
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRegio- and diastereoselective synthesis of multi-substituted silacyclopentanes by catalytic cyclosilylborylation of styryl(vinyl)silanes
Kanta Uejia and
Ryo Shintani *ab
*ab
aDivision of Chemistry, Department of Materials Engineering Science, Graduate School of Engineering Science, The University of Osaka, Toyonaka, Osaka 560-8531, Japan. E-mail: shintani.ryo.es@osaka-u.ac.jp
bInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), The University of Osaka, Suita, Osaka 565-0871, Japan
First published on 1st April 2026
Abstract
A NaOtBu-catalyzed regio- and diastereoselective cyclosilylborylation of styryl(vinyl)silanes with silylboronates has been developed for the synthesis of multi-substituted silacyclopentanes. 2-Silyl-5-boryl-1-silacyclopentanes obtained in the present catalysis can be further functionalized by taking advantage of the reactivity of the silyl and boryl groups.
Substituting a carbon atom of organic molecules by silicon is a useful strategy to modify or improve their original properties particularly in the field of pharmaceutical science.1 Since alicycles are ubiquitously found in various natural products and biologically active compounds, development of efficient synthetic methods of corresponding silacyclic compounds is of high importance.2 In particular, silacyclopentanes, silicon-switched analogues of widely found cyclopentanes, are expected to display promising biological activity,3 but synthesis of highly functionalized silacyclopentanes is currently still underexplored. One way to achieve this goal is to introduce functional groups to preformed silacyclopentenes by utilizing the reaction toward a carbon–carbon double bond,3a,3d,4 but direct synthesis of such silacyclopentanes is rather difficult presumably due to the limited available methods to construct the silacyclopentane framework from readily available precursors. Other than the classical reaction of 1,4-dimetalloalkanes with dichlorosilanes toward the synthesis of silacyclopentanes,3b,3c,5 ring expansion reactions of silacyclobutanes with carbenoids have been relatively well investigated,6 including a recent enantioselective carbene insertion under palladium catalysis7 (Scheme 1a). In addition, metal-catalyzed hydrosilylation of methylenecyclopropanes8 (Scheme 1b) as well as Lewis acid-catalyzed hydrosilylation of vinylcyclopropanes9 (Scheme 1c) have also been reported. However, highly substituted silacyclopentanes could not be prepared in these approaches.
 | ||
| Scheme 1 (a)–(d) Reported synthetic methods of silacyclopentanes through the formation of 5-membered rings and (e) cyclosilylborylation of styryl(vinyl)silanes (this work). | ||
To overcome this synthetic problem, we imagined that introduction of multiple substituents during the five-membered ring formation would be realized in a straightforward manner by cyclo-difunctionalization of linear dialkenylsilanes. In this regard, although a somewhat related example has been reported in the reaction of (4-butenyl)(vinyl)silanes with n-butyllithium (Scheme 1d), applicable substrate combinations are quite limited and only 2,3-dialkyl-1-silacyclopentanes can be accessed with moderate diastereoselectivity.10 Considering the ease of further derivatization reactions, we designed a reaction of silylboronates11 with dialkenylsilanes to give 2-silyl-5-boryl-1-silacyclopentanes12 and found that the both regioselectivity and diastereoselectivity can be effectively controlled by using styryl(vinyl)silanes as the substrate in the presence of a catalytic amount of NaOtBu under mild conditions (Scheme 1e), and herein we describe the details of this process.
Because styrene derivatives are known to undergo regioselective silylborylation by using a combination of silylboronates and alkali metal alkoxide catalysts,13 we initially employed (E)-styryl(vinyl)silane 1a as the model substrate and conducted a reaction with dimethylphenylsilylboronate 2a in the presence of KOtBu (20 mol%) in THF at 50 °C (Table 1, entry 1). Under these conditions, the desired silylation–cyclization–borylation effectively took place to give the silacyclopentane product having three stereocenters as a mixture of two diastereomers 3aa/3aa′ out of four possible diastereomers in 88% combined yield in the ratio of 80/20. The diastereoselectivity was found to be improved by using LiOtBu (84/16; entry 2) or NaOtBu (89/11; entry 3). The use of NaN(SiMe3)2 showed the same level of selectivity as NaOtBu (entry 4), but other sodium alkoxides such as NaOEt and NaOMe were found to be less effective or unreactive (entries 5 and 6). In the present reaction, the diastereomeric ratio (dr) between 3aa and 3aa′ was independent of the E/Z ratio of starting compound 1a (entry 7). Finally, by lowering the reaction temperature to 10 °C, somewhat higher diastereoselectivity of 91/9 was realized while keeping the good reactivity (entry 8).
|
|||
|---|---|---|---|
| Entry | Catalyst | Yield of 3aa + 3aa′a (%) | Ratio of 3aa/3aa′a |
| a Determined by 1H NMR.b The reaction was conducted using 1a with E/Z = 72/28.c The reaction was conducted at 10 °C for 16 h. | |||
| 1 | KOtBu | 88 | 80/20 |
| 2 | LiOtBu | 78 | 84/16 |
| 3 | NaOtBu | 90 | 89/11 |
| 4 | NaN(SiMe3)2 | 79 | 89/11 |
| 5 | NaOEt | 71 | 84/16 |
| 6 | NaOMe | 0 | — |
| 7b | NaOtBu | 88 | 89/11 |
| 8c | NaOtBu | 80 | 91/9 |
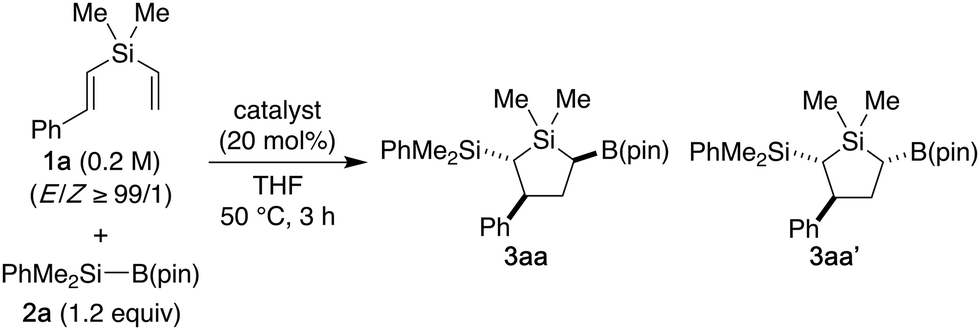
Under the conditions using NaOtBu (20 mol%), various other styryl(vinyl)silanes can be employed in the reaction with silylboronate 2a to give 2-silyl-3-aryl-5-boryl-1-silacyclopentanes 3 diastereoselectively (Scheme 2). For example, in addition to 1a having methyl groups on silicon, ethyl (1b) or phenyl (1c) substituted compounds gave the corresponding silacyclopentanes 3ba–ca as well, and the relative configuration of 3ba was firmly established by X-ray crystallographic analysis. With regard to the variation of the styryl group, several substituted aryl groups can be accommodated to give products 3da–ha with similar efficiency, and higher diastereoselectivity of up to 94/6 was observed for substrates having a bulky aryl group such as 1g and 1h. The reaction could also be applied to substrates having naphthyl (1i and 1j) or heteroaryl (1k and 1l) groups to give 3ia–la. In addition, other silylboronates such as 2b and 2c were compatible with the present reaction, leading to the formation of 2-trialkylsilylated products 3ab–ac. It is worth noting that the present reaction could also be conducted on a preparative scale using 2.5 mmol of 1a to give 1.0 g of 3aa (89% yield, dr = 91/9).
 | ||
| Scheme 2 Diastereoselective synthesis of 2,3,5-trisubtituted silacyclopentanes: scope. | ||
To further expand the scope of accessible multi-substituted silacyclopentanes, we examined the reaction of styryl(1-propenyl)silane 1m with silylboronate 2a for the synthesis of 2,3,4,5-tetrasubstituted 1,1-dimethyl-1-silacyclopentane 3ma (Scheme 3a). As a result, by conducting the reaction using 40 mol% of NaOtBu under a diluted condition, the desired product was successfully obtained in 83% yield as a mixture of two diastereomers in the ratio of 84/16 out of eight possible diastereomers. Similarly, 1n having ethyl groups on silicon as well as 1o and 1p having substituted styryl groups were converted to silacyclopentanes 3na–pa in 68–83% yield with dr = 82/18–84/16, and X-ray crystal structure of the major diastereomer of 3na revealed that the four substituents on the ring carbons were in all-trans relationship.
 | ||
| Scheme 3 (a) Diastereoselective synthesis of 2,3,4,5-tetrasubstituted silacyclopentanes. (b) Stereo-retentive transformations of 3aa. | ||
Silyl- and boryl-substituted silacyclopentane 3aa obtained in the present reaction can be further derivatized with retaining the stereochemical integrity. For example, Matteson homologation by treatment with n-butyllithium in the presence of bromochloromethane gave compound 4 having a borylmethyl substituent in 86% yield.14 In addition, the boryl group could be converted to a vinyl group in 43% yield by Zweifel olefination of 3aa with vinylmagnesium bromide followed by treating it with iodine in methanol.15 The boryl group of 3aa could also be oxidized to a hydroxy group by hydrogen peroxide under basic conditions to give silacyclopentanol 6 in a nearly quantitative yield.16 Swern oxidation of the hydroxy group of 6 led to silacyclopentanone 7,17 a type of underexplored cyclic acylsilane,18 in 53% yield as a single diastereomer, which confirms that 3aa and 3aa′ have a common relative configuration between dimethylphenylsilyl group and phenyl group with the opposite configuration at the boryl group. On the other hand, oxidation of the carbon–silicon bond of 6 selectively took place under Tamao–Fleming conditions to give trans-silacyclopentanediol 8 in 88% yield.19
A proposed catalytic cycle for the reaction of styryl(vinyl)silane 1a with silylboronate 2a is illustrated in Scheme 4. Initially, coordination of NaOtBu to 2a gives borate intermediate A, which would be in equilibrium with dissociated dimethylphenylsilylsodium and tBuOB(pin).13,20 The silicon nucleophile then attacks the β-position of styryl group of 1a to give benzyl anion intermediate B. Subsequent ring-closure of this intermediate takes place through intramolecular nucleophilic attack at the β-position of vinylsilane to give silacyclopentyl anion C, which undergoes borylation with tBuOB(pin) to give cyclosilylborylation product 3aa along with regeneration of NaOtBu. The relative configuration between the silyl group at 2-position and the phenyl group at 3-position would be controlled in trans relationship at the C–C bond-forming cyclization step, and the stereochemistry of the boryl group at 5-position is finally set to give major diastereomer 3aa possessing three substituents on the ring carbons all in pseudo- equatorial positions of the chair-like silacyclopentane structure.
 | ||
| Scheme 4 Proposed catalytic cycle for the reaction of 1a with 2a to give 3aa. | ||
To gain some insights into the above-proposed mechanism, we prepared dimethylphenylsilyllithium as a discrete nucleophile and conducted stoichiometric reactions with styryl(vinyl)silane 1a (Scheme 5a). The reactions were conducted at –78 °C and quenched by protonation using MeOH after different reaction times. When the reaction was stopped after 0.5 h, uncyclized (1-silyl-2-phenylethyl)(vinyl)silane 9aa and cyclized 2-silyl-3-phenyl-1-silacyclopentane 10aa were obtained in 45% yield and 35% yield, respectively. By extending the reaction time prior to the MeOH quench, the yield of 9aa became lower and that of 10aa became higher accordingly. These results suggest that the silacyclopentane formation takes place in a stepwise fashion consisting of addition of a silicon nucleophile to the β-position of the styrene moiety and subsequent intramolecular addition of the resulting benzyl nucleophile to the pendant vinyl group, supporting the proposed catalytic cycle in Scheme 4.
 | ||
| Scheme 5 Reactions of 1a with dimethylphenylsilyllithium followed by electrophilic quench: (a) effect of reaction time and (b) scope of electrophiles. | ||
By taking advantage of the formation of a silacyclopentyllithium (C′ in Scheme 5a) in a stoichiometric fashion, we also examined the use of some other electrophiles to introduce different substituents at 5-position (Scheme 5b). When the reaction was quenched with trimethyl borate followed by transesterification with pinacol, cyclosilylborylation product 10ab (=3aa) was obtained in 65% yield with dr = 87/13. In addition, the use of chlorodimethylphenylsilane gave 2,5-disilyl-1-silacyclopentane 10ac in a good yield of 70%, although the diastereoselectivity was low (dr = 55/45). Similarly, introduction of phenylthio group was also achieved by using diphenyl disulfide as the electrophile to give 10ad in 74% yield.
In summary, we developed a regio- and diastereoselective cyclosilylborylation of styryl(vinyl)silanes with silylboronates in the presence of a catalytic amount of NaOtBu to give multi-substituted silacyclopentanes. The reaction can be performed for various substrates including styryl(1-propenyl)silanes, giving 2,3,4,5-tetrasubstituted compounds with all-trans configuration. The obtained 2-silyl-5-boryl-1-silacyclopentanes could be further functionalized by utilizing the reactivity of the silyl and boryl groups, and the reaction mechanism was also probed by conducting model stoichiometric reactions.
Support has been provided in part by JSPS KAKENHI Grant Numbers JP25K01767 (Grant-in-Aid for Scientific Research (B); R.S.) and JP24H01856 (Grant-in-Aid for Transformative Research Areas (A) Green Catalysis Science; R.S.). We thank Prof. Jun Takaya, Dr Yuto Morita, Mr Junpei Murakami, and Mr Daigo Hayashi at The University of Osaka for the measurement of high-resolution mass spectra. Elemental analysis was performed using research equipment shared in MEXT Project for promoting public utilization of advanced research infrastructure (Grant Number JPMXS0441200025; Program for supporting construction of core facilities).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental procedures, characterization data, and NMR spectra. See DOI: https://doi.org/10.1039/d6cc00416d.CCDC 2524632 (3ba) and 2524633 (3na) contain the supplementary crystallographic data for this paper.21a,b
References
- For reviews: (a) R. Ramesh and D. S. Reddy, J. Med. Chem., 2018, 61, 3779 CrossRef CAS PubMed; (b) S. Fujii and Y. Hashimoto, Future Med. Chem., 2017, 9, 485 CrossRef CAS PubMed; (c) E. Rémond, C. Martin, J. Martinez and F. Cavelier, Chem. Rev., 2016, 116, 11654 CrossRef PubMed; (d) A. K. Franz and S. O. Wilson, J. Med. Chem., 2013, 56, 388 CrossRef CAS PubMed; (e) G. A. Showell and J. S. Mills, Drug Discovery Today, 2003, 8, 551 CrossRef CAS PubMed.
- For reviews: (a) W.-T. Zhao, F. Gao and D. Zhao, Synlett, 2018, 2595 CrossRef CAS; (b) L. Li, Y. Zhang, L. Gao and Z. Song, Tetrahedron Lett., 2015, 56, 1466 CrossRef CAS.
- (a) D. Damour, M. Barreau, G. Dutruc-Rosset, A. Doble, O. Piot and S. Mignani, Bioorg. Med. Chem. Lett., 1994, 4, 415 CrossRef CAS; (b) B. R. Yoo, M. Y. Suk, J. S. Han, Y.-M. Yu, S.-G. Hong and I. N. Jung, Pestic. Sci., 1998, 52, 138 CrossRef CAS; (c) J. O. Daiss, C. Burschka, J. S. Mills, J. G. Montana, G. A. Showell, J. B. H. Warneck and R. Tacke, Organometallics, 2006, 25, 1188 CrossRef CAS; (d) K. Igawa, D. Yoshihiro, Y. Abe and K. Tomooka, Angew. Chem., Int. Ed., 2016, 55, 5814 CrossRef CAS PubMed.
- (a) T. Araki, D. Terunuma and T. Fuse, Bull. Chem. Soc. Jpn., 1972, 45, 293 CrossRef CAS; (b) S. Mignani, M. Barreau, D. Damour, A. Renaudon, V. Dejean and G. Manuel, Synth. Commun., 1998, 28, 1163 CrossRef CAS; (c) K. Igawa, A. Kuroo, D. Yoshihiro, Y. Yamanaka and K. Tomooka, Synlett, 2017, 2445 CrossRef CAS; (d) A. Kuroo, K. Igawa and K. Tomooka, Chem. Lett., 2024, 53, upae039 CrossRef CAS.
- (a) H.-S. Dang, B. P. Roberts and D. A. Tocher, J. Chem. Soc., Perkin Trans. 1, 1995, 117 RSC; (b) U. M. Dzhemilev, A. G. Ibragimov, R. R. Gilyazev and L. O. Khafizova, Tetrahedron, 2004, 60, 1281 CrossRef CAS.
- (a) D. Seyferth, R. Damrauer and S. S. Washburne, J. Am. Chem. Soc., 1967, 89, 1538 CrossRef CAS; (b) K. Matsumoto, K. Oshima and K. Utimoto, Tetrahedron Lett., 1990, 31, 6055 CrossRef CAS; (c) K. Matsumoto, Y. Aoki, K. Oshima, K. Utimoto and N. A. Rahman, Tetrahedron, 1993, 49, 8487 CrossRef CAS.
- J. Huo, K. Zhong, Y. Xue, M. Lyu, Y. Ping, Z. Liu, Y. Lan and J. Wang, J. Am. Chem. Soc., 2021, 143, 12968 CrossRef CAS PubMed.
- (a) B. Fu, L. Wang, K. Chen, X. Yuan, J. Yin, S. Wang, D. Shi, B. Zhu, W. Guan, Q. Zhang and T. Xiong, Angew. Chem., Int. Ed., 2024, 63, e202407391 CAS; (b) L. Wu, L. Zhang, J. Guo, J. Gao, Y. Ding, J. Ke and C. He, Angew. Chem., Int. Ed., 2024, 63, e202413753 CrossRef CAS PubMed; (c) X. Xu, A. Gao, X. Xu, J. Li and C. Cui, J. Am. Chem. Soc., 2024, 146, 4060 CrossRef CAS.
- P.-W. Long and M. Oestreich, Org. Lett., 2021, 23, 4834 CrossRef CAS PubMed.
- X. Wei and R. J. K. Taylor, Tetrahedron Lett., 2003, 44, 7143 CrossRef CAS.
- For reviews: (a) J.-J. Feng, W. Mao, L. Zhang and M. Oestreich, Chem. Soc. Rev., 2021, 50, 2010 RSC; (b) M. Oestreich, E. Hartmann and M. Mewald, Chem. Rev., 2013, 113, 402 CrossRef CAS PubMed.
- For examples of cyclosilylborylation reactions: (a) Y.-C. Xiao and C. Moberg, Org. Lett., 2016, 18, 308 CrossRef CAS PubMed; (b) Q. Zhang, Q.-J. Liang, J.-L. Xu, Y.-H. Xu and T.-P. Loh, Chem. Commun., 2018, 54, 2357 RSC; (c) Y. Zhang, W. Xu, T. Gao, M. Guo, C.-H. Yang, H. Xie, X. Kong, Z. Yang and J. Chang, Org. Lett., 2022, 24, 7021 CrossRef CAS PubMed.
- H. Ito, Y. Horita and E. Yamamoto, Chem. Commun., 2012, 48, 8006 RSC.
- (a) D. S. Matteson and R. W. H. Mah, J. Am. Chem. Soc., 1963, 85, 2599 CrossRef CAS; (b) D. S. Matteson, J. Org. Chem., 2013, 78, 10009 CrossRef CAS PubMed.
- (a) G. Zweifel, H. Arzoumanian and C. C. Whitney, J. Am. Chem. Soc., 1967, 89, 3652 CrossRef CAS; (b) V. K. Aggarwal, M. Binanzer, M. Carolina de Ceglie, M. Gallanti, B. W. Glasspoole, S. J. F. Kendrick, R. P. Sonawane, A. Vázquez-Romero and M. P. Webster, Org. Lett., 2011, 13, 1490 CrossRef CAS PubMed; (c) R. J. Armstrong and V. K. Aggarwal, Synthesis, 2017, 3323 CAS.
- N. G. Bhat and A. Garza, Tetrahedron Lett., 2003, 44, 6833 CrossRef CAS.
- (a) A. J. Mancuso, S.-L. Huang and D. Swern, J. Org. Chem., 1978, 43, 2480 CrossRef CAS; (b) R. L. Danheiser, D. M. Fink, K. Okano, Y.-M. Tsai and S. W. Szczepanski, J. Org. Chem., 1985, 50, 5393 CrossRef CAS.
- (a) J. A. Soderquist and A. Hassner, J. Org. Chem., 1980, 45, 541 CrossRef CAS; (b) A. Studer, S. Amrein, H. Matsubara, C. H. Schiesser, T. Doi, T. Kawamura, T. Fukuyama and I. Ryu, Chem. Commun., 2003, 1190–1191 RSC.
- (a) K. Tamao, N. Ishida, T. Tanaka and M. Kumada, Organometallics, 1983, 2, 1694 CrossRef CAS; (b) I. Fleming, R. Henning and H. Plaut, J. Chem. Soc., Chem. Commun., 1984, 29 RSC.
- (a) K. Kojima, Y. Nagashima, C. Wang and M. Uchiyama, ChemPlusChem, 2019, 84, 277 CrossRef CAS PubMed; (b) A. Kawachi, T. Minamimoto and K. Tamao, Chem. Lett., 2001, 1216 CrossRef CAS; (c) R. Shintani, R. Fujie, M. Takeda and K. Nozaki, Angew. Chem., Int. Ed., 2014, 53, 6546 CrossRef CAS PubMed; (d) X.-W. Liu, C. Zarate and R. Martin, Angew. Chem., Int. Ed., 2019, 58, 2064 CrossRef CAS PubMed; (e) I. Fujii, H. Hirata, H. Moniwa and R. Shintani, Chem. Commun., 2024, 60, 6921 RSC.
- (a) CCDC 2524632: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qr2rb; (b) CCDC 2524633: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qr2sc.
| This journal is © The Royal Society of Chemistry 2026 |