
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Recent advancements in carbonylative synthesis of quinolinones
Mariangela
Novello
ab,
Raffaella
Mancuso
 *a,
Bartolo
Gabriele
*a and
Xiao-Feng
Wu
*bc
*a,
Bartolo
Gabriele
*a and
Xiao-Feng
Wu
*bc
aLaboratory of Industrial and Synthetic Organic Chemistry (LISOC), Department of Chemistry and Chemical Technologies, University of Calabria, Via Pietro Bucci 12/C, 87036 Arcavacata di Rende (CS), Italy. E-mail: raffaella.mancuso@unical.it; bartolo.gabriele@unical.it
bLeibniz-Institut für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: Xiao-Feng.Wu@catalysis.de
cDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 116023 Dalian, Liaoning, China. E-mail: xwu2020@dicp.ac.cn
First published on 18th February 2026
Abstract
Carbonylation reactions, which rely on the controlled incorporation of carbon monoxide into organic substrates, represent a key tool in both organic and industrial chemistry. Carbonylation offers an efficient route to carbonyl-containing compounds starting from simple and readily available substrates, and it remains a highly active area of research in organometallic chemistry and catalysis. Current efforts focus on developing new catalysts, more efficient and sustainable methodologies, and innovative applications in emerging areas such as green chemistry and asymmetric synthesis. Given the great importance of heterocyclic compounds, the carbonylative approach has become increasingly important for their synthesis. In this review, we summarize and discuss advancements in the synthesis of quinolinone derivatives, a class of benzo-fused nitrogen-containing heterocyclic compounds, via carbonylative approaches.
 Mariangela Novello | Mariangela Novello earned her Master's in Pharmaceutical Chemistry and Technology at the University of Calabria in 2018. She is currently a PhD student in Environmental, Energy and Construction Science and Engineering, conducting her research at the Laboratory of Industrial and Synthetic Organic Chemistry (LISOC). Her research focuses on the development and optimization of carbonylation and carboxylation processes for the preparation of molecules of industrial, pharmaceutical and applied interest through the use of recyclable catalysts and unconventional solvents. |
 Raffaella Mancuso | Raffaella Mancuso earned her PhD in “Methodologies for the Development of Molecules of Pharmacological Interest” from the University of Calabria (Italy) in 2026. She currently holds the position of Full Professor of Organic Chemistry at the same University. Her current research interests focus on the synthesis of heterocyclic molecules of pharmaceutical, industrial, or applicative interest, and the use of nonconventional solvents in organic synthesis. |
 Bartolo Gabriele | Bartolo Gabriele earned his PhD in Chemical Sciences from the University of Calabria (Italy) in 1994. He currently holds the position of Full Professor of Organic Chemistry at the same University. His primary research focuses on the conversion of carbon oxides (CO and CO2) into high valued added compounds by catalytic methods and on the development of new organic materials for applications in membrane science. |
 Xiao-Feng Wu | Xiao-Feng Wu was born and raised in China. After being educated and trained in China (Zhejiang Sci-Tech University), France (Rennes 1 University) and Germany (Leibniz-Institute for Catalysis), he started his independent research at LIKAT and ZSTU where he was promoted to professor in 2013 and afterwards defended his Habilitation from Rennes 1 University (2017). In 2020, he joined Dalian Institute of Chemical Physics (DICP) and established a group working on light carbon transformation and practical synthesis. Xiao-Feng has authored >700 publications, edited >10 books and filed many patents. Xiao-Feng received several awards and was selected as a Fellow of the Royal Society of Chemistry (FRSC) in 2024. |
1. Introduction
Cyclic compounds are defined by the presence of ring structures composed either exclusively of carbon atoms (carbocycles) or incorporating one or more heteroatoms (heterocycles), often in combination with various degrees of unsaturation. Their ubiquitous presence in biological systems and in numerous bioactive molecules has long made them a central topic of interest within the scientific community. Heterocyclic frameworks constitute essential structural motifs in a wide array of natural and synthetic biologically active compounds, and their synthesis continues to be a major focus in contemporary organic chemistry. Among the diverse families of heterocyclic compounds, quinolinones stand out due to their structural features—formed by the fusion of a benzene ring with a six-membered nitrogen-containing core. Their structural motifs are essential building blocks found widely across functional materials1–4 and occur in many naturally derived molecules exhibiting noteworthy biological activities including antibiotic,5,12–15 anticancer,6,8,12,14 antiviral,7,9–11 antimalarial,16,17 antiparasitic,18 antitubercolar,19 antiinflammatory,20,21 herbicidal,22 antifungal,23 antihypertensive and antipsycotic activities.24–27 Quinolinones, therefore, exhibit various pharmacological properties and play an important role in the development of new drugs. They have been and are fundamental in modern medicine because they offer high efficacy against a wide range of bacteria. It is no coincidence that the first was nalidixic acid, which was not a result of targeted research, but was discovered accidentally in 1962 as a byproduct during an attempt to synthesize chloroquina, an antimalarial drug.28,29Generally, quinolones can be synthesized by traditional approaches such as the Conrad–Limpach (Fig. 1a), the Gould–Jacobs (Fig. 1b), and the Camps cyclization (Fig. 1c).30 However, all these methods involve thermal cyclocondensation steps and typically require harsh conditions that employ high temperatures and strong bases during the synthesis. The development of innovative, efficient, and accessible synthetic routes to these heterocycles is therefore of great significance. Within this context, carbonylation reactions/carbonylative processes have emerged as an appealing strategy for the construction of high-value compounds from simple building blocks using CO or its surrogates. Carbonylation31 is emerging as a key technology for developing sustainable chemical processes, enabling the conversion of simple substrates into high-value molecules with excellent atom economy. The use of CO from renewable sources and non-noble catalysts is driving this chemistry toward green-chemistry paradigms. Current research aims to reduce environmental impact, improve process safety, and promote carbonylation as a fully circular synthetic strategy. Carbon monoxide is the simplest and most versatile C1 unit, meeting the principles of atom economy,32 step economy,33 and green chemistry.34 The ability to generate structurally complex, pharmacologically relevant molecules through carbonylative procedures provides an attractive alternative to traditional multistep synthetic pathways, particularly due to the efficient carbon-chain extension enabled by CO insertion (Fig. 1d). In this review, we present an overview of different and innovative methodologies for the synthesis of quinolinones via carbonylative transformations.
 | ||
| Fig. 1 General procedures for preparation of quinolinones. | ||
2. Synthesis of quinolinones by Pd(0)-catalyzed carbonylation reactions
Over the past decade, the transition-metal-catalyzed synthesis of quinolinone derivatives has gained considerable importance in organic chemistry. A wide range of transition-metal complexes, particularly those based on Pd, Cu, Rh, and Ir, have been successfully employed as catalysts for these transformations. In this context, transition-metal catalysis enables a variety of synthetically valuable reactions that are otherwise challenging or, in some cases, unattainable using conventional methodologies.Among the metal-catalyzed approaches available for the construction of quinolinone frameworks, palladium-catalyzed carbonylation of substituted o-haloanilines with aryl or alkyl terminal alkynes represents one of the most straightforward and efficient strategies. In 1991, Torii and co-workers reported the first example of Pd-catalyzed carbonylative coupling between 2-haloanilines, terminal alkynes, and carbon monoxide. Using 5 mol% of PdCl2(PPh3)2 as the catalyst in diethylamine under 20 bar of CO at 120 °C, the corresponding 2-substituted-4-quinolinones were obtained in good yields.35 The authors demonstrated that the nucleophilic cyclization proceeded efficiently when a primary amino group was present, and that iodoanilines were superior to bromoanilines in terms of product yields. By contrast, under identical conditions, alkylated anilines afforded the desired products only in very low yields.
Shortly thereafter, Kalinin and co-workers described a closely related protocol employing 10 mol% of PdCl2(dppf) in diethylamine under 20 bar of CO at 120 °C.36 This methodology was later applied by Haddad et al. in 2006 to the synthesis of BILN 2061 (the drug is known as Ciluprevir) derivatives37 (Scheme 1a), a compound class that includes the first effective inhibitor of the hepatitis C virus (HCV) NS3 protease.38 Specifically, the authors utilized Pd-catalyzed carbonylative Sonogashira coupling followed by cyclization between 2-iodo-5-methoxyaniline and thiazolylacetylene. Notably, higher yields of the desired quinolinone were achieved when PdCl2(dppf) was employed instead of PdCl2(PPh3)2 under optimized reaction conditions.
 | ||
| Scheme 1 Pd(0)-catalyzed carbonylative synthesis of quinolinones from 2-iodoaniline derivatives. | ||
In parallel, the Torii's group in 1990 developed a palladium-catalyzed carbonylation of 3-substituted 3-(2-haloarylamino)prop-2-enoates. This approach enabled the synthesis of a series of 2-substituted 1,4-dihydro-4-oxoquinoline-3-carboxylates in good yields. The reactions were carried out using 5 mol% of Pd(OAc)2, 20 mol% of PPh3, and three equivalents of K2CO3 in DMF at 120 °C for 20 h under 20 bar of CO. Prior to purification, the crude products were treated with diazomethane39 (Scheme 1b).
Further advances were reported by Ye and Alper, who demonstrated that 2-iodoanilines could also undergo carbonylative cyclization with allenes to afford structurally related quinolinone derivatives.40 In this case, the palladium-catalyzed cyclocarbonylation of o-iodoanilines with allenes and CO was conducted in the ionic liquid 1-butyl-3-methylimidazolium hexafluorophosphate (BMIM PF6). The reaction proceeded efficiently under a relatively low CO pressure (5 bar), using 2 mol% of Pd2(dba)3·CHCl3 and two equivalents of DIPEA at 90 °C for 20 h, affording 3-methylene-2,3-dihydro-1H-quinolin-4-ones in moderate to excellent yields (Scheme 1c). As highlighted by the authors, the ionic liquid significantly enhanced the efficiency of the cyclocarbonylation process, and the recyclability of the combined ionic liquid/catalyst/ligand system was successfully demonstrated.
Another palladium-catalyzed approach involving o-iodoanilines was published in 1992 by Yamanaka and co-workers for the synthesis of 4-quinolinone derivatives. In this study, 4-quinolinones were accessed through Pd-catalyzed carbonylative coupling between 2-iodophenyl carbamate and (Z)-3-(tri-tert-butyl-2-ethoxyvinyl)stannane. The reaction was carried out in chloroform using PdCl2(PPh3)2 as the catalyst under 5 atm of CO at 80 °C, affording ethyl (E)-2-(3-ethoxy-1-oxoprop-2-en-1-yl)phenyl carbamate as the key intermediate. Subsequent acid-mediated cyclization of this intermediate led to the formation of the desired 4-quinolinone scaffold (Scheme 2).41
 | ||
| Scheme 2 Synthesis of 4-quinolinones via Stille reaction followed by cyclization. | ||
In 2009, Djakovitch and co-workers presented a Pd-catalyzed carbonylative Sonogashira coupling between 2-iodoanilines, alkynes, and CO (5 bar), employing Et3N as the base and 1 mol% of PdCl2(dppp) [dppp = 1,3-bis(diphenylphosphino)propane] as the catalyst. Under these conditions, the corresponding carbonylative coupling intermediates were selectively formed. In a subsequent step, the addition of Et2NH promoted an organocatalyzed cyclization, ultimately affording the desired quinolin-4(1H)-ones with high selectivity and good to excellent yields (Scheme 3a).42 Despite the efficiency of this one-pot, two-step multicatalytic strategy, its main drawback lies in the use of homogeneous catalysts, which are difficult to remove and may lead to significant Pd and ligand residues in the final products—an important limitation for applications related to animal and human health.
 | ||
| Scheme 3 Synthesis of 2-substituted quinolin-4(1H)ones by a carbonylative Sonogashira coupling reaction under homogeneous and heterogeneous Pd-catalysis. | ||
Building on this work, the same group later addressed these issues by developing a heterogeneous catalytic approach. In 2011, they reported the use of a tandem system combining a supported palladium catalyst, ((Pd(PNP))@SBA-15), with a grafted amine catalyst, [NH2]@SBA-3, operating in a cooperative [Pd/amine] mode. This strategy enabled the selective synthesis of 2-phenylquinolin-4(1H)-one (Scheme 3b).43,44 Notably, the heterogeneous system led to a substantial reduction in palladium contamination of the final products, with only 3–5 ppm of Pd detected in the quinolin-4(1H)-ones, compared to approximately 40 ppm observed when homogeneous catalysts were used. In addition, the overall reaction time was significantly shortened from 7 to 3 days under otherwise identical reaction conditions. The [(Pd(PNP))@SBA-15]/[NH2@SBA-3] catalyst mixture could be successfully recycled for up to three consecutive runs without a significant loss of activity.
