
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Local environment neighbor sensitivity analysis: visualization of cation effect at liquid–solid interface
Ryosuke Jinnouchi * and
Masao Suzuki Shibata
* and
Masao Suzuki Shibata
Toyota Central R&D Labs., Inc, Nagakute 480-1192, Aichi, Japan. E-mail: jryosuke@mosk.tytlabs.co.jp
First published on 8th April 2026
Abstract
We present a method to extract the effects of environmental neighbors on local energies in heterogeneous systems from machine-learning force fields and demonstrate its ability to visualize cation-induced stabilization of hydroxyl groups on Pt in water.
In recent years, machine-learning force fields (MLFFs) have attracted significant attention as promising models for describing interatomic interactions, offering an alternative to conventional first-principles (FP) calculations1,2 and classical force fields.3,4 MLFFs are supervised machine-learning models trained on FP data for potential energies, atomic forces, and stress tensors to predict interatomic interactions.5–15 Unlike classical force fields, which rely on fixed functional forms based on physical laws such as Coulomb and dispersion interactions, MLFFs employ flexible functions developed in the machine-learning (ML) community to represent interatomic interactions. As a result, MLFFs can describe a wide range of interactions—including ionic, dispersive, metallic, covalent, and hydrogen bonding—within a single framework, achieving near FP accuracy while being several orders of magnitude faster than FP calculations. Owing to these properties, MLFFs enable quantitative predictions for complex multi-element organic and inorganic materials that are difficult to treat accurately with classical force fields and computationally prohibitive for FP methods. Their applications have therefore expanded to phenomena such as phase transitions, thermal and ionic transports, chemical reactions and catalytic processes in gases, liquids, solids, and their interfaces.5–12,14,15
The development of MLFFs has progressed rapidly, and a wide variety of functional forms has been proposed. Nevertheless, most MLFFs represent interatomic interactions based on two common concepts: descriptors of many-body interactions and nonlinear mapping. Because the potential energy E of a material originates from many-body interactions, it can be expressed using the following cluster expansion:16–19
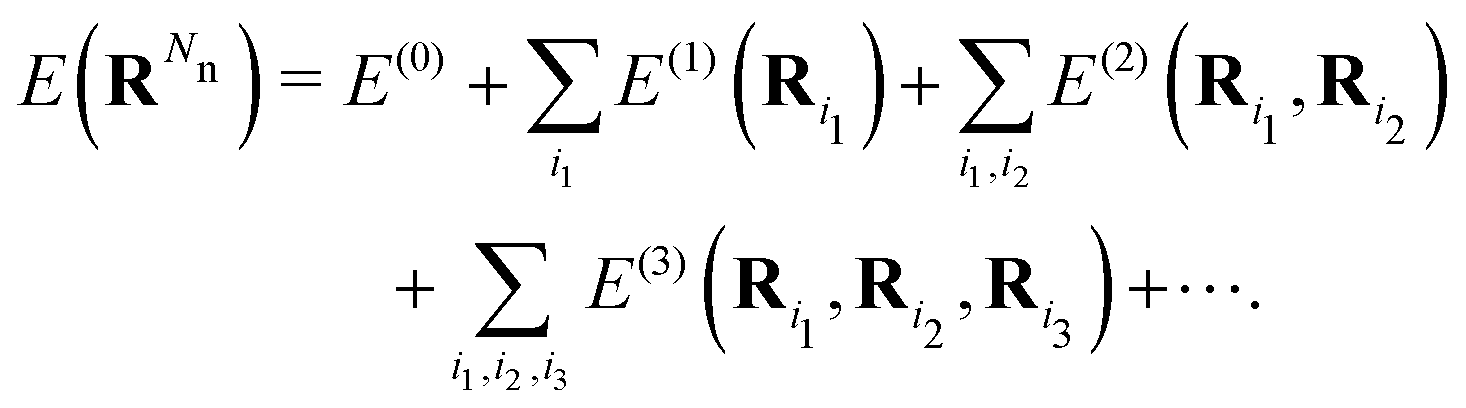 | (1) |
Here, Ri denotes the position vector of the i-th atom, and RN represents the set of position vectors of all N atoms in the system. E(ν) denotes a ν-body interaction, which is defined as a term whose (ν + 1)-th derivative with respect to atomic positions vanishes, as ∂ν+1E(ν)/∂Ri1⋯∂Riν+1 = 0.20 In MLFFs, descriptors representing ν-body local structures are introduced, and the potential energy E is expressed as a function of these descriptors. As the order ν of the descriptors is systematically increased, E can be represented more accurately. However, the number of descriptors grows exponentially with ν, leading to a corresponding increase in computational cost. To address this issue, many MLFFs restrict the descriptor order to ν ≈ 3 or 4 and instead represent many-body effects through higher-order terms generated by nonlinear functions of these descriptors. This strategy for efficiently incorporating high-order many-body interactions is known as the “density trick”.11 Neural networks and kernel functions are commonly used as the nonlinear functions.
