remain rare, and systematic strategies to increase charge density while simultaneously maintaining air stability and aqueous compatibility32 are still limited. In this context, we set out to extend ladder-type structures by increasing the number of cationic phosphorus centres within a single conjugated framework. Whereas Yoshifuji's diphosphaquinones are intrinsically limited to dicationic species, incorporation of four phosphorus atoms would enable access to a tetracationic material with substantially increased charge density.
In previous work,4,32 we investigated π-extended dicationic phosphonium-bridged ladder stilbenes. Notably, these exhibited both water solubility and air stability. Among these, one of the naphtho-annulated derivatives was found to undergo a competing transformation, generating an unprecedented bis(Δ2-phosphetene) dication (Fig. 1).32
 | ||
| Fig. 1 The focus of previous work was the synthesis of π-extended phospholo[3,2-b]phospholes,4,32 while this study presents the extension of the π-system via a bidirectional extension of the phospholo[3,2-b]phosphole motif. Structurally related diboraindeno[1,2-b]fluorene derivatives30 (Griffin and Gilliard, Jr.) and diphosphaquinones31 (Yoshifuji) are shown above. | ||
To construct a bidirectional phospholo[3,2-b]phosphole framework and to suppress the potential formation of a bis(Δ2-phosphetene) structure, a linear substitution pattern was therefore selected for the present study.
The first precursor (1) was synthesized in two steps from 1-bromo-2-iodobenzene (see Scheme 1), while the second precursor (2) was obtained in a five-step synthesis, both using well established synthetic methods (see SI for details). Sonogashira couplings of these precursors with 1,4-dibromo-2,5-diiodobenzene yielded compounds 333 (70%) and 4 (71%). After in situ lithiation with tert-butyllithium and subsequent treatment of the lithiated intermediates with ClP(iPr)2, the tetraphosphine targets 5 and 6 were obtained in high yields (73% and 70%). In contrast to the diphosphines reported in our previous work,32 the tetraphosphines 5 and 6 could be isolated and fully characterized, despite their pronounced sensitivity towards air and their thermal lability. The 31P{1H} NMR spectra display two singlet resonances for each compound (δ = 4.9 ppm, 3.0 ppm for 5; 10.0 ppm, 4.0 ppm for 6, see SI).
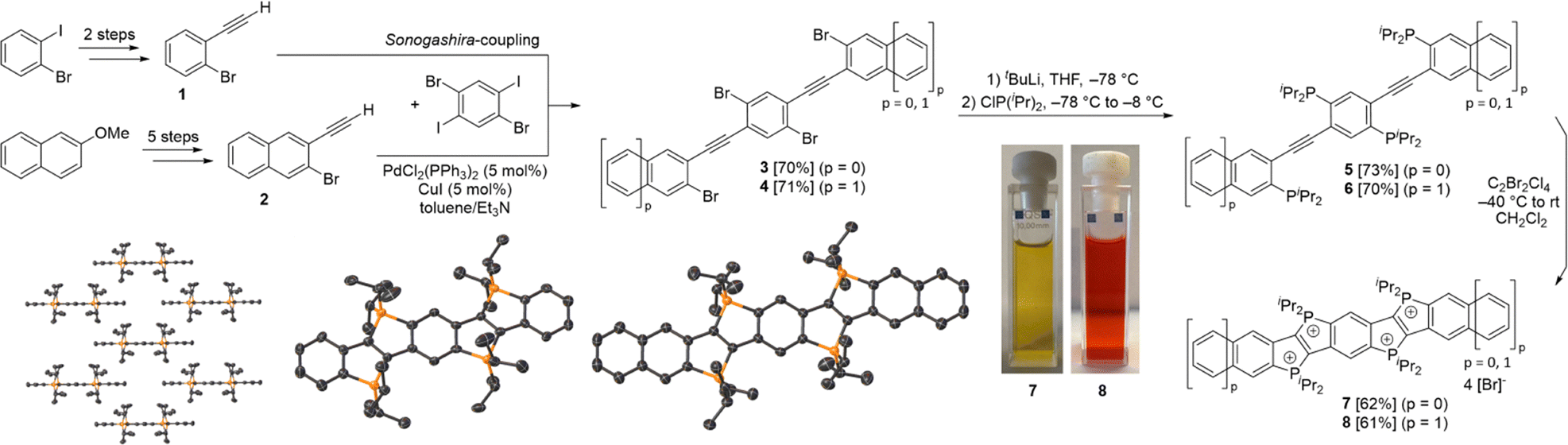 | ||
| Scheme 1 Synthesis of 7 and 8 and the crystallographically determined structures of 7 and 8 (co-crystallized and disordered solvent molecules, bromide counter ions, and hydrogen atoms omitted for clarity, thermal ellipsoids drawn at the 50% probability level). | ||
In the subsequent oxidation and cyclization, 5 and 6 were treated with C2Cl4Br2 in dichloromethane at −40 °C and then warmed to room temperature, which afforded the tetracationic P-heteropolycycles 7 and 8 as bromide salts. These were isolated as bright orange-red powders in moderate yields (62% and 61%) by filtration. Compounds 7 and 8 give yellow and orange solutions, respectively (Scheme 1). The UV/Vis absorption spectra display similar features (see SI). However, the absorption bands of 8 are bathochromically shifted relative to those of 7. This trend is consistent with the increased π-conjugation in 8 and is in agreement with previous observations for dicationic phosphonium-bridged ladder stilbenes.32 The lowest-energy absorption maximum of 8 (530 nm) is red-shifted by 42 nm compared to 7 (488 nm), whereas the higher-energy absorption bands show smaller shifts of approximately 10 nm (351 and 272 nm for 7, compared to 360 and 283 nm for 8).