In 2003, Larock and coworkers demonstrated an efficient synthesis of 2-quinolones from functionalized o-iodoaniline derivatives45,46 by reacting them with internal alkynes under 1 atm of CO in the presence of pyridine and catalytic amounts of Pd(OAc)2 (Scheme 4a). The reactions were conducted under the following optimized conditions: 0.5 mmol of the 2-iodoaniline carbamate derivative, 5 mol% Pd(OAc)2, 2 equiv. of pyridine, and 1 equiv. of Bu4NCl in 5 mL of DMF at 100 °C for 12 h. The crude products were treated for 30 min at room temperature with 1 M ethanolic NaOH to complete the hydrolysis of the carbamate protecting group. This unusual alkyne/CO insertion selectivity observed with internal alkynes prompted the authors to investigate analogous reactions involving terminal alkynes under the same catalytic conditions (Scheme 4b). Removal of the carbamate protecting group by treating the crude reaction with 1M ethanolic NaOH was necessary to avoid the formation of a mixture of deprotected and protected quinolin-2-ones.
 | ||
| Scheme 4 Pd(0)-catalyzed annulation of internal and terminal alkynes. | ||
The proposed mechanism, shown in Scheme 5, involves the in situ initial reduction of Pd(OAc)2 to Pd(0). Oxidative addition of the aryl iodide to Pd(0) affords an arylpalladium complex that subsequently reacts with the terminal alkyne to form a vinylpalladium intermediate. Insertion of CO into the vinyl–Pd bond then yields an acylpalladium species. Nucleophilic attack of the carbamate nitrogen on the acyl carbon leads to formation of the N-protected 2-quinolone and regenerates Pd(0), while the final hydrolysis of the carbamate group provides the desired product.
 | ||
| Scheme 5 Proposed mechanism for the synthesis of 2-quinolones from functionalized o-iodoaniline derivatives. | ||
An efficient synthetic route to 4,5-fused tricyclic 2-quinolones via palladium-catalyzed carbonylative annulation of alkyne-tethered N-substituted o-iodoanilines was shown by Zhang and co-workers (Scheme 6).47 The transformation proceeded under relatively mild conditions and showed broad functional group tolerance. Importantly, this methodology was successfully applied to the efficient synthesis of BI 224436, a known HIV integrase inhibitor.48 At the outset, the authors evaluated Larock's conditions for 2-quinolone synthesis—Pd(OAc)2 (0.1 equiv.), pyridine (2.0 equiv.), TBAC (tert-butyl acetate) (1.0 equiv.) in DMF (0.01 M) under 1 atm of CO at 100 °C for 24 h. However, under these conditions, the desired 4,5-fused tricyclic 2-quinolone was obtained in low yield, with substantial recovery of the starting material. Further optimization revealed that the presence of a phosphine ligand was crucial to improve the efficiency of the process. The optimal conditions were ultimately identified in 10 mol% Pd(OAc)2, 5 equiv. of pyridine, 1 equiv. of LiCl, and 0.2 equiv. of PPh3 in DMF at 100 °C for 24 h, which led to a marked improvement in product formation.
 | ||
| Scheme 6 Synthesis of 4,5-fused tricyclic 2-quinolinones. | ||
Palladium-catalyzed intermolecular aminocarbonylation followed by intramolecular amidation in a cascade fashion has been shown to provide an efficient and selective route to 2-quinolones from a broad range of 2-(2-haloalkenyl)aryl halide substrates (Scheme 7).49 In this study, the authors investigated the coupling of dibromostyrenes with amines under a balloon pressure of carbon monoxide in toluene. Under the optimized conditions, employing 3 mol% of Pd2(dba)3 as the catalyst and 3 equivalents of Cs2CO3 as the base, several phosphine ligands, including P(i-Pr)3, dppp, and P(t-Bu)3—were found to be effective. These systems enabled the efficient formation of the corresponding 2-quinolinones in good yields, highlighting the versatility and robustness of the cascade aminocarbonylation/amidation strategy.
 | ||
| Scheme 7 Synthesis of 2-quinolinones by Pd(0)-catalyzed aminocarbonylation/intramolecular amidation cascade sequence starting from 2-(2-haloalkenyl)aryl halides. | ||
Building on the carbonylative cyclization of 2-substituted N-(2-haloaryl)-2-propenoates reported by Torii et al.,39 Mori and co-workers initially applied the same reaction conditions—Pd(OAc)2, PPh3, K2CO3 in DMF under CO (20 atm) at 120 °C—to their own model system. In this case, the ester functionality employed by Torii was replaced by a ketone, with the aim of accessing the corresponding quinolinone analogues. However, under these conditions the desired product was obtained in low yield. Subsequent studies demonstrated that 2-substituted 3-aroylquinolin-4(1H)-ones, accessible through palladium-catalyzed carbonylative cyclization of N-(2-iodoaryl)enaminones,50 were potent inhibitors of the Hedgehog (Hh) signaling pathway via direct antagonism of both wild-type and drug-resistant forms of the smoothened receptor. Notably, these compounds effectively suppressed Hh-dependent growth processes and the proliferation of tumor cells characterized by aberrant activation of the Hh pathway,51 which plays a central role in development and tumorigenesis.
Motivated by these findings, Mori et al. envisioned that the palladium-catalyzed carbonylative cyclization of readily available N-(2-iodoaryl)enaminones could provide a practical and efficient entry to 2-substituted 3-aroylquinolin-4(1H)-ones. Optimal results were achieved using Pd2(dba)3 and Cs2CO3 in acetonitrile at 100 °C under 20 atm of carbon monoxide, in the presence of bulky monodentate ligands such as XPhos. Importantly, this transformation could also be performed without isolating the enaminone intermediates. Satisfactory yields were obtained by directly adding Pd2(dba)3, XPhos, Cs2CO3, MeCN, and CO (20 atm) to the crude reaction mixture generated from the coupling of 2-iodoanilines with α,β-ynones after removal of volatile components (Scheme 8).
 | ||
| Scheme 8 Synthesis of 3-aroylquinolin-4(1H)-ones by Pd(0)-catalyzed carbonylative cyclization of N-(2-iodoaryl)enaminones. | ||
In 2012, Alper and co-workers reported an efficient one-step protocol for the synthesis of 2,3,3-triethoxycarbonyl-2,3-dihydro-4(1H)-quinolinone derivatives via palladium-catalyzed intermolecular cyclocarbonylation of 2-iodoanilines with diethyl ethoxycarbonylbutendienoate.52 This transformation proceeds through an initial Michael addition followed by carbonylation, providing rapid access to structurally complex quinolinone frameworks. In their study, a series of 2-iodoanilines were allowed to react with diethyl ethoxycarbonylbutendienoate using a Pd2(dba)3/2-(di-tert-butylphosphino)biphenyl catalytic system in acetonitrile at 80 °C under 500 psi of carbon monoxide for 20 h (Scheme 9).
 | ||
| Scheme 9 Synthesis of 2,3-dihydroquinolin-4(1H)-ones by Pd-catalyzed intramolecular cyclocarbonylation of 2-iodoanilines and diethyl ethoxycarbonyl butendienoate. | ||
Quinolones were accessed through a palladium-catalyzed cascade involving oxidative addition of an aryl iodide, followed by carbonylation, allene insertion, and intramolecular trapping of the resulting allylpalladium(II) species by a nitrogen nucleophile. In contrast to the related cascade processes reported by Alper and co-workers, which typically require high carbon monoxide pressures (≈20 atm), Grigg et al. deliberately focused their studies on reactions conducted under atmospheric pressure of CO. Specifically, they investigated [3+1+2] cycloaddition reactions of N-tosyl-o-iodoanilines using allene (1 atm) and carbon monoxide (1 atm) in a Schlenk tube. The reactions were performed in toluene at 45–70 °C in the presence of Pd(PPh3)4 (5 mol%) and K2CO3 (2 equiv.) and were completed within 16–35 h, affording the desired products in good yields (Scheme 10).53 Mechanistically, the catalytic cycle begins with oxidative addition of the aryl iodide bond to Pd(0), followed by coordination and insertion of CO to form an acylpalladium(II) intermediate. Subsequent addition of this species to the central carbon of the allene generates an allylpalladium(II) intermediate, which is then attacked by the internal nitrogen nucleophile to furnish the enone-containing quinolone product. Notably, this (3+1+2) cycloaddition exhibits good tolerance toward substituents on both the allene and the aryl iodide, enabling access to a diverse range of heterocycles bearing s-cis enone motifs. A related one-pot quinolone synthesis involving a subsequent Michael addition was also explored, albeit under slightly modified conditions. In this variant, the [3+1+2] cycloaddition of N-tosyl-o-iodoaniline with CO (1 atm) and allene (1 atm) was allowed to proceed for intermediate species, and then CO was released prior to the addition of an external nucleophile. The ensuing Michael addition was completed over an additional 24 h (Scheme 11). Both aliphatic and heteroaromatic nitrogen nucleophiles were compatible with this protocol; notably, in the case of 1,2,4-triazole, exclusive formation of the 1-substituted triazole was observed.
 | ||
| Scheme 10 Synthesis of quinol-4-ones via a palladium-catalysed cascade carbonylation-allene insertion. | ||
 | ||
| Scheme 11 Catalytic cycle of Pd(0)-catalyzed cascade carbonylation-allene insertion for the synthesis of quinol-4-ones. | ||
By integrating palladium-catalyzed carbonylation with nonracemic Lewis base catalysis, this three-component cascade reaction of benzyl bromides, carbon monoxide, and vinyl benzoxazinanones provides an efficient and highly stereoselective route to chiral quinolinone derivatives. In this transformation, the palladium catalyst plays a dual role by mediating both the carbonylation process and the formation of a zwitterionic π-allyl palladium intermediate, which engages with the C1-ammonium enolate to furnish the final products.
Gong et al. proposed that the zwitterionic π-allyl palladium species D, generated from vinyl benzoxazinanones, could react with the C1-ammonium enolate intermediate C to furnish the chiral heterocycle. This concept ultimately enabled a three-component annulation of benzyl bromides, CO, and vinyl benzoxazinanones (Scheme 12).54 Initial studies focused on the model reaction between benzyl bromide, vinyl benzoxazinanone, and CO in the presence of an achiral palladium catalyst and a nonracemic Lewis base catalyst, benzotetramisole (BTM). Xantphos was selected as the ligand owing to its well-established efficiency in accelerating Pd-catalyzed carbonylation reactions.55–59 In the proposed catalytic system, palladium is involved both in the carbonylation step leading to intermediate C and in the oxidative addition process that generates the π-allyl palladium intermediate D. Notably, during formation of the π-allyl palladium species, the Pd catalyst is temporarily sequestered and cannot be recycled until the reaction sequence is completed. Consequently, the concentration of intermediate D must be carefully controlled to ensure that sufficient free palladium remains available for carbonylation. Guided by this consideration, a solution of vinyl benzoxazinanone in THF was slowly added over 5 h via syringe pump, which indeed led to efficient formation of the desired product. Importantly, the reaction could be readily scaled up to 1 mmol using a CO balloon as the carbon monoxide source, affording the product in 75% yield. A plausible reaction mechanism is outlined in Scheme 13. Oxidative addition of the C–Br bond of benzyl bromide to Pd(0) generates intermediate A′, which undergoes CO insertion to form the acylpalladium species A. Subsequent β-hydride elimination furnishes a ketene intermediate B, which is intercepted by the chiral Lewis base BTM to generate the C1-ammonium enolate C. In parallel, oxidative addition of vinyl benzoxazinanone to Pd(0) leads to a π-allyl palladium complex D′, which is converted into intermediate D upon decarboxylation. Nucleophilic attack of enolate C on the π-allyl palladium moiety affords intermediate E, followed by intramolecular cyclization to deliver the chiral quinolinone. Alternatively, direct reaction of intermediate D with ketene B may occur, resulting in the formation of the racemic product.
 | ||
| Scheme 12 Enantioselective synthesis of enantioenriched 1,2,3,4-tetrahydroquinolines via enantioselective cascade carbonylation/annulation of benzyl bromides, CO, and vinyl benzoxazinanones enabled by Pd/nonracemic Lewis-base relay catalysis. | ||
 | ||
| Scheme 13 Proposed mechanism for the construction of chiral quinolinone derivatives from benzyl bromides, CO, and vinyl benzoxazinanones. | ||
Chung and co-workers reported the use of aromatic tosylates as substrates for palladium-catalyzed aminocarbonylation to 3,4-dihydroisoquinolin-1(2H)-ones (Scheme 14).60 The reaction was carried out using Pd(OAc)2 (2 mol%) as the catalyst and dicyclohexylphosphinoethane (dcyphe) as the ligand in DMF at 100 °C under an atmospheric pressure of carbon monoxide. Notably, this transformation proceeds without the addition of an external amine source. Instead, intramolecular cyclization leads directly to lactam formation, accompanied by cleavage of the Boc protecting group during the process. This strategy provides a concise and efficient approach to 3,4-dihydroisoquinolinone frameworks under relatively mild carbonylation conditions.