In this way, MLFFs can predict interatomic interactions with high accuracy and efficiency. However, a major challenge for MLFFs lies in the difficulty of physically interpreting their predictions. In classical force fields, where interactions are predefined and decomposed into contributions such as Coulomb and dispersion interactions, predicted energies can be analyzed in terms of their underlying interaction components. Similarly, FP calculations provide deep physical insight through quantities such as electron densities, wave functions, and Hamiltonian matrices. In contrast, such analyses are not directly available in MLFFs.
Here, we present a method within the MLFF framework to visualize the surrounding species and their positions that influence the energy of a specific site in a system, and apply it to interfacial electrochemical reactions. As a case study, we focus on the effects of alkali cations on OH adsorbed on a Pt(111) surface in aqueous solution. Experiments have shown that small alkali cations, such as Li+, stabilize OH and slow the oxygen reduction reaction (ORR), whose elementary steps include OH reduction.22 This was the first observation demonstrating cation effects in interfacial electrocatalytic reactions and subsequently highlighted the importance of cation effects in reactions such as hydrogen evolution23,24 and CO2 reduction.24–26 However, assessing the stability of hydrated cations fluctuating in water at finite temperature remains challenging. This difficulty arises because interfacial solvent molecules reorganize on nanosecond time scales,27 which are difficult to access using conventional FP methods22,28,29 that rely on structural optimization at absolute zero or MD simulations limited to picosecond time scales. As a result, many aspects of cation effects on surface adsorbates remain unclear. In this work, we compute the effects of Li+, K+, and Cs+ on the redox potential of OH reduction using a recently developed MLFF-aided FP thermodynamic integration (TI) approach based on finite-temperature MD simulations. We show that smaller cations stabilize OH and lower the redox potential in silico. We then demonstrate, through conventional analyses of cation distributions and interfacial diffusion coefficients, that smaller cations are trapped near OH and exhibit reduced diffusivity. Finally, by applying the visualization method, we obtain further insight into the relationship between the localization of cation distributions and the stabilization of OH, suggesting that attractive interactions between alkali cations and OH play an important role.