The oxidized products exhibit two doublets in the 31P{1H} NMR spectra (δ = 63.0 ppm, 60.7 ppm for 7; 62.8 ppm, 58.2 ppm for 8). Both compounds were found to be air-stable for several weeks and could be purified by recrystallization from MeOH/Et2O. Their crystal structures established almost planar polycyclic π-systems with the iso-propyl groups pointing above and below the plane, thereby shielding the π-system laterally.
The redox behaviour of 7 and 8 was investigated by cyclic voltammetry. Three quasi-reversible and one irreversible reduction waves were observed for 7 (Fig. 2). Initiating the scan at 0 V vs. Fc/Fc+ and observing no current at the starting potential allows the redox wave at E1/2 = −0.39 V to be assigned to a reduction process (see SI for further details). The irreversible reduction wave at E = −2.02 V is most likely due to ring-opening. In contrast, for compound 8 a series of weak and irreversible redox features were found, suggesting decomposition (see SI for spectrum and further details). This observation is consistent with the reduced stability of 8 compared to 7, as observed in ctDNA interaction experiments.
 | ||
| Fig. 2 Cyclic voltammogram of 7 in MeCN, supp. electrolyte Bu4NOTf (0.1 M), reference Fc/Fc+ (black vertical line and black arrow indicate the start and direction of scans). | ||
Based on these findings, the isolation of reduced states of compound 7 was targeted. The dication 9 could be obtained by two synthetic routes: (i) reduction of the tetracation 7, and (ii) oxidation of semicyclised neutral P-diylide precursor 10 (Scheme 2). The latter was obtained by heating 5 in solution for 5.5 hours, whereas the corresponding doubly cyclized P-diylide was not detected even for higher temperatures, longer reaction times, or alternative solvents, all of which resulted in decomposition of the starting material. Oxidation of 10 with CDCl3 afforded the air-sensitive dication 9, while reductive conversion of tetracation 7 was achieved by treatment with CoCp2 in acetonitrile at room temperature. As to be expected for a quinoidal structural unit, the bond lengths in the central ring (dC9–C10 = 1.401(3) Å; dC9–C11 = 1.452(3) Å; dC10–C11 = 1.381(3) Å) deviate from the typical C–C bond length in benzene (1.397 Å).34 In contrast, the bond lengths of the tetracation 7 (dC9–C10 = 1.387(9) Å; dC9–C11 = 1.389(8) Å; dC10–C11 = 1.418(10) Å) are more evenly distributed and thus more consistent with the assigned benzene-like electronic structure. Additionally, a deviation from a typical P–C single-bond length (ca. 1.85 Å)35 is observed for dication 9 (dP2–C11 = 1.793(2) Å and dP2–C7 = 1.758(2) Å). In contrast, in tetracation 7 the P2–C7 bond (1.817(7) Å) is notably longer than the P2–C11 bond (1.792(2) Å).