 | ||
| Scheme 14 Pd(0)-catalyzed aminocarbonylation of aryl tosylates. | ||
By using the continuous flow technology, Ley and colleagues developed a palladium catalyzed method for carbonyl insertion to synthesize 3,4-dihydroisoquinilin-1(2H)-ones.61 The reaction was carried out in a semipermeable amorphous fluoropolymer Teflon® AF-2400 Tube-in-Tube assembly, reactors that permit the controlled transport of carbon monoxide into the solution at elevated pressure to generate homogeneous flow streams, thus avoiding some potential issues associated with segmented flow gas–liquid reactors. In the presence of 2 mol% Pd(OAc)2, 6 mol% xantphos, 30 mol% of hydrazine, 1 M THF, and 1.1 equiv. of Et3N, the desired product was formed in excellent yield (93%) under 15 bar of CO at 100 °C (Scheme 15).
 | ||
| Scheme 15 Continuous flow Pd(0)-catalyzed carbamoylation of aryl halide. | ||
Okuro and Alper developed a stereoselective protocol for the synthesis of 4-ethoxycarbonyl-3,4-dihydroisoquinolin-1(2H)-ones through palladium-catalyzed intermolecular cyclocarbonylation of diethyl (2-iodoaryl)acetates with N-tosylimines (Scheme 16).62 This transformation proceeds through a cascade sequence involving an initial Mannich addition, followed by cyclocarbonylation and a final decarboxylation step. A plausible mechanism for the formation of 3,4-dihydroisoquinolin-1(2H)-ones involves several key stages. First, a Mannich addition between diethyl (2-iodoaryl)acetate and the N-tosylimine furnishes the corresponding Mannich adduct. Subsequent oxidative addition of the C–I bond of this adduct to Pd(0) generates a palladium(II) intermediate, which undergoes insertion of carbon monoxide into the Pd–C bond to form an aroylpalladium species. Intramolecular nucleophilic attack by the nitrogen atom on this aroylpalladium intermediate then delivers the initial cyclized product A, with concomitant regeneration of the Pd(0) catalyst. Product A subsequently undergoes base-induced decarboxylation to afford the final dihydroisoquinolinone with a trans configuration. The stereochemical outcome is established during this decarboxylation step, in which exclusive formation of the thermodynamically more stable trans isomer is observed (Scheme 17).
 | ||
| Scheme 16 Synthesis of 4-alkoxycarbonyl-3,4-dihydroisoquinolin-1(2H)-ones. | ||
 | ||
| Scheme 17 Possible reaction mechanism for the formation of 4-alkoxycarbonyl-3,4-dihydroisoquinolin-1(2H)-ones. | ||
In 2017, Lei and co-workers reported the synthesis of 4-quinolones via a palladium-catalyzed oxidative carbonylation process (Scheme 18).63 In their protocol, variously substituted amines and ketones were reacted under atmospheric CO in the presence of Pd(dba)2 and CuBr(Me2S) as the Pd(II) reoxidation system, affording the corresponding 4-quinolone derivatives in good yields, also using Xantphos as a ligand. PhCO2Na and KI were also necessary for the reaction and the product was obtained under nonexplosive conditions (CO/O2 = 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1).
:1).
 | ||
| Scheme 18 Pd(0)-catalyzed carbonylative synthesis of 4-quinolinones from ketones and anilines. | ||
3. Synthesis of quinolinones by Pd(II)-catalyzed carbonylation reactions
Zhao and co-workers reported a palladium-catalyzed C–H carbonylation strategy assisted by an oxalyl amide-directing group for the synthesis of 3,4-dihydroisoquinolinones. In this protocol, β-phenylethylamines protected as oxalyl amides were exposed to carbon monoxide (1 atm) in toluene at 100 °C for 5 h, using Pd(OAc)2 (10 mol%) as the catalyst and AgOAc (2.5 equiv.) as the oxidant (Scheme 19).64 A plausible mechanism for the oxalyl amide–assisted carbonylation is depicted in Scheme 20. In pathway a, the palladium(II) complex II is generated via a concerted metalation–deprotonation (CMD) process. Coordination of CO to the Pd(II) center is followed by migratory insertion of CO into the Pd–C bond, leading to the formation of the key palladium intermediate IV. Alternatively, the catalytic cycle may proceed through pathway b, in which palladium complex I first coordinates CO, followed by CO insertion to form intermediate III. Subsequent C–H activation then furnishes the same key seven-membered palladacyclic intermediate IV. Reductive elimination from intermediate IV delivers the desired 3,4-dihydroisoquinolinone product and completes the catalytic cycle. This carbonylation protocol displays broad functional-group tolerance and enables the synthesis of a variety of 3,4-dihydroisoquinolinone derivatives in good to excellent yields. | ||
| Scheme 19 Pd(II)-catalyzed carbonylation of β-arylethylamides. | ||
 | ||
| Scheme 20 Proposed catalytic cycle for the synthesis of 3,4-dihydroisoquinolinone derivatives. | ||
A Pd(II)-catalyzed NH2-directed carbonylation of quaternary aromatic α-amino esters has been reported as an effective route to 3,4-dihydroisoquinolin-1(2H)-ones, delivering the desired products in moderate to good yields (Scheme 21).65 The reaction is carried out using Pd(OAc)2 (2 mol%) as the catalyst in the presence of benzoquinone as the oxidant, under 1 atm of carbon monoxide at 120 °C. Mechanistic studies indicate that the transformation proceeds via initial ortho-palladation mediated by Pd(II), leading to the formation of a cyclopalladated intermediate, which can be isolated in some cases. Benzoquinone plays a crucial role in reoxidizing Pd(0) back to Pd(II), thereby sustaining the catalytic cycle. Notably, bulky substituents surrounding the amino group were found to be beneficial for the carbonylation step, likely by facilitating effective cyclopalladation. In addition, the presence of a quaternary carbon at the α-position of the amino ester was shown to be essential, as it significantly promotes the cyclopalladation process and subsequent carbonylation.
 | ||
| Scheme 21 Pd(II)-catalyzed NH2 directed carbonylation of quaternary aromatic α-amino esters. | ||
Orito and co-workers synthesized dihydroisoquinolinones by palladium(II)-catalyzed direct carbonylation of arylethylamine using 5 mol% of Pd(OAc)2 and 50 mol% of Cu(OAc)2 in an CO atmosphere containing air. This is an example of a phosphine ligand-free protocol for carbonylation (Scheme 22).66
 | ||
| Scheme 22 Synthesis of dihydroisoquinolinones by phosphine ligand free Pd(II)-catalyzed direct carbonylation. | ||
A straightforward approach to quinoline-4-one derivatives has been reported based on a palladium-catalyzed cyclization–alkoxycarbonylation of 1-[(2-trimethylsilyl-ethynyl)phenyl]ureas (Scheme 23).67,68 The reactions are typically performed in a MeOH/CH3CN mixture (7:1, v/v) at 75 °C for 24 h, using 10 mol% of Pd/C as the catalyst in the presence of Bu4NI (1 equiv.) and KF (1.5 equiv.). The carbonylation is conducted under 2.4 MPa of a 3:1 mixture of CO and air, leading to the desired quinoline-4-one products in good efficiency.
 | ||
| Scheme 23 Synthesis of quinolin-4(1H)-one via Pd(II)-catalyzed oxidative carbonylation of 2-ethynylaniline. | ||
In 2013, Alper and co-workers reported a palladium-catalyzed oxidative cyclocarbonylation of N-monosubstituted-2-vinylanilines for the efficient synthesis of 2(1H)-quinolinones. The reaction was carried out in CH3CN at 110 °C for 20 h under 30 psi of CO and 10 psi of air, using a catalytic system composed of Pd(OAc)2 (10 mol%) and Cu(OAc)2 (50 mol%) (Scheme 24).69 A proposed mechanism for this transformation is illustrated in Scheme 25. The process was proposed to begin with coordination of the aniline nitrogen to an active Pd(II) species, forming a Pd–N bond with concomitant elimination of acetic acid. Subsequent coordination and insertion of CO generate a Pd-carbamoyl intermediate. Insertion of the vinyl group into the Pd–CO bond then forms an alkylpalladium intermediate, which undergoes β-hydride elimination to release the 2(1H)-quinolinone product. The resulting Pd(II) hydride species is reduced to Pd(0) via loss of acetic acid, and Pd(II) is regenerated by Cu(II) or molecular oxygen, thus completing the catalytic cycle. Alternatively, the reaction may proceed through initial attack of the electron-rich alkene on Pd(II) to form a pallacyclic intermediate, which could undergo reductive elimination to give the product. In this pathway, carbon monoxide insertion may occur either before or after palladacycle formation, highlighting the mechanistic flexibility of this oxidative cyclocarbonylation.
 | ||
| Scheme 24 Synthesis of 2-quinolinones via Pd(II)-catalyzed oxidative cyclocarbonylation of 2-vinylanilines. | ||
 | ||
| Scheme 25 Possible mechanism of oxidative cyclocarbonylation of 2-vinylaniniles to prepare 2(1H)-quinolinones. | ||
Recently, Gabriele and co-workers reported the PdI2/KI-catalyzed oxidative carbonylation of 4-(2-aminophenyl)-3-yn-1-ols for the selective synthesis of dihydrofuroquinolinone derivatives (Scheme 26).70 Their simple catalytic system, PdI2/KI,71 which has been successfully employed in various carbonylative heterocyclization processes,72 effectively promotes the sequential oxidative cyclization and cyclocarbonylation of suitably difunctionalized acetylenic substrates, leading to carbonylated polycyclic heterocycles. The group investigated the reactivity of 4-(2-aminophenyl)-3-yn-1-ols. Under the optimized conditions, 3,5-dihydrofuro[3,2-c]quinolin-4(2H)-one derivatives were obtained selectively in high yields. The optimized reaction conditions involved the use of MeOH as the solvent (substrate concentration = 0.10 mmol mL−1) at 100 °C for 24 h, in the presence of 0.33 mol% PdI2 and 3.3 mol% KI, under 60 atm of a 4:1 CO/air mixture. Mechanistically, an initial O-cyclization, involving an intramolecular 5-endo-dig nucleophilic attack of the hydroxyl group on the coordinated triple bond, forms a stable six-membered palladacycle intermediate, which then undergoes CO insertion followed by reductive elimination to obtain desired product (Scheme 27). Pd(0) is then reoxidised to catalytically active PdI2 by the action of O2 and the HI generated during the process.
 | ||
| Scheme 26 Synthesis of 3,5dihydrofuro[3,2-c]quinolin-4(2H)-ones by PdI2/KI-catalyzed oxidative carbonylative double cyclization of 4-(2-amino-phenyl)-3-yn-1-ols. | ||
 | ||
| Scheme 27 Mechanistic hypothesis for the selective formation of 3,5dihydrofuro[3,2-c]quinolin-4(2H)-ones. | ||
Della Cà and co-workers published a series of highly selective PdI2/KI-catalyzed oxidative carbonylation processes that provide rapid access to fused quinolinone frameworks and polyheterocyclic systems of remarkable structural complexity. These transformations are particularly noteworthy for their ability to enable the regioselective insertion of one to three molecules of carbon monoxide, while simultaneously promoting the formation of up to eight new bonds (one C–O, two C–C, and up to five C–N) in a single operation. A key feature of these reactions is their pronounced selectivity toward six-membered ring formation: indeed, the processes proceed through four sequential 6-endo-dig cyclizations. A detailed computational study clarified the origin of this site selectivity, revealing that both amides and ureas exhibit the same chemoselective behavior, reacting exclusively through O-cyclization. Building on previous results for Pd-catalyzed syntheses of indole-fused furanones73 and furofuranone derivatives74via double carbonylative cyclization cascades, the authors demonstrated that o-alkynylanilines bearing a secondary amino group, with R = COR4 or R = H, led to unexpected products. In the former case, the reaction selectively afforded oxazino[5,6-c]quinolin-5-ones (Scheme 28a), whereas substrates containing a free amino group provided, in a single operation, access to non-symmetrical fused polyheterocyclic architectures (Scheme 28b).75 The reactions were carried out in an autoclave using the PdI2 (1 mol%)/KI (10 mol%) catalytic system, under a CO pressure of 1.6 MPa and an air pressure of 0.4 MPa, in dry acetonitrile at 80 °C for 24 h. DFT calculations indicated that the key step involves the chemoselective nucleophilic attack of the amide carbonyl oxygen onto the Pd(II)-activated alkyne, via a 6-endo-dig cyclization leading to intermediates I–III. This pathway is strongly favored over alternative routes involving indolization followed by CO insertion and, most importantly, it completely suppresses the competing 5-exo-dig cyclization. Subsequent CO insertion and intramolecular nucleophilic displacement by the amine ultimately furnish the tricyclic product. Both electron-donating and electron-withdrawing substituents on the aromatic ring were well tolerated, as were ortho and meta substitutions. Even in the absence of substituents, the desired product was obtained in satisfactory yield. With regard to the amide R4 group, aryl substituents delivered the best results, whereas alkyl groups led to slightly lower yields.