The visualization method is constructed on the basis of a kernel-based MLFF approach30–32 implemented in the Vienna Ab initio Simulation Package (VASP).33,34 Similar to the Gaussian Approximation Potential (GAP) proposed by Bartók and co-workers,17,35 this method expresses the total potential energy as a sum of atomic energies:
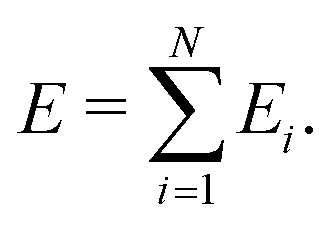 | (2) |
The atomic energy Ei is represented as a functional of the atom density distribution ρi surrounding atom i as Ei = F[ρi(r)]. In the present study, the density ρi is represented by the three-body angular distribution function ρi(3)(r,s,θ),17,32 which gives the probability of finding one atom at a distance between r and r + dr from atom i, and another atom at a distance between s and s + ds from atom i, such that the angle ∠kij lies between θ and θ + dθ. This probability is expressed as ρi(3)(r,s,θ)r2s2sinθdrdsdθ. Accordingly, r denotes the rotationally invariant coordinate (r,s,θ) [see details in Section S1 of SI]. In practice, ρi is expanded in an orthonormal basis set {ϕj|j= 1,…,ND} as
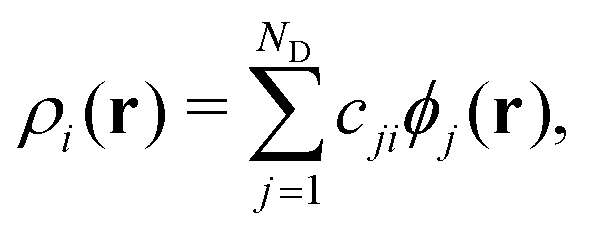 | (3) |
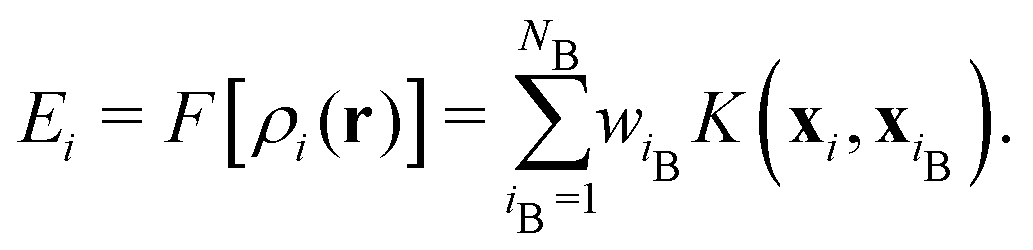 | (4) |
The reference atoms are selected from structures providing the training data, and the regression coefficients wiB are determined to optimally reproduce the training data of energies, forces and stress tensor components. For the kernel basis functions, we use the smooth overlap of atomic positions (SOAP).17
In this MLFF, the contribution of each atom to the total potential energy E can be readily obtained as Ei. However, since Ei is expressed as a linear combination of nonlinear kernel basis functions of the high-dimensional descriptor xi, it is not immediately obvious which aspects of the local environment influence Ei. Nevertheless, by exploiting the fact that Ei is a functional of the local density distribution ρi, and that the descriptors are expansion coefficients of ρi in an orthonormal basis, as described in eqn (3), the influence of the local environment can be quantified through the variation of Ei with respect to ρi, which can be derived as the simple expression given below (see its derivation in Section S2 in SI):
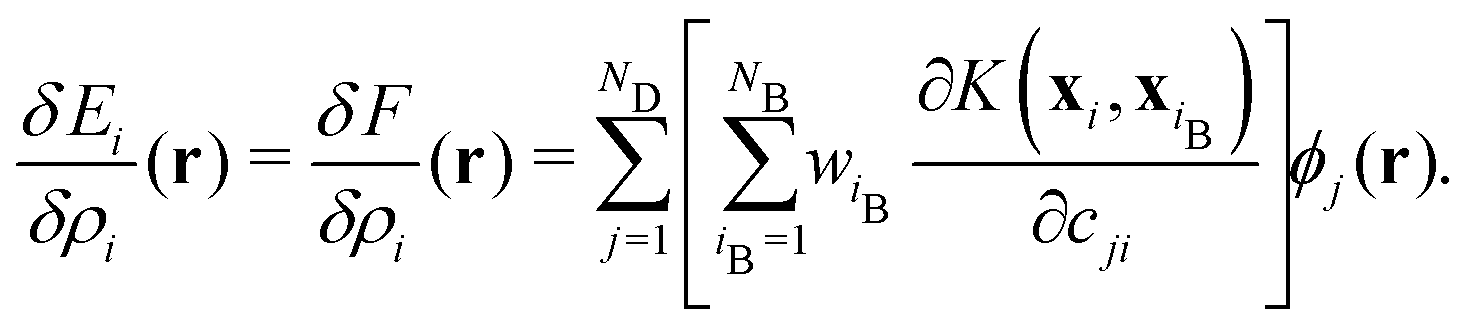 | (5) |
The functional derivative δEi/δρi(r) characterizes the local response of the energy Ei to an infinitesimal change in ρi at position r relative to atom i. With this functional, the local energy is explicitly decomposed to identify which species at which positions most strongly influence it. Notably, the exact variation in eqn (5) can be derived owing to the simple form of the local energy in eqn (4) and the expansion in a complete orthonormal basis set in eqn (3). Although eqn (5) is formulated for the kernel-based representation, a similar exact expression remains applicable to the atomic cluster expansion (ACE)19 and neural-network representation5,9,36 of F when many-body descriptors generated from orthonormal basis sets are used as inputs.11,12,15 We refer to this approach as local environment neighbor sensitivity (LENS) analysis. Details of the computational protocol are provided in Section S3 in SI.