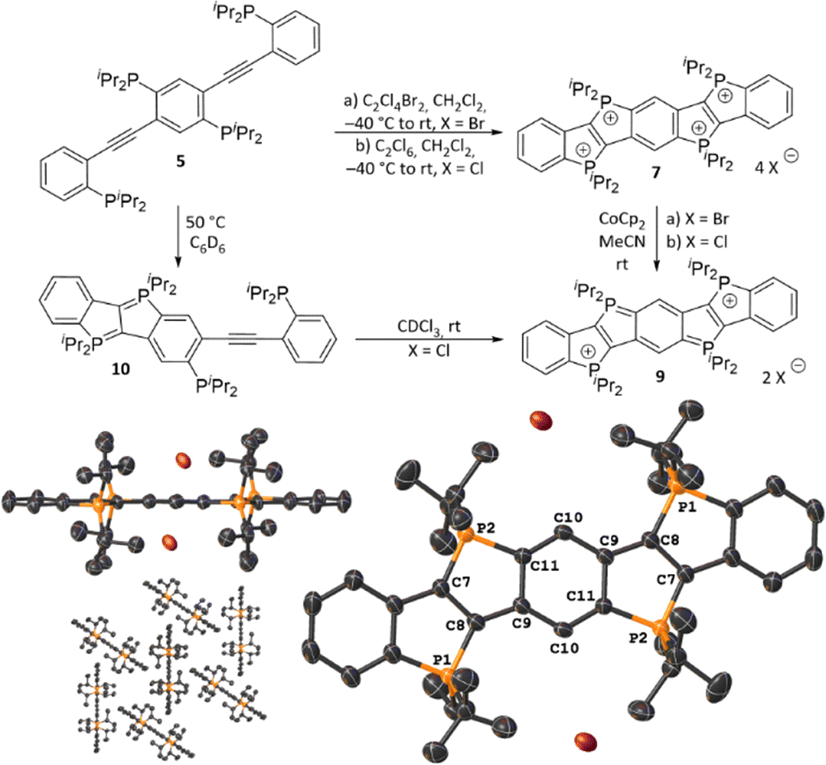 | ||
| Scheme 2 Synthesis of 9 via two different routes: oxidation of 10, or reduction of 7. The crystallographically determined molecular structure of 9 indicates decreased aromaticity in the central benzene unit (co-crystallized solvent molecules and hydrogen atoms omitted for clarity, thermal ellipsoids drawn at the 50% probability level). | ||
NICS(1)zz-XY scans (nucleus-independent chemical shifts)36,37 of 7 and 10 (Fig. 3) support this observation and, in particular, suggest that the dicationic phospholo[3,2-b]phosphole core (b, b*, c and c* rings) of 7 is electronically decoupled from the annulated arene rings on the sides. This is consistent with the findings of our previous studies of π-extended dicationic phosphonium-bridged ladder stilbenes32 which also showed that the small positive NICS(1)zz values for the 5-membered rings are not indicative of antiaromaticity38 but similar to those of a constrained trans-stilbene.32
 | ||
| Fig. 3 Top: NICS(1)zz-XY36,37 scans for 7 and 9 in comparison (B3LYP, def2-TZVPP, D3BJ, CPCM for CH2Cl2),41–49 starting in the centre of (d). Bottom: Anisotropy of the induced current density39,40 (ACID) plots for 7 (top) and 9 (bottom) in comparison (B3LYP, def2-TZVPP, GD3BJ, SCRF for CH2Cl2, isovalue = 0.05 (default), see SI for further details).41–48 | ||
Anisotropy of the induced current density39,40 (ACID) plots were computed to visualize the magnetic response of the π-system (Fig. 3). For compound 7, the current-density vectors within the central ring are oriented clockwise, thereby indicating a diatropic ring current (red circular arrows). In contrast, compound 10 exhibits current-density vectors of the central ring directed towards the observer (blue circles), consistent with a substantially reduced diatropic contribution. The calculated attenuation of the diatropic ring current correlates well with the NICS(1)zz values and the crystallographically determined bond-length pattern.