 | ||
| Scheme 28 Pd(II) catalyzed carbonylation of ortho-alkynylanilines to oxazino[5,6-c]quinoline-5-ones (a) and condensed heterocycles (b). | ||
An even more striking behavior emerged from the investigation of substrates bearing a free amino group in the α-position relative to the triple bond. Under the same standard conditions, the formation of a condensed polyheterocyclic structure arising from a triple carbonylative cascade was observed. In a single operation, eight new bonds and four fused heterocycles are formed, with good yields obtained in the presence of both electron-donating and electron-withdrawing substituents. The mechanisms leading to these compounds were proposed in Scheme 29, paths a and b. The most favorable pathway involves the chemoselective nucleophilic attack of the amide oxygen onto the palladium-activated alkyne via a 6-endo-dig cyclization, affording the σ-vinylpalladium complex III. After CO insertion and formation of intermediate IV, a second cyclization occurs, delivering the final product along with Pd(0), which is subsequently reoxidized (Scheme 29, path a). By contrast, in the presence of a primary amine (R = H; Scheme 29, path b), the formation of a symmetric urea catalyzed by palladium is likely, via an isocyanate intermediate. This highly reactive, multifunctional urea can undergo a sequence of 6-endo-dig O-cyclization, CO insertion, and reductive elimination. A subsequent nucleophilic attack of the iminic nitrogen of the isourea moiety onto the second alkyne generates palladium complex VIII, again following a 6-endo-dig pathway. Sequential insertion of a third CO molecule ultimately leads to the final cyclization step, yielding the target compound and regenerating Pd(0). Notably, it was demonstrated that a non-symmetrical urea exclusively furnishes the O-cyclization product, with no evidence of competing N-cyclization processes.
 | ||
| Scheme 29 Proposed pathways to formation of oxazino[5,6-c]quinoline-5-ones (a) and condensed heterocycles (b). | ||
Years later, Nicola Della Cà and colleagues presented a Pd-catalyzed carbonylative cyclization of o-alkynyl anilines bearing an amide functionality, enabling the efficient synthesis of fused oxazino-quinolinone derivatives under mild conditions. Notably, the transformation proceeds at room temperature and atmospheric CO pressure, employing 1,4-benzoquinone (BQ) as an external oxidant and affords the desired tricyclic heterocycles in good to excellent yields.76 A key strength of this methodology lies in its compatibility with CO surrogates, in particular benzene-1,3,5-triyl triformate (TFBen) and the recently developed calix[6]arene hexaformates (CLX[6]CO). Both systems are capable of releasing carbon monoxide in situ without the need for external activators. Notably, calix[6]arene hexaformates are reported for the first time as solid and recoverable carbonylating agents, representing a significant advance in the sustainability of carbonylation chemistry (Scheme 30). An N-benzyl aniline bearing a propargylamide moiety in the ortho position was selected as the model substrate. After extensive optimization, the optimal reaction conditions—Pd(OAc)2 (5 mol%), 1,10-phenanthroline (5 mol%), 1,4-benzoquinone (1.2 equiv.), CO (1 atm, balloon), in acetonitrile at room temperature for 6 h—led to complete conversion, highlighting the high efficiency of the catalytic system (Scheme 30 method A). Building on these results, the use of TFBen as a CO surrogate was explored. Owing to the presence of three formate groups, TFBen can theoretically release three equivalents of CO, making it a particularly attractive alternative to monoformates. Under these conditions, the yield increased to 95% when the reaction was conducted at 80 °C for 24 h under an inert atmosphere (N2) (Scheme 30 method B). A wide range of electron-donating and electron-withdrawing substituents on the benzyl fragment (R1) were well tolerated. Likewise, substituents of different nature in the para (Me, i-Pr, F, Br), meta (Cl), and ortho (Me) positions relative to the aniline nitrogen showed excellent compatibility. In contrast, strongly electron-withdrawing groups such as CF3 and CO2Me, which reduce the nucleophilicity of the nitrogen atom, resulted in diminished product formation. Variation of the amide fragment was also well accommodated, with para-substituted aryl groups affording the desired products in good yields. Importantly, the introduction of heteroaryl fragments was fully compatible with the carbonylation conditions.
 | ||
| Scheme 30 Synthesis of fused oxazino-quinolinone derivatives under mild conditions. | ||
With the aim of identifying new and efficient CO surrogates, a series of calix[6]arene hexaformates (CLX[6]CO) were prepared. These compounds are characterized by high chemical and thermal stability, allowing long-term storage even at room temperature; they have six formate groups capable of releasing up to six CO molecules; easy recovery of the decarbonylated material due to the low solubility of calix[6]arenes in organic solvents, enabling isolation by simple filtration; and tunability of the CO-release rate by modification of the para-substituents on the aromatic rings. The synthesized CLX[6]CO derivatives were found to be highly insoluble in acetonitrile, while they dissolve in 1,4-dioxane upon heating, a solvent commonly employed in carbonylation reactions.77 In contrast to TFBen, CLX[6]CO does not generate CO in situ upon simple heating. Therefore, under the optimized conditions used for the previous methodology—Pd(OAc)2 (5 mol%), 1,10-phenanthroline (5 mol%), 1,4-benzoquinone (1.2 equiv.), 80 °C, 24 h, N2—a solvent switch from acetonitrile to 1,4-dioxane, along with the addition of a base (Et3N), was required to promote decarbonylation at moderate temperatures (Scheme 30 method C). These conditions afforded the desired product together with trace amounts of a decarbonylated by-product. A clear substituent effect was observed: the tert-butyl-substituted derivative proved less efficient, whereas the brominated surrogate delivered synthetically useful yields (58%) with high selectivity. Although the yield is lower than that obtained with TFBen (≈90%), the principal advantage of CLX[6]CO lies in the facile separation and recovery of the decarbonylated by-product by simple filtration. The proposed reaction mechanism (Scheme 31) involves initial coordination of the substrate alkyne to Pd(II), forming complex I. The most favorable pathway proceeds through nucleophilic attack of the amide oxygen onto the activated triple bond via a 6-endo-dig cyclization, leading to the formation of the σ-vinyl-palladium intermediate II. Carbon monoxide, thermally generated from TFBen (or in situ from CLX[6]CO), subsequently enters the palladium coordination sphere to form complex III. Subsequent CO migratory insertion, followed by reductive elimination or nucleophilic displacement, affords the fused oxazino-quinolinone product along with a Pd(0) species. Finally, oxidation of Pd(0) by 1,4-benzoquinone regenerates the catalytically active Pd(II) species.
 | ||
| Scheme 31 Proposed reaction pathway for Pd(II)-catalyzed carbonylative cyclization of o-alkynyl anilines. | ||
4. Synthesis of quinolinones by carbonylation reactions catalyzed by other transition metals
In 2015, Jiao and coworkers reported an efficient Rh-catalyzed carbonylative annulation of simple anilines with CO and internal alkynes, providing direct access to 2-quinolinones. The transformation was carried out using Cu(OAc)2 as the terminal oxidant and Li2CO3 as the base (Scheme 32).78 During the initial optimization studies, several Pd-based catalysts, usually effective in C–H activation and aniline carbonylation chemistry, were evaluated in xylene under 1 atm of CO. However, all of these systems proved completely ineffective. By contrast, the use of the Wilkinson catalyst, Rh(PPh3)3Cl, in the presence of 0.5 equiv. of Li2CO3 enabled the smooth formation of the desired 2-quinolinone derivatives, with yields reaching up to 95%. The scope of the reaction with respect to the alkyne component was then examined. A broad range of internal alkynes could be employed, and both aliphatic and aromatic substrates were well tolerated under the optimized conditions, highlighting the versatility of the method. Based on experimental observations, the authors proposed a plausible catalytic cycle, shown in Scheme 33. Initially, oxidation of the Rh(I) precatalyst by Cu(OAc)2 generates the active Rh(III) species A, which upon ligand exchange with CO affords the Rh(III)–CO complex B. Coordination of the aniline substrate to B, followed by CO insertion, leads to the Rh(III) intermediate C. A subsequent concerted metalation–deprotonation step gives rise to the key rhodacyclic intermediate D. Ligand exchange between D and the internal alkyne furnishes the Rh(III)–alkyne complex E, which undergoes alkyne insertion to form the seven-membered rhodacycle F. Final reductive elimination from F delivers the 2-quinolinone product, while the resulting Rh(I) species is reoxidized by Cu(OAc)2 to regenerate the catalytically active Rh(III) complex A, thus closing the catalytic cycle. | ||
| Scheme 32 Rh-catalyzed construction of 2-quinolinone derivatives via C–H annulation of anilines with alkynes and CO. | ||
 | ||
| Scheme 33 Proposed mechanism for the synthesis of quinoline-2(1H)-ones. | ||
In 2017, Jiao and co-workers further expanded this chemistry by reporting a Rh-catalyzed aerobic oxidative approach to 2-quinolones from anilines, internal alkynes, and carbon monoxide. In this protocol, the dimeric complex [Rh(CO)2Cl]2 was employed as the catalyst, with Cu(OPiv)2 serving as a key additive under an atmosphere of CO and O2 (Scheme 34).79 Despite the well-known sensitivity of amines toward strong oxidants, both primary and tertiary anilines could be successfully engaged in this Rh-catalyzed aerobic oxidative cyclization. Overall, anilines bearing either electron-donating or electron-withdrawing substituents are well tolerated, demonstrating a broad substrate scope. Notably, the selected shown example, a known potential p38α MAP kinase inhibitor, could be directly assembled in synthetically useful yield using this methodology (Scheme 34b). A plausible catalytic cycle was proposed in which the Rh(I) species is first oxidized to Rh(III) by Cu(II). The resulting Cu(I) species is then reoxidized to Cu(II) by molecular oxygen in the presence of carbon monoxide, thereby sustaining the overall redox balance of the system. Density functional theory (DFT) calculations indicated that a rhodium catalyst is essential for an efficient transformation; alternative metals such as palladium or iridium were found to be ineffective due to prohibitively high energy barriers along the reaction pathway.
 | ||
| Scheme 34 Rh-catalyzed aerobic oxidative cyclization of anilines with alkynes. It should be noted that the reaction conditions (CO/O2 2/1 or 4/1 at 130 °C) are potentially explosive [C. M. Bartish, G. M. Drissel, in Kirk-Othmer Encyclopedia of Chemical Technology, 3rd ed., vol. 4, p. 774 (M. Grayson, D. Eckroth, G. J. Bushey, L. Campbell, A. Klingsberg, L. van Nes, eds.), Wiley, New York, 1978]. | ||
During optimization of the catalytic system, the ratio of CO to O2 was found to be crucial. The optimal CO/O2 ratio was 2:1 (1 atm total pressure) for tertiary anilines, whereas a higher ratio of 4:1 (1 atm) was required for primary and secondary anilines. It has to be noted, however, that these mixture are potentially explosive, as the flammability range of CO in pure O2 is 16.7–93.5% at room temperature and it becomes even larger at higher temperatures [See: C. M. Bartish, G. M. Drissel, in Kirk-Othmer Encyclopedia of Chemical Technology, 3rd ed., vol. 4, p. 774 (M. Grayson, D. Eckroth, G. J. Bushey, L. Campbell, A. Klingsberg, L. van Nes, eds.), Wiley, New York, 1978]. It is also noteworthy that the addition of a catalytic amount of dppb was necessary when tertiary anilines were used, further underscoring the fine balance between ligand environment and substrate class in this aerobic oxidative process.