We applied this analysis to cation effects on a hydroxyl group (OH) adsorbed on a Pt(111) surface in water. Prior to the LENS analysis, we computed the redox potential UOH for the OH reduction reaction, OH* + H+ + e− → * + H2O, where * denotes a surface site. The calculation was performed using an MLFF-aided FP-TI scheme based on finite-temperature MD calculations. Details of this method are described in Section S4 of the SI and in ref. 15,37–40. Here, we briefly outline the approach. This approach yields the reaction free energy ΔA by integrating the derivative of Hamiltonian 〈∂H/∂λ〉λ along a coupling path λ from the reactant state at λ = 0 to the product state at λ = 1 using MLFFs. Owing to its high computational efficiency, the MLFF approach enables statistical sampling over timescales of tens of nanoseconds, which is necessary to achieve satisfactory convergence of the reaction free energy.38 However, MLFFs deviate from FP potential energy surfaces (PESs) due to incomplete descriptions of short-range interactions and the neglect of long-range interactions. These deficiencies can be systematically corrected by performing an additional TI from the MLFF to the FP-PES. Although this second TI step requires computationally demanding FPMD calculations, the energetic differences between the MLFF and FP descriptions are small (see Section S6 in the SI). As a result, the free energy difference between the two methods converges within a few picoseconds. To make the FPMD calculations feasible, it was necessary to employ a relatively small interfacial model, specifically a system consisting of 48 water molecules corresponding to a concentration of 1.2 mol L−1 (see Section S5 in the SI). Although this concentrated aqueous environment may limit direct quantitative comparison with experimental results obtained at 0.1 mol L−1, the two-step TI scheme corrects potential errors inherent in the MLFF while accounting for nanosecond-scale solvent reorganization dynamics, thereby enabling accurate determination of FP free energy differences.
The effects of three alkali cations, Li+, K+, and Cs+, on UOH were examined using TI calculations. As a diffuse limit of the cation distribution, the same calculations were also performed for a system containing a homogeneous background charge instead of explicit cations. Following the TI calculations, LENS was applied to visualize the interactions arising from the cations.