With the aim of generating the radical mono- and trications related to 7 and 9, the precursor 5 was reacted with, respectively, one and three molar equivalents of ferrocenyl chloride. While attempts to isolate these radical cations were unsuccessful, the formation of the radical trication was confirmed in situ by EPR spectroscopy (Fig. 4 and SI for further details).
 | ||
| Fig. 4 Experimental and simulated EPR spectra obtained after reaction of 5 with 3 eq. [FeCp2]Cl or 3 eq. Ph3CCl, in CH2Cl2 at rt. | ||
Charged heteropolycyclic compounds have found applications as DNA-intercalators,50–52 and it was thus of interest to probe the interaction of the previously published dicationic phosphonium-bridged ladder stilbenes as well as the newly obtained tetracations with DNA. The two dicationic reference compounds represented in Fig. 5 provided relevant benchmarks in this context.4,32 Linear dichroism53 (LD) spectroscopy was performed to evaluate their non-covalent interaction with DNA. Whereas compound [ref. A]2+ showed no evidence of intercalation, probably due to insufficient π-extension, compound [ref. B]2+ exhibited clear spectroscopic signatures of DNA intercalation while maintaining good solution stability, demonstrating that effective DNA binding requires a balanced combination of π-extension, charge state, and aqueous stability (see SI for spectra and details).
 | ||
| Fig. 5 Previously published reference systems for benchmark of ctDNA intercalation with tetracations.4,32 | ||
Subsequently, the tetracationic ladder stilbene 7 was tested. Measurements showed no negative LD signal at the absorption wavelength of the compound, indicating the absence of DNA intercalation,53 in good agreement with [ref. A]2+ with the same extent of π-extension. The correlated decrease of the ctDNA LD band at ca. 260 nm suggests DNA coiling induced by electrostatic interactions rather than π–π intercalation.54 Compound 8 intercalates into DNA when mixed with ctDNA immediately after dissolution in pure water but its rapid decomposition in aqueous media in the absence of DNA precludes any reasonable application as intercalator. Overall, these preliminary results show that, in contrast to the dicationic reference [ref. B]2+, the tetracationic ladder stilbenes are not suitable as DNA intercalators. Insufficient π-extension, limited aqueous stability, and possibly excessive charge density appear to prevent sustained DNA binding.
In summary, the targeted synthesis of the tetracationic ladder stilbenes reported herein establishes a versatile molecular platform for π-extended, redox-active phosphorus-containing systems with exceptionally high charge density. Ongoing efforts are directed toward further structural modification to enhance redox stability and environmental compatibility, with potential applications in redox catalysis6,55 particularly as redox mediators.12,56–58
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details, NMR spectra, computational data, details on ctDNA-intercalation measurements, electrochemical measurements, crystallographic data. See DOI: https://doi.org/10.1039/d6cc00544f.CCDC 2525134–2525136 contain the supplementary crystallographic data for this paper.59a–c
Acknowledgements
The authors acknowledge support by the state of Baden-Württemberg through bwHPC and the DFG through grants INST 40/575-1 FUGG (JUSTUS 2 cluster) and 417637295. S. S. thanks the Landesgraduiertenförderung (LGF) for a PhD fellowship.References
- N. Asok, J. R. Gaffen and T. Baumgartner, Acc. Chem. Res., 2023, 56, 536–547 CrossRef CAS PubMed.
- H. Seo, MRS Commun., 2023, 13, 994–1008 Search PubMed.
- P. Wang, Angew. Chem., Int. Ed., 2025, 64, e202515639 Search PubMed.
- P. Federmann, H. K. Wagner, P. W. Antoni, J.-M. Mörsdorf, J. L. Pérez Lustres, H. Wadepohl, M. Motzkus and J. Ballmann, Org. Lett., 2019, 21, 2033–2038 Search PubMed.
- M. H. Wong, Q. An, J. Kress, J.-M. Mörsdorf, J. Ballmann and Y. Vaynzof, Appl. Phys. Lett., 2021, 119, 233903 CrossRef CAS.
- T. Baumgartner and F. Jäkle, Main group strategies towards functional hybrid materials, John Wiley & Sons, 2018 Search PubMed.
- T. Baumgartner, Acc. Chem. Res., 2014, 47, 1613–1622 Search PubMed.
- D. Joly, P. A. Bouit and M. Hissler, J. Mater. Chem. C, 2016, 4, 3686–3698 RSC.
- M. A. Shameem and A. Orthaber, Chem. – Eur. J., 2016, 22, 10718–10735 CrossRef CAS PubMed.
- Y. Xu, W. Chen, R. Pu, J. Ding, Q. An, Y. Yang, W. Liu and Z. Zuo, Nat. Commun., 2024, 15, 9366 CrossRef CAS PubMed.
- D. S. Thakur, Sushmita, S. A. Meena and A. K. Verma, Chem. Rec., 2024, 24, e202400058 CrossRef CAS PubMed.
- J. Kim, J. Ling, Y. Lai and P. J. Milner, ACS Mater. Au, 2024, 4, 258–273 CrossRef CAS PubMed.
- M.-S. Bertrams, K. Hermainski, J.-M. Mörsdorf, J. Ballmann and C. Kerzig, Chem. Sci., 2023, 14, 8583–8591 RSC.
- T. Baumgartner and R. Réau, Chem. Rev., 2006, 106, 4681–4727 CrossRef CAS PubMed.
- F. Jäkle, Chem. Rev., 2010, 110, 3985–4022 CrossRef.
- J. Ding, S. Luo, Y. Xu, Q. An, Y. Yang and Z. Zuo, Chem. Commun., 2023, 59, 4055–4058 RSC.
- T. A. Welsh and E. R. Draper, RSC Adv., 2021, 11, 5245–5264 Search PubMed.
- K. M. Dean and A. E. Palmer, Nat. Chem. Biol., 2014, 10, 512–523 CrossRef CAS.
- D. Zhang, Z. Chen, Z. Du, B. Bao, N. Su, X. Chen, Y. Ge, Q. Lin, L. Yang, Y. Hua, S. Wang, X. Hua, F. Zuo, N. Li, R. Liu, L. Jiang, C. Bao, Y. Zhao, J. Loscalzo, Y. Yang and L. Zhu, Cell Discovery, 2023, 9, 56 Search PubMed.
- O. Hinsberg, Justus Liebigs Ann. Chem., 1901, 319, 257–286 Search PubMed.
- O. Fischer and E. Hepp, Chem. Ber., 1890, 23, 2789–2793 CrossRef.
- M. Märken, B. D. Lindner, A. L. Appleton, F. Rominger and U. H. F. Bunz, Pure Appl. Chem., 2014, 86, 483–488 Search PubMed.
- O. Tverskoy, F. Rominger, A. Peters, H.-J. Himmel and U. H. F. Bunz, Angew. Chem., Int. Ed., 2011, 50, 3557–3560 CrossRef CAS PubMed.
- A. L. Appleton, S. M. Brombosz, S. Barlow, J. S. Sears, J.-L. Bredas, S. R. Marder and U. H. F. Bunz, Nat. Commun., 2010, 1, 91 CrossRef PubMed.
- K. Zhang, X. Wang, Z. Zhou, J. Guo, H. Liu, Y. Zhai, Y. Yao, K. Yang and Z. Zeng, Angew. Chem., Int. Ed., 2025, 64, e202418520 CrossRef CAS.
- X. Zhang, Y. Jiang, Q. Ma, S. Hu and S. Liao, J. Am. Chem. Soc., 2021, 143, 6357–6362 CrossRef CAS PubMed.
- Z. Yang, J. Chen and S. Liao, ACS Macro Lett., 2022, 11, 1073–1078 CrossRef CAS PubMed.
- Y. Ren and T. Baumgartner, Dalton Trans., 2012, 41, 7792–7800 RSC.
- A. Fukazawa, H. Yamada and S. Yamaguchi, Angew. Chem., Int. Ed., 2008, 47, 5582–5585 CrossRef CAS PubMed.
- B. Y. E. Tra, Y. Zhao, A. Molino, Y. Ouyang, C.-L. Deng, C. McAloon, N. D. McMillion, H. Kim, C. Zhang, P. Müller, R. G. Griffin and R. J. Gilliard Jr., J. Am. Chem. Soc., 2025, 147, 18431–18437 CrossRef CAS.
- F. Murakami, S. Sasaki and M. Yoshifuji, Angew. Chem., Int. Ed., 2002, 41, 2574–2576 CrossRef CAS.
- S. Senn, J.-M. Mörsdorf, M.-S. Bertrams, C. Kerzig and J. Ballmann, Chem. Sci., 2025, 16, 20517–20526 RSC.
- S. Yamaguchi, C. Xu and K. Tamao, J. Am. Chem. Soc., 2003, 125, 13662–13663 CrossRef CAS PubMed.
- L. Pauling, The nature of the chemical bond and the structure of molecules and crystals: an introduction to modern structural chemistry, Cornell University Press, Ithaca, 1960 Search PubMed.
- CRC Handbook of Chemistry and Physics, ed. D. R. Lide, CRC Press, Boca Raton, FL, 84th edn, 2003 Search PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v R. Schleyer, Chem. Rev., 2005, 105, 3842–3888 CrossRef CAS PubMed.
- R. Gershoni-Poranne and A. Stanger, Chem. – Eur. J., 2014, 20, 5673–5688 CrossRef CAS PubMed.
- J. Poater, M. Solà, R. G. Viglione and R. Zanasi, J. Org. Chem., 2004, 69, 7537–7542 CrossRef CAS PubMed.
- D. Geuenich, K. Hess, F. Köhler and R. Herges, Chem. Rev., 2005, 105, 3758–3772 Search PubMed.
- R. Herges and D. Geuenich, J. Phys. Chem. A, 2001, 105, 3214–3220 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3094 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378–6396 CrossRef CAS PubMed.
- M. Garcia-Ratés and F. Neese, J. Comput. Chem., 2020, 41, 922–939 CrossRef.
- V. Sharma, M. Gupta, P. Kumar and A. Sharma, Curr. Pharm. Des., 2021, 27, 15–42 CAS.
- M. K. Goftar, N. M. Kor and Z. M. Kor, Int. J. Adv. Biol. Biomed. Res., 2014, 2, 812–822 Search PubMed.
- K. Mišković, M. Bujak, M. Baus Lončar and L. Glavaš-Obrovac, Arh. Hig. Rada Toksikol., 2013, 64, 593–602 Search PubMed.
- A. Rodger, Sci. Prog., 2008, 91, 377–396 Search PubMed.
- J. Widom and R. L. Baldwin, J. Mol. Biol., 1980, 144, 431–453 Search PubMed.
- C. Xie, A. J. Smaligo, X.-R. Song and O. Kwon, ACS Cent. Sci., 2021, 7, 536–558 Search PubMed.
- E. Steckhan, Angew. Chem., Int. Ed. Engl., 1986, 25, 683–701 Search PubMed.
- Y. Lai, A. Halder, J. Kim, T. J. Hicks and P. J. Milner, Angew. Chem., Int. Ed., 2023, 62, e202310246 Search PubMed.
- R. Francke and R. D. Little, Chem. Soc. Rev., 2014, 43, 2492–2521 RSC.
- (a) CCDC 2525134: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qrly1; (b) CCDC 2525135: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qrlz2; (c) CCDC 2525136: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qrm04.
Footnote |
| † J. B. deceased on June 7, 2025. |
| This journal is © The Royal Society of Chemistry 2026 |