In 2016, the Wu group disclosed the first example of an iridium-catalyzed carbonylative annulation of simple anilines with internal alkynes, enabling the synthesis of quinolin-2(1H)-one derivatives (Scheme 35).80 The reaction was carried out with a catalytic amount of [Ir(COD)Cl]2 in xylene at 120 °C under a CO atmosphere (30 bar), using Cu(OAc)2 (2 equiv.) as the terminal oxidant. During optimization, a range of ligands and silver salts were evaluated as additives, including AgBF4, AgSbF6, AgOTf, and AgOOCCF3. Among these, sodium trifluoroacetate (0.5 equiv.) proved to be the most effective additive, leading to the highest efficiencies. It is worth noting that, under these conditions, terminal alkynes were not suitable substrates, and only internal alkynes participated successfully in the annulation process. A plausible catalytic cycle for this transformation is outlined in Scheme 36. The process begins with oxidation of the Ir(I) precatalyst to an Ir(III) species, which then enters the productive catalytic cycle. Ligand exchange with the aniline substrate generates a nitrogen-coordinated iridium intermediate. Subsequent coordination and insertion of CO affords an iridium formamide species, which undergoes ortho C–H bond activation to form a five-membered iridacycle. The internal alkyne then coordinates to the metal center and inserts into the Ir–C bond, giving rise to a seven-membered iridacyclic intermediate. Finally, reductive elimination furnishes the quinolin-2(1H)-one product, while the resulting Ir(I) species is reoxidized to Ir(III) by Cu(OAc)2, thereby completing the catalytic cycle.
 | ||
| Scheme 35 Synthesis of 2-quinolinones by an Ir-catalyzed three component reaction. | ||
 | ||
| Scheme 36 Proposed reaction mechanism of quinolin-2(1H)-ones from internal alkynes and anilines. | ||
5. Synthesis of quinolinone derivatives through metal-catalyzed carbonylation reactions using CO surrogates
5.1. Mo(CO)6 as the carbon monoxide source
The insertion of a carbon monoxide molecule, which represents a fundamental C1 building block in organic and inorganic chemistry, is generally achieved using high CO pressure but nowadays the severe toxicity and flammability of this gas have led to the use of CO surrogates which are non-toxic and easy to handle. Many different surrogates have been evaluated according to their nature and their mechanistic aspects. A search in the literature revealed that several compounds like formic acid,81 formats,82 chloroform,83 aldehydes84 and other CO surrogates85 have been used as CO sources. In addition, metal carbonyls have been investigated as CO-releasing agents in carbonylative reactions.86 The first example of metal carbonyls employed as CO delivering agents was described by the Larhed group in 2002.87 As a solid CO source, the authors took into consideration different metal carbonyls, such as [Cr(CO)6], [Mo(CO)6], [Ni(CO)4], [Fe(CO)5], and [Co2(CO)8]. While [Ni(CO)4] is known to be highly toxic, and [Fe(CO)5] and [Co2(CO)8] are almost inactive, [Cr(CO)6] and [Mo(CO)6] are the most promising candidates. In fact, Mo(CO)6 is one of the most efficient surrogates since it does not require acidic conditions, it can release up to six molecules of carbon monoxide and generates metal molybdenum as the main by-product.In 2018, Ghosh and co-workers reported a CO-gas-free strategy for the synthesis of 4-quinolones based on a carbonylative Sonogashira annulation sequence catalyzed by a Pd–NHC complex (Scheme 37).88 In this approach, substituted 2-iodoanilines were coupled with a variety of acetylenes, using a Pd–NHC catalyst and Mo(CO)6 as an in situ carbon monoxide source at 95 °C under a nitrogen atmosphere. Notably, a broad range of 2-iodoaniline derivatives performed equally well in the carbonylative Sonogashira annulation, affording the corresponding 4-quinolones in good to excellent yields. Under the optimized conditions [Pd–NHC (2 mol%), Me2NH (4 equiv.), and Mo(CO)6 (2 equiv.) in DMF at 95 °C] the coupling of 2-iodoaniline with phenylacetylene proceeded smoothly, delivering 2-phenyl-4-quinolone in 94% yield after 15 h.
 | ||
| Scheme 37 Synthesis of 4-quinolinones by Pd-catalyzed using Mo(CO)6 as the CO source. | ||
In 2021, the Wu group developed a new palladium-catalyzed carbonylative cyclization of benzyl chlorides with o-nitrobenzaldehydes, providing an efficient route to 3-arylquinolin-2(1H)-ones (Scheme 38).89 In this transformation, Mo(CO)6 plays a dual role, acting both as a carbon monoxide surrogate and as a reductant. During optimization, a range of ligands, bases, and palladium catalysts were evaluated, and Pd(OAc)2 emerged as the optimal catalyst. Under the optimized conditions, prolonging the reaction time to 30 h allowed the desired product to be isolated in up to 92% yield. With these conditions in hand, the scope and generality of the method were systematically investigated. A variety of benzyl chlorides and o-nitrobenzaldehydes could be successfully employed, furnishing the corresponding quinolin-2(1H)-one derivatives in moderate to excellent yields, thereby demonstrating the robustness of this carbonylative protocol. A plausible reaction mechanism is depicted in Scheme 39. The catalytic cycle begins with oxidative addition of Pd0Ln to benzyl chloride, affording a benzyl–palladium intermediate A. Subsequent coordination and insertion of CO (released in situ from Mo(CO)6), generates the acylpalladium complex B. In parallel, o-nitrobenzaldehyde is reduced by Mo(CO)6 in the presence of water to give o-aminobenzaldehyde, which then reacts with acylpalladium intermediate B to form intermediate C, with concomitant release of HCl that is neutralized by DBU. Reductive elimination from intermediate C furnishes intermediate D and regenerates the Pd0Ln species for the next catalytic cycle. Finally, intramolecular condensation of intermediate D, assisted by DBU, delivers the target 3-arylquinolin-2(1H)-ones.
 | ||
| Scheme 38 Pd-catalyzed carbonylative synthesis of 3-arylquinolin-2(1H)-ones from benzyl chlorides, o-nitrobenzaldehydes and Mo(CO)6 as the CO source. | ||
 | ||
| Scheme 39 Possible reaction mechanism for the preparation of 3-arylquinolin-2(1H)-ones. | ||
In 2014, Kashania and co-workers reported an efficient palladium-catalyzed carbonylative annulation strategy for the synthesis of (dihydro)quinolones from readily available 2-iodoanilines, internal alkynes or alkenes, with Mo(CO)6 as a solid carbon monoxide source (Scheme 40).90 This ligand-free protocol enables the construction of biologically relevant (dihydro)quinolin-2(1H)-one frameworks under relatively simple catalytic conditions, while avoiding the practical and safety issues associated with the use of gaseous CO. Under the optimized reaction conditions [o-iodoaniline, internal alkynes or alkenes (5 equiv.), pyridine (3 equiv.), Pd(OAc)2 (10 mol%), and Mo(CO)6 (1.5 equiv.) in DMF at 160 °C for 12 h] the corresponding 3,4-disubstituted (dihydro)quinolin-2(1H)-ones were obtained in good yields. The combination of operational simplicity, ligand-free catalysis, and the use of a bench-stable CO surrogate highlights the practicality of this approach for assembling structurally diverse quinolinone derivatives.
 | ||
| Scheme 40 Synthesis of 2(1H)-quinolones by carbonylative annulation of unsaturated compounds using molybdenum hexacarbonyl. | ||
In 2015, Larhed and co-workers reported a palladium-catalyzed, CO-gas-free carbonylative Sonogashira/cyclization sequence for the synthesis of functionalized 4-quinolones from 2-iodoanilines and alkynes (Scheme 41).91 Two complementary protocols were developed, both relying on molybdenum hexacarbonyl as a solid and convenient source of carbon monoxide. In protocol A, the carbonylative coupling and cyclization occur rapidly under microwave (MW) irradiation, delivering the cyclized 4-quinolone products after only 20 min at 120 °C. In contrast, protocol B operates as a gas-free, one-pot, two-step process at room temperature, which enables the use of more sensitive functional groups, such as nitro and bromide substituents, that might not tolerate elevated temperatures or microwave conditions. Both approaches exhibit a broad substrate scope. A wide range of 2-iodoanilines and alkynes can be employed, including electron-rich and electron-poor anilines, as well as aryl, aliphatic, and heterocyclic alkynes, affording the corresponding quinolone derivatives in moderate to good yields. The operational simplicity, functional group tolerance, and avoidance of gaseous CO make these protocols particularly attractive for the modular synthesis of structurally diverse 4-quinolones.
 | ||
| Scheme 41 Synthesis of 4-quinolones via carbonylative Sonogashira cross-coupling using molybdenum hexacarbonyl as the CO source. | ||
In 2022, Wu and co-workers reported the first carbonylative synthesis of 3-alkenylquinolin-2(1H)-ones from o-iodophenol-derived allyl ethers, using o-nitrobenzaldehydes as nitrogen sources under relatively mild conditions (Scheme 42).92 In this protocol, Mo(CO)6 served dually as the CO surrogate and reductant, enabling the efficient formation of a broad range of 3-alkenylquinolin-2(1H)-ones in good to excellent yields. Initial optimization using Pd(OAc)2 as the catalyst, RuPhos as the ligand, and Cs2CO3 as the base in CH3CN at 100 °C for 30 h afforded the model product in only low yield. Solvent screening showed that toluene, DMF, and 1,4-dioxane led to trace or no product, whereas DMSO provided the desired product in 10% yield. Evaluation of different bases (Na2CO3, K2CO3, DBU, Et3N) did not significantly improve the outcome. However, the addition of 3 equiv. of tetrabutylammonium iodide (TBAI) dramatically increased the yield to 99%, highlighting its crucial role. The generality of the optimized protocol was then explored with a variety of o-iodophenol-derived allyl ethers. Substrates bearing alkyl substituents (linear, branched, or cyclic) underwent smooth conversion to the corresponding quinolinones in consistently high yields. Aryl-substituted allyl ethers were also well tolerated, regardless of whether the aromatic ring carried electron-donating or electron-withdrawing groups such as cyano or trifluoromethyl. Importantly, halogen substituents (F, Cl) remained intact throughout the transformation, providing the desired quinolinones in high yields. The scope was further extended to a range of o-nitrobenzaldehydes, which performed well under the standard conditions.
 | ||
| Scheme 42 Palladium-catalyzed reductive aminocarbonylation of o-iodophenol-derived allyl ethers with o-nitrobenzaldehydes to 3-alkenylquinolin 2(1H)-ones. | ||
In 2014, Wu and co-workers reported a palladium-catalyzed carbonylative [3+2+1] annulation of N-arylpyridin-2-amines with internal alkynes to access 2-quinolinones under CO-gas-free conditions, using Mo(CO)6 as a convenient solid CO surrogate (Scheme 43).93 A broad range of internal alkynes was examined. Alkynes bearing electron-donating substituents on the aromatic ring reacted smoothly, while those with electron-withdrawing groups—including F, Br, acetyl, and trifluoromethyl—were also well tolerated, affording the corresponding 2-quinolinones in good yields. The scope of N-arylpyridin-2-amines was also broad. Substrates featuring electron-donating groups on the aniline moiety generally performed well, providing the annulated products efficiently. A plausible catalytic mechanism, proposed by the authors, is outlined in Scheme 44. The reaction is initiated by coordination of the pyridine ring of the substrate to Pd(II), followed by C–H activation to form intermediate A. Subsequent insertion of the internal alkyne generates intermediate B. Coordination and migratory insertion of CO (released in situ from Mo(CO)6) into B affords intermediates C or C′. Reductive elimination from these intermediates furnishes the desired 2-quinolinone product and generates Pd(0). The active Pd(II) species is then regenerated via oxidation of Pd(0) by benzoquinone (BQ) and/or AgOAc, completing the catalytic cycle. This CO-gas-free protocol provides a practical and versatile route to 2-quinolinones, accommodating a variety of functional groups on both the alkyne and the N-arylpyridin-2-amine substrate.
 | ||
| Scheme 43 Synthesis of 2-quinolinones via Pd-catalyzed carbonylative [3+2+1] annulation of internal alkynes. | ||
 | ||
| Scheme 44 Proposed mechanism for the synthesis of 2-quinolinones. | ||
In 2022, Wu and co-workers reported a palladium-catalyzed carbonylative cyclization of 2-bromonitrobenzenes with alkynes for the synthesis of quinolin-4(1H)-one scaffolds, using Pd supported on graphitic carbon nitride (Pd/g-C3N4) as a heterogeneous catalyst (Scheme 45).94 Employing a low loading of Pd/g-C3N4, Mo(CO)6 as both CO surrogate and reductant, and nitroarenes as the nitrogen source, the reaction proceeded efficiently, delivering a range of quinolin-4(1H)-ones in good to excellent yields. The substrate scope included various 2-bromonitrobenzenes and alkynes bearing para-, meta-, and ortho-substituents with both electron-donating and electron-withdrawing groups, all of which afforded the corresponding quinolone derivatives in excellent yields. In line with the green chemistry principles, the reusability of the catalytic system was evaluated. Under the optimized conditions, 0.5% Pd/g-C3N4 and 10% Pd/C were tested in the reaction of 2-bromonitrobenzene with phenylacetylene. Remarkably, the use of 0.5% Pd/g-C3N4 catalyst retained high activity even after five cycles, whereas the Pd/C catalyst lost its activity completely after a single cycle, highlighting the advantage of the graphitic carbon nitride support.
 | ||
| Scheme 45 Supported Pd-catalyzed carbonylative cyclization of 2-bromonitrobenzenes, alkynes and Mo(CO)6 to access quinolin-4(1H)-ones. | ||
6. Other carbon monoxide sources
In 2018, Das and co-workers reported a multicomponent strategy for the regioselective synthesis of 3-substituted 2-quinolones, based on the coupling of 2-iodoaniline derivatives, terminal alkynes, and (CO2H)2·2H2O as a bench-stable carbon monoxide source under microwave irradiation (Scheme 46).95 The transformation is catalyzed by recyclable heterogeneous Pd@PS nanoparticles, offering an attractive combination of operational simplicity and sustainability. | ||
| Scheme 46 Regio-selective synthesis of 3-aryl/alkyl-2-quinolones. | ||
The Pd@PS catalyst was prepared from Pd(OAc)2 using Amberlite IRA 900 Cl− resin as the polystyrene (PS) support. A range of solvents was evaluated, with DMF emerging as the most suitable medium. Further optimization involved screening both organic and inorganic bases. Inorganic bases were ineffective, whereas organic bases such as Et3N and tetrabutylammonium iodide (TBAI) showed comparable activity. TBAI was ultimately preferred because of its broader substrate scope and its ability to selectively afford 3-substituted 2-quinolones as the major products, with only negligible formation of 4-quinolone isomers. Other palladium catalysts, including Pd(OAc)2, Pd/C, and Pd/Al2O3, were also examined but proved inferior. The optimal conditions consisted of 2-iodoaniline (1 equiv.), phenylacetylene (2.5 equiv.), TBAI (2 equiv.), Pd@PS (5 mol%), and (CO2H)2·2H2O (4 equiv.) in DMF at 135 °C, delivering the desired regioselective product in up to 73% yield.