Fig. 1 shows the computed interfacial redox potential UOH as a function of the Shannon radius of the cation, rion.41 For comparison, the redox potential UOH obtained for a system containing a homogeneous positive background charge instead of an explicit cation is also shown. The redox potential UOH follows the order Li+ < K+ < Cs+ ≈ background charge, indicating a decrease in OH* stability with increasing cation size. This observation matches with the experiment.22 Another notable feature is the strong correlation between UOH and the interfacial cation diffusion coefficient Dint (see Section S7 of the SI). As Dint increases, UOH approaches the value for a diffusive homogeneous background charge. This trend suggests that smaller cations are trapped near OH* through attractive interactions and stabilize OH*. The localization of small cations is evident from the distributions shown in Fig. 2. The radial and three-dimensional concentration distributions indicate that Li+ incorporates the O atom of OH* into its first hydration shell and preferentially resides near the adsorbate, whereas the corresponding peak is significantly weaker for K+ and Cs+. None of the cations directly contact the Pt(111) surface; water molecules or OH* always separate them from the surface, consistent with FP structure optimizations by Greeley et al.22
 | ||
| Fig. 1 Computed redox potential UOH of the OH reduction, OH* + H+ + e− → * + H2O, versus the Shannon radius of cation rion. Solid line indicates computed UOH for the system involving a homogeneous background charge. Error bars are determined from the block averaging analysis.3,4 Graphics show the snapshots of interfacial structures obtained from the MD simulations at 300 K, and the numbers are the Bader charges of cation and OH*. Small white, medium red, medium blue, medium light green, large purple and large light green spheres show H, O in H2O, O in OH*, Li+, K+ and Cs+. Graphics are obtained by using VESTA.21 | ||
 | ||
| Fig. 2 Radial distribution functions between X+ (X = Li, K or Cs) and O in OH*. Insets show side and top views of three-dimensional density isosurfaces of the alkaline cation at 0.002 Å−3 for blue and 0.007 Å−3 for red. Graphics are obtained by using VESTA.21 | ||
Bader charge42 and electronic structure analyses show that OH* is stably negatively charged via electron donation from Pt 5d to O 2p orbitals,43 while the cations remain close to +1e, indicating that the interaction is predominantly electrostatic (see Fig. 1 and Section S8 in the SI). Although the kernel-based MLFF does not explicitly include long-range interactions, their effects are effectively incorporated into short-range interactions within the cutoff radius—such as the cation-OH* distances considered in this study—and are reproduced for the same system used in training within the deviations reported in Fig. S2.
All of the above results suggest that smaller cations interact attractively with OH* and stabilize it. Additional insight for this interaction is obtained from the LENS analysis. Fig. 3(a)–(c) visualize the functional derivative δEi/δρi(r). Here, we focus on the three-body angular distribution involving H atoms, the O atom in OH*, and a cation X+ (X = Li, K, or Cs). The relative coordinate r is defined by the distance r between H and O, the distance s between X+ and O, and the angle θ = ∠HOX. In Fig. 3, the angle is fixed at 110°, which is close to the direction of the sp3 orbital of the central O atom and corresponds to a region where the functional derivative takes a low value. The LENS analysis reveals a distinct, highly localized negative response of Ei to increasing ρi at r ≈ 1 Å and s ≈ 2 Å for Li+, a broader negative response centered at r ≈ 1 Å and s ≈ 3 Å for K+, and an more diffuse and weaker negative response around r ≈ 1 Å and s ≈ 3.5 Å for Cs+. Notably, the variational response appears deeper for K+. However, the negative peak is spatially delocalized, leading to a correspondingly delocalized distribution of K+ (Fig. 2 and Section S9 in the SI) and a reduced stabilizing contribution to OH*. A rough linear-response estimate of the local energy change, 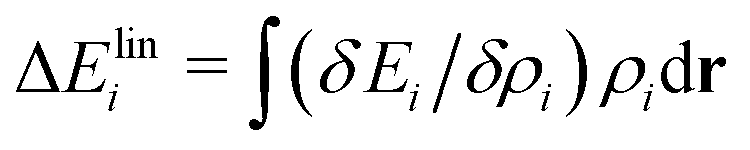 , shows that Li+ stabilizes OH* more strongly than K+ [Fig. 3(d)]. Although the magnitude of this local energetic response differs from the change in UOH, which reflects the free-energy difference of the entire system, and the difference between K+ and Cs+ cannot be resolved within this approximation, the LENS analysis nevertheless captures key features of the attractive interaction between OH* and small cations.
, shows that Li+ stabilizes OH* more strongly than K+ [Fig. 3(d)]. Although the magnitude of this local energetic response differs from the change in UOH, which reflects the free-energy difference of the entire system, and the difference between K+ and Cs+ cannot be resolved within this approximation, the LENS analysis nevertheless captures key features of the attractive interaction between OH* and small cations.
 | ||
| Fig. 3 Visualization of the functional derivative δEi/δρi obtained using LENS (a)–(c) and the stabilization energy ΔEilin estimated from the linear-response approximation (see text) (d). Here, Ei is the energy of the O atom in OH*, and ρi denotes the probability density of finding an H atom at a distance r to r + dr from this O atom and a cation X+ (X = Li, K, or Cs) at a distance s to s + ds from the same O atom, with ∠ HOX = θ = 110°, as illustrated in the insets. LENS yields smooth functional-derivative landscapes below ca. 1 Å for Li+ and 2 Å for the other ions; however, these regions are not sampled in the MD trajectories. The values in these regions arise from kernel extrapolation and are therefore physically meaningless. | ||
In summary, our MLFF-aided TI scheme combined with the interaction visualization method LENS shows that small alkaline cations are trapped by OH adsorbed on Pt(111) in water and stabilize this interfacial species via attractive interactions, consistent with experiments.22 Notably, the kernel-based representation of interatomic interactions, which does not explicitly encode physical laws, learns from FP data to capture the attractive interaction between small cations and OH without human intervention. Furthermore, LENS reveals information beyond the total-energy summation in eqn (4) by decomposing atomic energy contributions in terms of local species distributions. These results show that MLFFs combined with LENS can non-empirically identify species that stabilize or destabilize atoms or atomic groups in complex heterogeneous environments, highlighting their potential for materials design.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). The supplementary information includes derivation of eqn (5), details of models and computational procedures. The data and LENS code used for this work are provided in a zipped file to facilitate reproduction of the results. See DOI: https://doi.org/10.1039/d6cc00436a.References
- R. M. Martin, Electronic Structure: Basic Theory and Practical Methods, Cambridge University Press, Cambridge, 2004 Search PubMed.
- D. Marx and J. Hutter, Ab Initio Molecular Dynamics: Basic Theory and Advanced Methods, Cambridge University Press, 2009 Search PubMed.
- M. P. Allen and D. J. Tildesley, Computer Simulation of Liquids, Oxford University Press, 2017 Search PubMed.
- D. Frenkel and B. Smit, Understanding Molecular Simulation, Academic Press, Inc., USA, 2nd edn, 2001 Search PubMed.
- J. Behler, J. Chem. Phys., 2016, 145, 170901 CrossRef.
- V. L. Deringer, M. A. Caro and G. Csányi, Adv. Mater., 2019, 31, 1902765 CrossRef CAS.
- R. Jinnouchi, K. Miwa, F. Karsai, G. Kresse and R. Asahi, J. Phys. Chem. Lett., 2020, 11, 6946–6955 CrossRef CAS.
- T. W. Ko, J. A. Finkler, S. Goedecker and J. Behler, Nat. Commun., 2021, 12, 398 CrossRef CAS PubMed.
- J. Behler, Chem. Rev., 2021, 121, 10037–10072 CrossRef CAS PubMed.
- O. T. Unke, S. Chmiela, H. E. Sauceda, M. Gastegger, I. Poltavsky, K. T. Schütt, A. Tkatchenko and K.-R. Müller, Chem. Rev., 2021, 121, 10142–10186 CrossRef CAS PubMed.
- F. Musil, A. Grisafi, A. P. Bartók, C. Ortner, G. Csányi and M. Ceriotti, Chem. Rev., 2021, 121, 9759–9815 CrossRef CAS PubMed.
- V. L. Deringer, A. P. Bartók, N. Bernstein, D. M. Wilkins, M. Ceriotti and G. Csányi, Chem. Rev., 2021, 121, 10073–10141 CrossRef CAS PubMed.
- D. M. Anstine and O. Isayev, J. Phys. Chem. A, 2023, 127, 2417–2431 CrossRef CAS PubMed.