Recently, in 2024, Zhang and co-workers reported an efficient and mild protocol for the synthesis of 4-quinolone derivatives that integrates controlled CO release with palladium catalysis and base-promoted cyclization (Scheme 47).96 This strategy combines three key processes: CO liberation from Fe(CO)5, Pd-catalyzed carbonylative Sonogashira coupling, and intramolecular cyclization enabled by a dual-base system consisting of piperazine and triethylamine. Iron pentacarbonyl (Fe(CO)5) serves as a practical liquid CO source in this transformation. Owing to its controlled and tunable CO-releasing behavior, Fe(CO)5 offers several advantages, including safe syringe-based handling, formation of non-toxic byproducts, and a high CO utilization efficiency. As a result, this liquid CO surrogate effectively supports palladium-catalyzed carbonylative C–C bond formation followed by intramolecular cyclization. The overall process proceeds through a three-component condensation of sub-stoichiometric amounts of Fe(CO)5 with 2-iodoanilines and terminal alkynes. As illustrated in Scheme 48, the reaction can be divided into two interconnected stages: the Pd-catalyzed carbonylative Sonogashira coupling (left) and the subsequent cyclization of the ynone intermediate (right). In the first catalytic cycle, mechanistic studies indicate that oxidative addition of 2-iodoaniline to the active Xantphos–Pd(0) species A affords palladium complex B. Intermediate B then reacts with CO generated in situ from the Fe(CO)5/triethylamine system to give the acylpalladium species C. Subsequent nucleophilic attack of phenylacetylene, assisted by triethylamine, forms acylpalladium intermediate D, which undergoes reductive elimination to furnish the carbonylated ynone intermediate E. In the second stage, piperazine promotes intramolecular Michael addition of the 2-aminoynone E to afford intermediate F, which further evolves into the cyclic enolate G. Final elimination of the piperazine moiety from either F or G delivers the desired 4-quinolone product, thus completing the transformation.
 | ||
| Scheme 47 Synthesis of 4(1H)-quinolones via non-gaseous carbonylative Sonogashira cyclization using Fe(CO)5. | ||
 | ||
| Scheme 48 Proposed mechanism for Pd-catalyzed synthesis of 4(1H)-quinolones. | ||
It is well established that formic acid can decompose into carbon monoxide and water at elevated temperatures under acidic conditions.97 Building on this concept, Ferretti and co-workers have explored the use of formate derivatives as CO surrogates for the synthesis of heterocyclic compounds. In a more recent contribution, they demonstrated that a HCO2H/Ac2O mixture can be efficiently employed as a CO source for the synthesis of 4-quinolones via the reductive cyclization of 2′-nitrochalcones, thereby overcoming the drawback associated with phenol formation observed with other formate-based surrogates (Scheme 49).98 In this methodology, the active catalyst is generated in situ from palladium and 1,10-phenanthroline (Phen) as the ligand. In the presence of the HCO2H/Ac2O system, a wide range of 2′-nitrochalcones could be smoothly converted into the corresponding 4-quinolones in high yields, with acetic acid and CO2 as the only stoichiometric byproducts. Notably, higher selectivity was achieved when acetonitrile was used as the solvent at the same reaction temperature (130 °C). Inspired by the structure of graveoline, they also applied this protocol to its synthesis, successfully obtaining the natural product in high yield (88%) (Scheme 50). Graveoline is an alkaloid isolated from Ruta graveolens L.99,100 and is known to exhibit a range of pharmacological activities,101 including the ability to induce autophagy in skin melanoma cancer cells.102
 | ||
| Scheme 49 Synthesis of 4-quinolones by reductive cyclization of 2'-nitrochalcones. | ||
 | ||
| Scheme 50 Synthesis of graveoline. | ||
A plausible mechanism for the reductive cyclization of o-nitrostyrenes to indole derivatives has been proposed and extended to the quinolone system (Scheme 51). The process begins with coordination of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the nitrostyrene moiety to palladium, bringing the olefin into close proximity to the metal center and contributing to the observed selectivity. Following coordination, the nitro group is reduced to a nitrosoarene, which can undergo intramolecular reaction with the olefinic moiety. The observation of N-hydroxyquinolones when palladium is used without added ligands strongly supports cyclization at this stage. An alternative pathway involving full reduction of the nitro group to an aniline, followed by Michael addition to give a dihydroquinolone and subsequent oxidation, appears unlikely. Indeed, previous studies have shown that only the quinolone derived directly from the nitrochalcone is formed under similar conditions, effectively ruling out an aniline-based mechanism. In the final step, the N-hydroxyquinolone undergoes deoxygenation by CO, a transformation known to be catalyzed by palladium complexes capable of reducing nitroarenes and therefore expected to proceed readily under these conditions. Although imido complexes are often proposed as intermediates in nitroarene cyclization reactions, no solid experimental evidence supports their involvement here, and this possibility was not considered further. Overall, this study introduced an efficient and practical protocol for the synthesis of 2-aryl-4-quinolones that exploits carbon monoxide as a reducing agent without requiring gaseous CO. Instead, inexpensive formic acid activated by acetic anhydride serves as a convenient CO surrogate. The reaction could be performed in simple pressure tubes or flasks of various sizes, and the only stoichiometric byproducts (CO2 and acetic acid) are easily removed. The method tolerates a broad range of substituents and delivers higher isolated yields than analogous reactions conducted under pressurized CO, underscoring the effectiveness of this CO-surrogate strategy.
C double bond of the nitrostyrene moiety to palladium, bringing the olefin into close proximity to the metal center and contributing to the observed selectivity. Following coordination, the nitro group is reduced to a nitrosoarene, which can undergo intramolecular reaction with the olefinic moiety. The observation of N-hydroxyquinolones when palladium is used without added ligands strongly supports cyclization at this stage. An alternative pathway involving full reduction of the nitro group to an aniline, followed by Michael addition to give a dihydroquinolone and subsequent oxidation, appears unlikely. Indeed, previous studies have shown that only the quinolone derived directly from the nitrochalcone is formed under similar conditions, effectively ruling out an aniline-based mechanism. In the final step, the N-hydroxyquinolone undergoes deoxygenation by CO, a transformation known to be catalyzed by palladium complexes capable of reducing nitroarenes and therefore expected to proceed readily under these conditions. Although imido complexes are often proposed as intermediates in nitroarene cyclization reactions, no solid experimental evidence supports their involvement here, and this possibility was not considered further. Overall, this study introduced an efficient and practical protocol for the synthesis of 2-aryl-4-quinolones that exploits carbon monoxide as a reducing agent without requiring gaseous CO. Instead, inexpensive formic acid activated by acetic anhydride serves as a convenient CO surrogate. The reaction could be performed in simple pressure tubes or flasks of various sizes, and the only stoichiometric byproducts (CO2 and acetic acid) are easily removed. The method tolerates a broad range of substituents and delivers higher isolated yields than analogous reactions conducted under pressurized CO, underscoring the effectiveness of this CO-surrogate strategy.
 | ||
| Scheme 51 Proposed reaction mechanism for the synthesis of 4-quinolones using formic acid as a CO surrogate. | ||
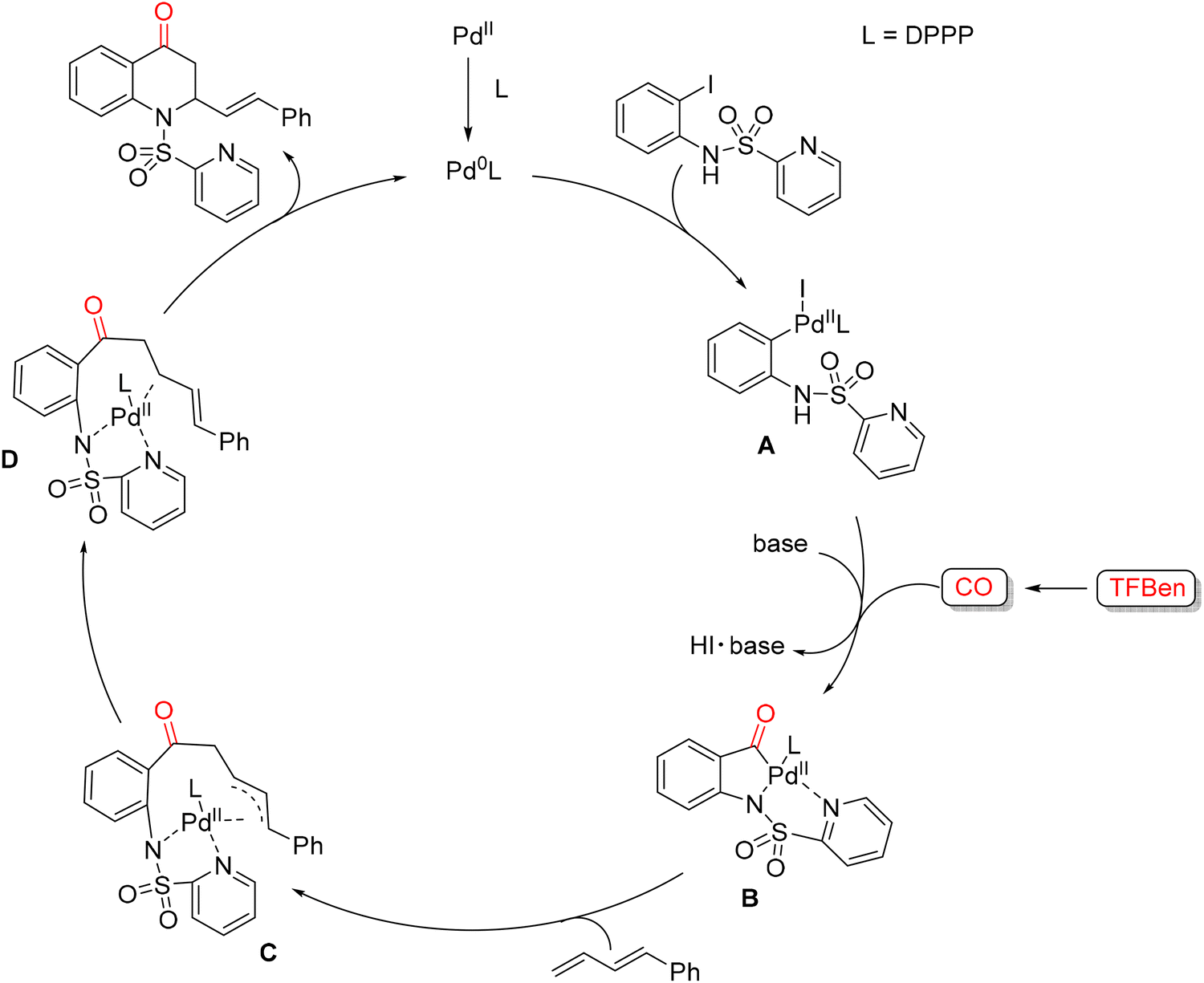
In 2021, Wu and co-workers reported a palladium-catalyzed 1,2-amino carbonylation of 1,3-dienes with (N-SO2Py)-2-iodoanilines for the synthesis of 2,3-dihydroquinolin-4(1H)-one scaffolds (Scheme 52).103 This methodology employs benzene-1,3,5-triyl tri-formate (TFBen)104 as a CO source, developed by the authors, and utilizes the N-SO2Py moiety as a directing group to facilitate selective carbonylation. Under these conditions, a variety of 2,3-dihydroquinolin-4(1H)-ones were obtained in good yields, up to 88%. Optimization studies began with the model reaction between (N-SO2Py)-2-iodoaniline and diene in the presence of Pd(OAc)2 and various phosphine ligands at 110 °C for 24 h. Among the ligands tested, DPPP gave the desired product in 39% yield. Screening different palladium sources revealed that Pd(TFA)2 significantly enhanced the yield to 60%. Solvent evaluation showed that toluene, dioxane, MeCN, and DMSO performed poorly, while the initially used solvent gave the best results. Extending the reaction time further improved the yield to 79%, and subsequent base screening identified K3PO4 as optimal, affording the product in 84% yield. Lowering the catalyst loading, however, led to diminished efficiency. With the optimized conditions established, the authors explored the substrate scope. Various (N-SO2Py)-2-iodoanilines bearing electron-donating or electron-withdrawing substituents were well tolerated, providing products in 86% yields. A broad range of 1,3-dienes was also compatible: phenyl-substituted dienes with para-electron-donating or -withdrawing groups, meta-substituted dienes and even sterically hindered ortho-substituted dienes smoothly delivered products in high yields. The crucial role of the directing group was highlighted by control experiments. Replacement of the N-SO2Py group with N-benzenesulfonyl-2-iodoaniline led to complex mixtures, demonstrating the importance of the pyridyl nitrogen. Furthermore, treatment of 1 with Pd(TFA)2 (1.0 equiv.) and PPh3 (2.0 equiv.) under standard conditions generated the acylpalladium intermediate in 98% yield, supporting the proposed catalytic sequence. Based on these observations, a plausible catalytic mechanism was proposed (Scheme 53). Pd(0), generated in situ from Pd(TFA)2 and DPPP, undergoes oxidative addition of (N-SO2Py)-2-iodoaniline to form Pd(II) complex A. CO, released from TFBen in the presence of a base, inserts into A to generate the acylpalladium intermediate B. Subsequent insertion of the 1,3-diene produces a π-allyl-Pd(II) intermediate C, which rearranges to Pd(II) complex D. Reductive elimination from D then furnishes the 2,3-dihydroquinolin-4(1H)-one product and regenerates Pd(0), completing the catalytic cycle.
 | ||
| Scheme 52 Palladium-catalyzed 1,2-amino carbonylation of 1,3-dienes with (N-SO2Py)-2-iodoanilines: 2,3-dihydroquinolin-4(1H)-ones synthesis. | ||
 | ||
| Scheme 53 Proposed mechanism for Palladium-catalyzed 1,2-amino carbonylation of 1,3-dienes with (N-SO2Py)-2-iodoanilines. | ||
In 2022, a cobalt-catalyzed cyclization of N-(2-vinylphenyl)picolinamides with carbon monoxide was developed by Wu and co-workers for the synthesis of (NH)-quinolin-2(1H)-ones.105 Good yields of the desired products were formed with benzene-1,3,5-triyl triformate (TFBen) as the CO source. The picolinamide was removed after the reaction to give (NH)-quinolin-2(1H)-ones.
7. Conclusions
In this review, we summarized the pivotal role of carbon monoxide as a versatile C1 building block in promoting a wide range of transformations for the synthesis of quinolinone scaffolds. Various synthetic strategies were discussed, including approaches based on both homogeneous and heterogeneous catalysis, often operating under mild conditions or even at room temperature. Particular attention was devoted to palladium-catalyzed methodologies, which represent the most extensively investigated systems in this field. In addition, alternative CO-gas-free approaches were highlighted, as they offer practical advantages and facilitate reaction scalability.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.References
- L. Deng, J. A. Deichert, S. Nguyen, I. S. Young and C. Han, Org. Lett., 2023, 25, 6710–6714 CrossRef CAS PubMed.
- S. Hu, J. Chen, J. X. Cao, S. S. Zhang, S. X. Gu and F. E. Chen, Bioorg. Chem., 2023, 136, 106549 CrossRef CAS PubMed.
- V. Sharma, R. Das, D. K. Mehta, S. Gupta, K. N. Venugopala, R. Mailavaram, A. B. Nair, A. K. Shakya and P. K. Deb, Bioorg. Med. Chem., 2022, 59(116674), 1–17 Search PubMed.
- D. Jiang, J. Heterocycl. Chem., 2018, 55, 2003–2018 CrossRef CAS.
- Z. Zhang, X. Xiao, T. Su, J. Wu, J. Ren and J. Zhu, Eur. J. Med. Chem., 2017, 140, 239–251 CrossRef CAS PubMed.
- J. Li, T. Zheng, Y. Jin, J. Xu, J. Yu and Y. Lv, Chem. Pharm. Bull., 2018, 66, 55–60 CrossRef CAS PubMed.
- R. Wang, K. Xu and W. Shi, Arch. Pharm., 2019, 1–17 Search PubMed.
- X. Xu, Z. Luo, K. He and M. Zhan, Adv. Mater. Res., 2013, 634–638, 1116–1119 Search PubMed.
- J. D. Yoneda, M. G. Albuquerque, K. Z. Leal, F. D. Costa Santos, P. N. Batalah, L. Brozeguinia, S. R. Peter, R. B. Alencastro, A. C. Cunhaa, M. C. de Souza, V. F. Ferreira, V. A. Giongo, C. C. Sanos and G. C. P. Paixas, J. Mol. Struct., 2014, 1074, 263–270 CrossRef CAS.
- S. Massari, D. Daelemans, G. Manfroni, T. Sabatini, O. Tabarrini, C. Pannecouque and V. Cecchetti, Bioorg. Med. Chem., 2009, 17, 667–674 CrossRef CAS PubMed.
- D. Sriram, P. Yogeeswari, N. Srichakravarthy and T. R. Bal, Bioorg. Med. Chem. Lett., 2005, 14, 1085–1087 CrossRef PubMed.
- A. Chrzanowska, P. Roszkowski, A. Bielenica, W. Olejarz, K. Stepien and M. Struga, Eur. J. Med. Chem., 2020, 185, 111810 CrossRef CAS PubMed.
- H. M. Faidallah, A. S. Girgis, A. D. Tiwari, H. H. Honkanadavar, S. J. Thomas, A. Samir, A. Kalmouch, K. A. Alamry, K. A. Khan and T. S. Ibrahim, Eur. J. Med. Chem., 2018, 143, 1524–1534 CrossRef CAS PubMed.
- I. M. Abdel-Rahman, M. Mustafa, S. A. Mohamed, R. Yahia, M. Abdel-Aziz, G. E.-D. A. Abuo-Rahma and A. M. Hayallah, Bioorg. Chem., 2021, 107, 104629 CrossRef CAS PubMed.
- K. Itoh, Y. Kuramoto, H. Amano, D. Kazamori and A. Yazaki, Eur. J. Med. Chem., 2015, 103, 354–360 CrossRef CAS PubMed.
- Y. Zhang, W. A. Guiguemde, M. Sigal, F. Zhu, M. C. Connelly, S. Nwaka and R. K. Guy, Bioorg. Med. Chem., 2010, 18, 2756–2766 CrossRef CAS PubMed.
- W. Rolf, X. K. Jane, J. S. Martin, H. David, R. K. Dennis and K. R. Michael, Exp. Parasitol., 2011, 127, 545–551 CrossRef PubMed.
- A. R. Millanao, A. Y. Mora, N. A. Villagra, S. A. Bucarey and A. A. Hidalgo, Molecules, 2021, 26, 7153–7195 CrossRef CAS PubMed.
- A. Ahmeda and M. Daneshtalaba, J. Pharm. Pharm. Sci., 2012, 15, 52–72 Search PubMed.
- C. Qi, T. Guo, W. Xiong, L. Wang and H. Jiang, ChemistrySelect, 2017, 2, 4691–4695 CrossRef CAS.
- L. Wu, Y. Hao, Y. Liu and Q. Wang, Org. Biomol. Chem., 2019, 7, 6762–6770 RSC.
- M. Maji, D. Panja, I. Borthakur and S. Kundu, Org. Chem. Front., 2021, 8, 2673–2709 RSC.
- F. E. Ward, D. L. Garling, R. T. Bickler, D. M. Lawer and D. P. Cummings, J. Med. Chem., 1981, 24, 1073–1077 CrossRef CAS PubMed.
- V. A. Glushkov and Yu. V. Shklyaev, Chem. Heterocycl. Compd., 2001, 37, 663–687 CrossRef CAS.
- Z. Ashraf and A. Saeed, Int. J. Org. Chem., 2014, 27, 146–152 Search PubMed.
- A. A. Aly, M. Ramadan, G. E. D. A. Abuo-Rahma, Y. A. M. M. Elshaier, M. A. I. Elbastawesy, A. B. Brown and S. Bräse, Adv. Heterocycl. Chem., 2021, 135, 147–196 CrossRef.
- P. S. Dube, L. J. Legoabe and R. M. Beteck, Mol. Divers., 2023, 27, 1501–1526 CrossRef CAS PubMed.
- M. I. Andersson and A. P. MacGowan, J. Antimicrob. Chemother., 2003, 51, 1–11 CrossRef CAS PubMed.
- G. Y. Lsher, E. J. Froelich, M. D. Gruett, J. H. Bailey and R. P. Brundage, J. Med. Chem., 1962, 5, 1063–1065 CrossRef PubMed.
- A. A. Boteva and O. P. Krasnykh, Chem. Heterocycl. Compd., 2009, 45, 757–785 CrossRef CAS.
- For recent books and reviews, see: (a) ed. B. Gabriele, Carbon Monoxide in Organic Synthesis – Carbonylation Chemistry, Wiley-VCH, Weinheim, 2022 Search PubMed; (b) ed. X.-F. Wu, B. Han, K. Ding and Z. Liu, The chemical transformations of C1 compounds, Wiley, 2022 Search PubMed; (c) ed. L. Kollár, Modern Carbonylation Methods, Wiley-VCH, Weinheim, 2008 Search PubMed; (d) ed. M. Beller, Catalytic Carbonylation Reactions, Springer, Berlin, 2006 Search PubMed; (e) K. Sun, G.-P. Lu, W. Han and M. Beller, Angew. Chem., Int. Ed., 2025, 64, e202512346 CrossRef CAS PubMed; (f) Z.-P. Bao and X.-F. Wu, EES Catal., 2025, 3, 1179–1195 RSC; (g) P. Mehara, P. Sharma and P. Das, Chem. Rec., 2025, 25, e202500071 CrossRef CAS PubMed; (h) B. Pant, D. Prakash and D. S. Dhami, ChemistrySelect, 2024, 9, e202401825 CrossRef CAS; (i) C. Árvai and L. T. Mika, Chin. J. Chem., 2024, 42, 406–429 CrossRef; (j) Á. Molnár, Coord. Chem. Rev., 2024, 504, 215668 CrossRef; (k) B. Gabriele, R. Mancuso, N. Della Ca, L. Veltri and I. Ziccarelli, Catalysts, 2023, 13, 1025 CrossRef CAS; (l) J.-B. Peng, H. Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CrossRef CAS.
- (a) B. M. Trost, Science, 1991, 254, 1471–1477 Search PubMed; (b) B. M. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259–281 CrossRef CAS; (c) B. M. Trost, Acc. Chem. Res, 2002, 35, 695–705 CrossRef CAS PubMed.
- (a) P. A. Wender and B. L. Miller, Nature, 2009, 460, 197–201 CrossRef CAS PubMed; (b) P. A. Wender, V. A. Verma, T. J. Paxton and T. H. Pillow, Acc. Chem. Res., 2008, 41, 40–49 CrossRef CAS PubMed.
- (a) P. Anastas and N. Eghbali, Chem. Soc. Rev., 2010, 39, 301–312 Search PubMed; (b) R. A. Sheldon, I. Arends and H. Ulf,Green Chemistry and Catalysis, Wiley-VCH, Weinheim, 2008 Search PubMed; (c) R. A. Sheldon, Chem. Commun., 2008, 3352–3365 Search PubMed; (d) R. A. Sheldon, Green Chem., 2007, 9, 1273–1283 Search PubMed.
- S. Torii, H. Okumoto and L. H. Xu, Tetrahedron Lett., 1991, 32, 237–240 CrossRef CAS.
- V. N. Kalinin, M. V. Shostakovsky and A. B. Ponomaryov, Tetrahedron Lett., 1992, 33, 373–376 CrossRef CAS.
- N. Haddad, J. Tan and V. Farina, J. Org. Chem., 2006, 71, 5031–5034 CrossRef CAS PubMed.
- M. Reiser, H. Hinrichsen, Y. Benhamou, R. Sentjens, H. Wede-meyer, L. Calleja, X. Forns, J. Croenlein, C. Yong, G. Nehmiz and G. Steinmann, Hepatology, 2003, 37, S135–S144 Search PubMed.
- S. Torii, H. Okumoto and L. H. Xu, Tetrahedron Lett., 1990, 31, 7175–7178 Search PubMed.
- F. Ye and H. Alper, J. Org. Chem., 2007, 72, 3218–3222 CrossRef CAS PubMed.
- T. Sakamoto, A. Yasuhara, Y. Kondo and H. Yamanaka, Chem. Pharm. Bull., 1992, 40, 1137–1139 CrossRef CAS.
- M. Genelot, A. Bendjeriou, V. Dufaud and L. Djakovitch, Appl. Catal., A, 2009, 369, 125–132 Search PubMed.
- M. Genelot, V. Dufaud and L. Djakovitch, Tetrahedron, 2011, 67, 976–981 CrossRef CAS.
- N. Batail, M. Genelot, V. Dufaud, L. Joucla and L. Djakovitch, Catal. Today, 2011, 173, 2–14 CrossRef CAS.
- D. V. KadniKov and R. C. Larock, J. Organomet. Chem., 2003, 687, 425–435 CrossRef CAS.
- D. V. Kadnikov and R. C. Larock, J. Org. Chem., 2004, 69, 6772–6780 CrossRef CAS PubMed.
- X. Zhang, H. Liu and Y. Jia, Chem. Commun., 2016, 52, 7665–7667 RSC.
- (a) K. R. Fandrick, et al. , Angew. Chem., Int. Ed., 2015, 54, 7144–7148 CrossRef CAS PubMed; (b) L. D. Fader, et al. , ACS Med. Chem. Lett., 2014, 5, 422–427 CrossRef CAS PubMed; (c) X. Bu, L. W. Deady, G. J. Finlay, B. C. Baguley and W. A. Denny, J. Med. Chem., 2001, 44, 2004–2014 CrossRef CAS PubMed; (d) T. Ozturk and A. McKillop, Can. J. Chem., 2000, 78, 1158 CrossRef CAS; (e) H. Fujiwara and I. Okabayashi, Chem. Pharm. Bull., 1993, 41, 1163–1165 CrossRef CAS.
- A. C. Tadd, A. Matsuno, M. R. Fielding and M. C. Willis, Org. Lett., 2009, 11, 583–586 CrossRef CAS PubMed.
- R. Alfonsi, B. Botta, S. Cacchi, L. Di Marcotullio, G. Fabrizi, R. Faedda, A. Goggiamani, A. Iazzetti and M. Mori, J. Med. Chem., 2017, 60, 1469–1477 CrossRef CAS PubMed.
- A. R. i Altaba, P. Sánchez and N. Dahmane, Nat. Rev. Cancer, 2002, 2, 361–372 CrossRef PubMed.
- K. Okuro and H. Alper, J. Org. Chem., 2012, 77, 4420–4424 CrossRef CAS PubMed.
- R. Grigg, A. Liu, D. Shaw, S. Suganthan, D. E. Woodalla and G. Yoganathana, Tetrahedron Lett., 2000, 41, 7125–7128 Search PubMed.
- W. W. Ding, Y. Zhou, Z. Y. Han and Z. Gong, J. Org. Chem., 2023, 88, 5187–5193 CrossRef CAS PubMed.
- J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. Buchwald, J. Org. Chem., 2008, 73, 7102–7107 Search PubMed.
- A. Barré, M.-L. Ţînţaş, F. Alix, V. Gembus, C. Papamicaël and V. Levacher, J. Org. Chem., 2015, 80, 6537–6544 Search PubMed.
- L.-J. Han, C.-S. Rao, S.-S. Ma, G.-Y. Sheng, J.-P. Zhang and B.-H. Xu, J. Catal., 2021, 404, 283–290 Search PubMed.
- C.-S. Kuai, B.-H. Teng and X.-F. Wu, Angew. Chem., Int. Ed., 2024, 63, e202318257 Search PubMed.
- L. Pietrobon, L. Ronchin and A. Vavasori, Catalysts, 2024, 14, 660–673 CrossRef CAS.
- S. Chung, N. Sach, C. Choi, X. Yang, S. E. Drozda, R. A. Singer and S. W. Wright, Org. Lett., 2015, 17, 2848–2851 CrossRef CAS PubMed.
- U. Gross, P. Koos, M. O’Brien, A. Polyzos and S. V. Ley, Eur. J. Org. Chem., 2014, 6418–6430 CrossRef CAS.
- K. Okuro and H. Alper, Tetrahedron Lett., 2012, 53, 4816–4818 CrossRef CAS.
- J. Wu, Y. Zhou, T. Wu, Y. Zhou, C. W. Chiang and A. Lei, Org. Lett., 2017, 19, 6432–6435 CrossRef CAS PubMed.
- L. Zhang, C. Wang, J. Han, Z. B. Huang and Y. Zhao, J. Org. Chem., 2016, 81, 5256–5262 Search PubMed.
- J. Albert, X. Ariza, T. Calvet, M. Font-Bardia, J. Garcia, J. Granell, A. Lamela, B. Lopez, M. Martinez, L. Ortega, A. Rodriguez and D. Santos, Organometallics, 2013, 32, 649–659 Search PubMed.
- K. Orito, A. Horibata, T. Nakamura, H. Ushito, H. Nagasaki, M. Yuguchi, S. Yamashita and M. Tokuda, J. Am. Chem. Soc., 2004, 126, 14342–14343 CrossRef CAS PubMed.
- M. Costa, N. D. Cà, B. Gabriele, C. Massera, G. Salerno and M. Soliani, J. Org. Chem., 2004, 69, 2469–2477 CrossRef CAS PubMed.
- B. Gabriele, G. Salerno and M. Costa, Synlett, 2004, 2468–2483 CrossRef CAS.
- J. Ferguson, F. Zeng, N. Alwis and H. Alper, Org. Lett., 2013, 15, 1998–2001 CrossRef CAS PubMed.
- R. Mancuso, A. De Salvo, P. Russo, A. Falcicchio, N. Della Ca, L. P. Munoz and B. Gabriele, Chin. J. Chem., 2023, 41, 2801–2809 CrossRef CAS.
- (a) B. Gabriele, M. Costa, G. Salerno and G. P. Chiusoli, J. Chem. Soc., Chem. Commun., 1992, 1007–1008 RSC; (b) B. Gabriele, M. Costa, G. Salerno and G. P. Chiusoli, J. Chem. Soc., Perkin Trans. 1, 1994, 83–87 RSC; (c) B. Gabriele and G. Salerno, PdI2. In e-EROS (Electronic Encyclopedia of Reagents for Organic Synthesis), ed D. Crich, Wiley-Inter science, New York, 2006 Search PubMed.
- (a) B. Gabriele, Tetrahedron Chem, 2024, 12, 100107 CrossRef CAS; (b) R. Mancuso, N. Della Ca, L. Veltri, I. Ziccarelli and B. Gabriele, Catalysts, 2019, 9, 610 CrossRef; (c) B. Gabriele, Targets Heterocycl. Syst., 2018, 22, 41–55 CAS; (d) B. Gabriele, G. Salerno and M. Costa, Synlett, 2004, 2468–2483 CrossRef CAS; (e) B. Gabriele, G. Salerno, M. Costa and G. P. Chiusoli, J. Organomet. Chem., 2003, 687, 219–228 CrossRef CAS.
- A. Acerbi, C. Carfagna, M. Costa, R. Mancuso, B. Gabriele and N. Della Ca, Chem. – Eur. J., 2018, 24, 4835–4840 CrossRef CAS PubMed.
- (a) R. Mancuso, I. Ziccarelli, A. Chimento, N. Marino, N. Della Ca, R. Sirianni, V. Pezzi and B. Gabriele, iScience, 2018, 3, 279–288 CrossRef CAS PubMed; (b) R. Mancuso, R. Milie, A. P. Piccionello, D. Olivieri, N. Della Ca, C. Carfagna and B. Gabriele, J. Org. Chem., 2019, 84, 7303–7311 CrossRef CAS PubMed.
- F. Pancrazzi, N. Sarti, P. P. Mazzeo, A. Bacchi, C. Carfagna, R. Mancuso, B. Gabriele, M. Costa, A. Stirling and N. Della Ca, Org. Lett., 2020, 22, 1569–1574 CrossRef CAS PubMed.
- A. Voronov, F. Pancrazzi, A. M. Constantin, R. Maggi, R. Mancuso, B. Gabriele, D. Olivieri, C. Carfagna, A. Casnati, F. Rispoli, L. Baldini and N. Della Ca, Chin. J. Chem., 2023, 41, 3223–3228 CrossRef CAS.
- (a) G. Chiusoli, Carbon, J. Mol. Catal. A, 2003, 204–205, 133–142 CrossRef CAS; (b) B. Gabriele, G. Salerno, M. Costa and G. P. Chiusoli, Tetrahedron Lett., 1999, 40, 989–990 CrossRef CAS.
- X. Li, X. Li and N. Jiao, J. Am. Chem. Soc., 2015, 137, 9246–9249 CrossRef CAS PubMed.
- X. Li, J. Pan, H. Wu and N. Jiao, Chem. Sci., 2017, 8, 6266–6273 RSC.
- F. Zhu, Y. Li, Z. Wang and X.-F. Wu, Adv. Synth. Catal., 2016, 358, 3350–3354 CrossRef CAS.
- S. Perrone, L. Troisi and A. Salomone, Eur. J. Org. Chem., 2019, 4626–4643 CrossRef CAS.
- X. Q. Hu, Z. K. Liu and W. J. Xiao, Catalysts, 2020, 10, 1054–1080 CrossRef CAS.
- Z. Yin, W. Shi and X.-F. Wu, J. Org. Chem., 2023, 88, 4975–4994 CrossRef CAS PubMed.
- D. Bhattacherjee, M. Rahman, S. Ghosh, A. K. Bagdi, G. V. Zyryanov, O. N. Chupakhin, P. Das and A. Hajra, Adv. Synth. Catal., 2021, 363, 1597–1624 CrossRef CAS.
- L. A. Aronica, A. M. Caporusso, G. Tuci, C. Evangelisti, M. Manzoli, M. Botavina and G. Martra, Appl. Catal., 2014, 480, 1–9 CrossRef CAS.
- S. P. Chavana, G. B. B. Varadwaj, K. Parida and B. M. Bhanage, Appl. Catal., 2015, 506, 237–245 CrossRef.
- N. F. K. Kaiser, A. Hallberg and M. Larhed, J. Comb. Chem., 2002, 4, 109–111 CrossRef CAS PubMed.
- P. Ghosh, A. K. Nandi and S. Das, Tetrahedron Lett., 2018, 59, 2025–2029 Search PubMed.
- J.-L. Liu, C.-Y. Hou, X. Qi and X.-F. Wu, Molecular Catal, 2021, 514, 111842–111846 CAS.
- F. Jafarpour and A. Otaredi-Kashania, ARKIVOC, 2014, 4, 193–203 Search PubMed.
- L. Åkerbladh, P. Nordeman, M. Wejdemar, L. R. Odell and M. Larhed, J. Org. Chem., 2015, 80, 1464–1471 CrossRef PubMed.
- J.-L. Liu, W. Wang, X. Qi and X.-F. Wu, Org. Lett., 2022, 24, 2248–2252 CrossRef CAS PubMed.
- J. Chen, K. Natte, A. Spannenberg, H. Neumann, M. Beller and X.-F. Wu, Chem. – Eur. J., 2014, 20, 14189–14193 Search PubMed.
- J.-S. Wang, C. Li, J. Ying, T. Xu, W. Lu, C.-Y. Li and X.-F. Wu, J. Catal., 2022, 408, 81–87 CrossRef CAS.
- V. Thakur, A. Sharma, Yamini, N. Sharma and P. Dasa, Adv. Synth. Catal., 2019, 361, 426–431 CrossRef CAS.
- M. Guo, D. Wu, H. Yang, X. Zhang, D.-X. Xue and W. Zhang, Molecules, 2024, 29, 850–861 CrossRef CAS PubMed.
- S. Morimoto and K. Kakiuchi, Angew. Chem., Int. Ed., 2004, 43, 5580–5588 Search PubMed.
- F. Ferretti, M. A. Fouad, C. Abbo and F. Ragaini, Molecules, 2023, 28, 5424–5437 CrossRef CAS PubMed.
- E. Stashenko, R. Acosta and J. R. Martınez, J. Biochem. Biophys. Methods, 2000, 43, 379–390 CrossRef CAS PubMed.
- Z.-Y. An, Y.-Y. Yan, D. Peng, T.-M. Ou, J.-H. Tan, S.-L. Huang, L.-K. An, L.-Q. Gu and Z.-S. Huang, Eur. J. Med. Chem., 2010, 45, 3895–3903 CrossRef CAS PubMed.
- T. S. Wu, L. S. Shi, J. J. Wang, S. C. Lou, H. C. Chang, Y. P. Chen, Y. H. Kuo, Y. L. Chang and C. M. Tenge, J. Chin. Chem. Soc., 2003, 50, 171–178 CrossRef CAS.
- S. Ghosh, K. Bishayee and A. R. Khuda-Bukhsh, Phytother. Res., 2014, 28, 1153–1162 Search PubMed.
- J.-S. Wang, Y. Na, J. Ying and X.-F. Wu, Org. Chem. Front., 2021, 8, 2429–2433 RSC.
- (a) Q. Gao, J.-M. Lu, L. Yao, S. Wang, J. Ying and X.-F. Wu, Org. Lett., 2021, 23, 178–182 CrossRef CAS PubMed; (b) J. Ying, Z. Le and X.-F. Wu, Org. Lett., 2020, 22, 194–198 CrossRef CAS PubMed; (c) J. Ying, L.-Y. Fu, G. Zhong and X.-F. Wu, Org. Lett., 2019, 21, 5694–5698 CrossRef CAS PubMed.
- Y. Zhu, J. Ying and X.-F. Wu, Mol. Catal., 2022, 524, 112267 CAS.
| This journal is © The Royal Society of Chemistry 2026 |