- I. Batatia, S. Batzner, D. P. Kovács, A. Musaelian, G. N. C. Simm, R. Drautz, C. Ortner, B. Kozinsky and G. Csányi, Nat. Mach. Intell., 2025, 7, 56–67 CrossRef.
- R. Jinnouchi and S. Minami, ACS Nano, 2025, 19, 22600–22644 CrossRef CAS.
- J. E. Mayer, J. Chem. Phys., 1937, 5, 67–73 CrossRef CAS.
- A. P. Bartók, R. Kondor and G. Csányi, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 184115 CrossRef.
- A. V. Shapeev, Multiscale Model. Simul., 2016, 14, 1153–1173 CrossRef.
- R. Drautz, Phys. Rev. B, 2019, 99, 014104 CrossRef CAS.
- A. Glielmo, C. Zeni and A. De Vita, Phys. Rev. B, 2018, 97, 184307 CrossRef CAS.
- K. Momma and F. Izumi, J. Appl. Crystallogr., 2008, 41, 653–658 CrossRef CAS.
- D. Strmcnik, K. Kodama, D. van der Vliet, J. Greeley, V. R. Stamenkovic and N. M. Marković, Nat. Chem., 2009, 1, 466–472 CrossRef CAS.
- A. Goyal, S. Louisia, P. Moerland and M. T. M. Koper, J. Am. Chem. Soc., 2024, 146, 7305–7312 CrossRef CAS.
- M. S. Shibata, Y. Morimoto, A. Z. Weber and I. V. Zenyuk, ACS Energy Lett., 2025, 10, 3922–3930 CrossRef CAS.
- J. Resasco, L. D. Chen, E. Clark, C. Tsai, C. Hahn, T. F. Jaramillo, K. Chan and A. T. Bell, J. Am. Chem. Soc., 2017, 139, 11277–11287 CrossRef CAS.
- S. Ringe, E. L. Clark, J. Resasco, A. Walton, B. Seger, A. T. Bell and K. Chan, Energy Environ. Sci., 2019, 12, 3001–3014 RSC.
- D. T. Limmer, A. P. Willard, P. Madden and D. Chandler, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 4200–4205 CrossRef CAS.
- I. T. McCrum and M. J. Janik, J. Phys. Chem. C, 2016, 120, 457–471 CrossRef CAS.
- H. H. Kristoffersen, K. Chan, T. Vegge and H. A. Hansen, Chem. Commun., 2020, 56, 427–430 RSC.
- R. Jinnouchi, J. Lahnsteiner, F. Karsai, G. Kresse and M. Bokdam, Phys. Rev. Lett., 2019, 122, 225701 CrossRef CAS.
- R. Jinnouchi, F. Karsai and G. Kresse, Phys. Rev. B, 2019, 100, 014105 CrossRef CAS.
- R. Jinnouchi, F. Karsai, C. Verdi, R. Asahi and G. Kresse, J. Chem. Phys., 2020, 152, 234102 CrossRef CAS PubMed.
- G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- A. P. Bartók, M. C. Payne, R. Kondor and G. Csányi, Phys. Rev. Lett., 2010, 104, 136403 CrossRef.
- J. Behler and M. Parrinello, Phys. Rev. Lett., 2007, 98, 146401 CrossRef.
- R. Jinnouchi, F. Karsai and G. Kresse, npj Comput. Mater., 2024, 10, 107 CrossRef CAS.
- R. Jinnouchi, J. Chem. Phys., 2024, 161, 194110 CrossRef CAS PubMed.
- R. Jinnouchi, F. Karsai and G. Kresse, Chem. Sci., 2025, 16, 2335–2343 RSC.
- R. Jinnouchi and S. Minami, J. Phys. Chem. Lett., 2025, 16, 265–273 CrossRef CAS PubMed.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- W. Tang, E. Sanville and G. Henkelman, J. Phys.: Condens. Matter, 2009, 21, 084204 CrossRef CAS PubMed.
- M. Arce, P. Quaino and E. Santos, Catal. Today, 2013, 202, 120–127 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |