
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Optimizing the chloroallene pathway toward the one-pot synthesis of rubrene
Alberto Ongaro†
 a,
Matteo Benetazzo†a,
Giulia Tecchioa,
Simone Silvestrini‡
a,
Federico Rastrellia,
Luca Vaghib,
Antonio Papagnib,
Tommaso Carofiglioa and
Michele Maggini*ac
a,
Matteo Benetazzo†a,
Giulia Tecchioa,
Simone Silvestrini‡
a,
Federico Rastrellia,
Luca Vaghib,
Antonio Papagnib,
Tommaso Carofiglioa and
Michele Maggini*ac
aDepartment of Chemical Sciences, University of Padova, Via F. Marzolo 1, 35131, Padova, Italy. E-mail: michele.maggini@unipd.it
bDepartment of Materials Science, University of Milano Bicocca, Via R. Cozzi 53, 20125, Milano, Italy
cInstitute of Condensed Matter Chemistry and Technologies for Energy ICMATE-CNR, Corso Stati Uniti 4, 35127, Padova, Italy
First published on 19th February 2026
Abstract
Rubrene, a benchmark organic semiconductor, is commonly synthesised from 1,1,3-triphenylpropargyl alcohol (TPPA) via the key chloroallene intermediate (TPCA) through a one-pot protocol whose efficiency is highly sensitive to the TPPA–TPCA conversion conditions. We show that treatment of TPPA with PCl5 in the presence of a base affords up to 85% TPCA formation (HPLC), but optimization of the subsequent high-temperature step is hampered by competing pathways leading to cyclobutene by-products, which complicate purification and limit the robustness of a truly one-pot process. Guided by these observations and by reports on trimethylsilyl chloride (TMSCl)-mediated propargyl–allenyl rearrangements, we developed an alternative TMSCl-based protocol. The combination of TMSCl with a sterically hindered base enables a markedly cleaner one-pot synthesis, delivering rubrene in 68% yield by UV-Vis analysis and 61% yield of the isolated product, with complete suppression of cyclobutene formation and a greatly simplified workup. This study provides a rational framework for controlling the chloroallene reaction manifold and establishes a more practical and reproducible one-pot route to rubrene.
Rubrene 1 (5,6,11,12-tetraphenyl tetracene, Chart 1), first reported in the early twentieth century,1 has attracted wide attention for its potential use in OLED technology2,3 and for its remarkable charge-carrier mobility of up to 40 cm2 V−1 s−1.4 In addition, its ease of crystal growth and resistance to oxidation have further strengthened its value as an organic semiconductor compared to other acenes.1g,5
 | ||
| Chart 1 Molecular structure of rubrene. | ||
Despite its importance in materials science, only a limited number of methods are available for synthesising the rubrene skeleton.6 The parent rubrene is typically obtained from 1,1,3-triphenylpropargyl alcohol 5 (TPPA, Scheme 1), using a well-established, though dated, one-pot protocol,1 whose mechanism has long been debated.
 | ||
| Scheme 1 Selected evolution pathways of TPCA 2 (see ref. 12a for details). | ||
Triphenylchloroallene 2 (1,3,3-triphenyl-1-chloropropadiene, TPCA), formed by reacting TPPA 5 with a chloride-based activating reagent, was proposed as the key intermediate in the process.7 The first reliable mechanism of rubrene formation was proposed by Rigaudy and Capdevielle in 1977.8 In line with the known reactivity of allenes, TPCA 2 undergoes thermal dimerization to give the bis-allene diradical 3 (Scheme 1). Studies by Roberts and Sharts,9 further supported by Gajewski and Shi,10 demonstrated that the formation of 3 is non-reversible. The diradical subsequently collapses into a mixture of bismethylidene-cyclobutane isomers, which ultimately rearrange to give rubrene. The room-temperature solvolysis of TPCA 2 has been reported to afford 1,3,3-triphenyl-2-propen-1-one (TPE) 4, providing an alternative reaction pathway available to the chloroallene intermediate.11 Previous studies have also shown that diradical 3, besides evolving into rubrene, can also undergo chlorine elimination to form cyclobutene derivative 6, which becomes the predominant product when electron-donating substituents are present on the phenyl rings of the parent propargyl alcohol.12 These findings highlight the pivotal role of the chloroallene intermediate and the experimental conditions that direct its evolution toward rubrene, which under the reported one-pot protocol is obtained in moderate yields.12a In this work, we report a study aimed at improving rubrene yields through a truly one-pot protocol by enhancing the conversion of TPPA into the key TPCA intermediate and controlling its subsequent evolution into rubrene. Achieving this goal requires careful attention to three aspects: (i) precise control of the experimental conditions governing TPCA formation; (ii) efficient promotion of its dimerization, the crucial step leading to rubrene; and (iii) minimisation of competing pathways, such as hydrolysis of TPCA back to TPPA or formation of TPE. The conversion of TPPA into TPCA can be achieved using different chloride-based reagents, each operating through a distinct substitution mechanism (Scheme 2). With mesyl chloride (CH3SO2Cl, MsCl) in the presence of triethylamine (TEA), chloride is released and the highly electrophilic sulfene intermediate 7 is generated.13 This reacts with the hydroxyl group of TPPA to form the mesyl ester,14 which is then attacked by chloride at the β-alkynyl carbon, followed by elimination of the mesylate to give TPCA 2 (Scheme 2, mechanism A). In contrast, thionyl chloride (SOCl2) forms a chlorosulfite intermediate that undergoes an internal nucleophilic substitution with extrusion of SO2, directly affording TPCA 2 (Scheme 2, mechanism B). Although no kinetic data are available, mechanism A is generally viewed as a chloride-mediated displacement on an activated intermediate, whereas mechanism B follows an intramolecular SNi-type pathway. The essential distinction between these pathways lies in the role of chloride: mechanism A requires chloride in solution, whereas mechanism B does not. TPPA-to-TPCA conversion via pathways consistent with route B has been reported to proceed rapidly and in high yield under conditions that disfavor free chloride in solution, which would be incompatible with a purely intermolecular chloride addition mechanism.1f This has direct implications for the reaction conditions: in mechanism A, the amine hydrochloride salt must be soluble in the reaction solvent,12b while in principle this requirement does not apply to mechanism B.
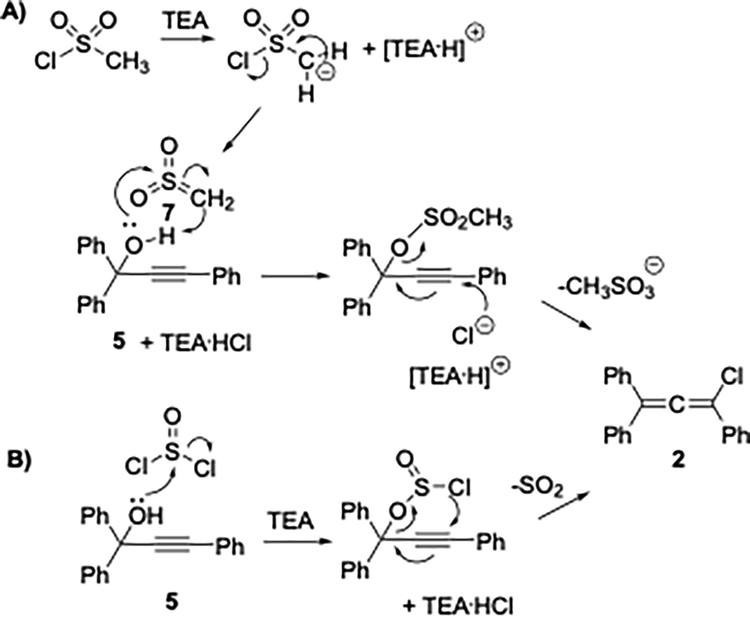 | ||
| Scheme 2 Reaction of TPPA 5 with CH3SO2Cl (A) and SOCl2 (B). | ||
We first examined TPCA formation to identify the most effective reagent for converting TPPA into TPCA across both mechanistic pathways. For pathway A, we tested MsCl. For pathway B, we selected oxalyl chloride, the Vilsmeier reagent (chloromethylene dimethyliminium chloride), obtained from N,N-dimethylformamide and POCl3, PCl5, and dichlorotriphenylphosphorane (Ph3PCl2) that was generated in situ by reacting triphenylphosphine with triphosgene, using in all cases a solvent in which the hydrochloride salt of the base employed was soluble. SOCl2 was not investigated further, as it represents the standard reagent for rubrene synthesis.1 This systematic investigation not only broadened our understanding of the TPPA–TPCA transformation but also enabled us to identify the experimental conditions that most effectively enhance rubrene yield. Reactions were carried out in dichloromethane at 0 °C for 20 min, using a TPPA/chloride-based reagent/base ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1.3:2.5 at a TPPA concentration of 0.14 M. Unreacted TPPA and the side product TPE were qualitatively identified by HPLC-MS with diode-array detection at 250 nm, while TPCA was quantified against coronene as an internal standard at 300 nm. The presence of TPCA and/or TPE was further confirmed by UDEFT 13C NMR spectroscopy in the 190–210 ppm region, with diagnostic resonances at 204 ppm for the sp-hybridised allenic carbon of TPCA and at 192 ppm for the carbonyl carbon of TPE. UDEFT (uniform driven equilibrium Fourier transform) 13C NMR is particularly useful for crude mixtures rich in aromatic carbons, where long T1 relaxation times cause signal saturation in conventional 13C NMR. Originally employed in 29Si NMR spectroscopy, the DEFT experiment is designed to drive the magnetization of slowly relaxing nuclei back to equilibrium, reducing T1-dependent variations in signal intensity and enabling the use of short recycling delays. The use of adiabatic pulses further ensures accurate re-equilibration after each transient, minimising cumulative errors in magnetisation trajectories. Full experimental details, including chromatograms and spectra, are provided in the SI. UDEFT 13C NMR enabled detection and qualitative identification of an additional allenaminium chloride by-product, (TTPAC; see SI, Scheme S2), likely arising from nucleophilic attack of TEA on TPCA.15 To avoid this competitive substitution, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was therefore selected as a non-nucleophilic base to suppress this pathway and enable cleaner TPCA formation. Table 1 summarises the reactivity of TPPA with selected chloride-based reagents.
:1.3:2.5 at a TPPA concentration of 0.14 M. Unreacted TPPA and the side product TPE were qualitatively identified by HPLC-MS with diode-array detection at 250 nm, while TPCA was quantified against coronene as an internal standard at 300 nm. The presence of TPCA and/or TPE was further confirmed by UDEFT 13C NMR spectroscopy in the 190–210 ppm region, with diagnostic resonances at 204 ppm for the sp-hybridised allenic carbon of TPCA and at 192 ppm for the carbonyl carbon of TPE. UDEFT (uniform driven equilibrium Fourier transform) 13C NMR is particularly useful for crude mixtures rich in aromatic carbons, where long T1 relaxation times cause signal saturation in conventional 13C NMR. Originally employed in 29Si NMR spectroscopy, the DEFT experiment is designed to drive the magnetization of slowly relaxing nuclei back to equilibrium, reducing T1-dependent variations in signal intensity and enabling the use of short recycling delays. The use of adiabatic pulses further ensures accurate re-equilibration after each transient, minimising cumulative errors in magnetisation trajectories. Full experimental details, including chromatograms and spectra, are provided in the SI. UDEFT 13C NMR enabled detection and qualitative identification of an additional allenaminium chloride by-product, (TTPAC; see SI, Scheme S2), likely arising from nucleophilic attack of TEA on TPCA.15 To avoid this competitive substitution, 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) was therefore selected as a non-nucleophilic base to suppress this pathway and enable cleaner TPCA formation. Table 1 summarises the reactivity of TPPA with selected chloride-based reagents.
| Reagent | Base | TPPAa | ATPCA/AISa | ATPEa | TTPACb | |
|---|---|---|---|---|---|---|
| a HPLC-MS; IS = internal standard.b UDEFT 13C NMR.c Oxalyl chloride.d Vilsmeier reagent. | ||||||
| 1 | MsCl | TEA | Traces | 0.29 | No | Yes |
| 2 | OxClc | TEA | Yes | 0.58 | No | No |
| 3 | VRd | TEA | Yes | 0.20 | No | Yes |
| 4 | PCl5 | TEA | Traces | 0.11 | Traces | Yes |
| 5 | Ph3PCl2 | TEA | Yes | 0.12 | Traces | No |
| 6 | MsCl | DBU | Yes | 0.41 | No | No |
| 7 | PCl5 | DBU | Traces | 0.85 | Traces | No |
Under optimised conditions (PCl5/DBU in CH2Cl2 at 0 °C for 20 min), TPCA was formed in 85% yield, as determined by HPLC analysis. Since the thermal evolution of TPCA into rubrene requires temperatures above 100 °C, dichloromethane was replaced by 1,2-dichlorobenzene (ODCB) to enable both the TPPA–TPCA rearrangement and the subsequent high-temperature steps using a single solvent. During optimisation, partial decomposition of DBU was observed under prolonged heating, prompting its replacement with collidine, a more thermally robust base. In the optimised procedure, TPPA and PCl5 in ODCB were first reacted at 0 °C for 30 min, after which collidine was added and the mixture heated at 140 °C. Rubrene formation was monitored by UV-Vis spectroscopy and showed a gradual increase over time, reaching a maximum yield of 56% after extended heating (24 h) at 140 °C. Analysis of the crude reaction mixture after solvent removal and MeOH trituration revealed the presence of rubrene together with rubrene endoperoxide and cyclobutene 6, consistent with the mechanistic framework outlined in Scheme 1, with endoperoxide formation likely arising from adventitious oxygen and light exposure. Importantly, the rubrene endoperoxide can be quantitatively reverted to rubrene upon heating at 160 °C,16 indicating that this impurity is reversible and does not represent an intrinsic limitation of the process. In contrast, cyclobutene 6 arises from an irreversible diversion of the chloroallene manifold, thereby directly impacting both the yield and purification of rubrene. These observations highlight the intrinsic complexity of the TPCA reaction network, whose thermal evolution proceeds through diradical intermediates that can irreversibly collapse into both rubrene and cyclobutene 6, with a marked but not exclusive preference for rubrene. Alternative heating strategies were therefore explored. Continuous-flow experiments conducted at 140–160 °C for short residence times did not provide advantages over batch processing. In contrast, microwave irradiation efficiently accelerated the transformation. Screening at 140–180 °C identified 160 °C for 3 h as optimal, affording rubrene in 59% yield by UV-Vis analysis, comparable to prolonged conventional heating but with a markedly reduced reaction time (see the SI). Although these experiments confirmed the robustness of the TPCA-based pathway, they also underscored practical limitations of PCl5-based conditions when applied to the one-pot process, primarily due to the formation of cyclobutene by-products that complicate purification and reduce yields. This prompted us to consider alternative chloride-based promoters capable of maintaining efficient chloroallene formation while improving the downstream product profile. In this context, we were inspired by literature reports describing trimethylsilyl chloride (TMSCl)-mediated propargyl-allenyl rearrangements,17 where TMSCl acts as both an alcohol-activating promoter and a chloride source. Accordingly, TMSCl was evaluated under mild, non-interfering basic conditions, using the sterically hindered, non-nucleophilic base 2,6-di-tert-butylpyridine (DTBP). Addition of DTBP to TPPA in ODCB at room temperature, followed by 1.1 equivalents of TMSCl and heating at 140 °C for 8 h, resulted in a remarkably clean one-pot conversion to rubrene. Under these conditions, rubrene was obtained in 66% yield by UV-Vis analysis and 59% yield of isolated product after simple MeOH trituration of the reaction crude and filtration. Only trace amounts of rubrene endoperoxide were detected, and cyclobutene 6 was not observed. After filtration of rubrene, TPE was identified as the major component in the filtrate, likely formed via hydrolytic interception of TPCA by trace adventitious water (see the SI). Previous studies have highlighted the delicate balance governing the TPCA reaction manifold, in which the competition between rubrene and cyclobutene formation depends on subtle changes in reaction conditions and substrate-dependent factors. To assess the robustness of the TMSCl-mediated one-pot protocol beyond the initial small-scale experiments, we performed additional reactions under modified conditions. Scale-up of the process starting from 1 g of TPPA afforded rubrene in 56% yield of isolated product (69% by UV-Vis analysis), confirming that the observed pathway selectivity is not a small-scale artefact. Moreover, doubling the initial TPPA concentration at 140 °C under otherwise identical scale-up conditions afforded a comparable yield (68% by UV-Vis analysis and 61% yield of isolated product) within the same reaction time, without evidence for increased cyclobutene formation (see the SI). These results indicate that, under TMSCl-mediated conditions, the evolution of the TPCA intermediate toward rubrene remains robust upon scale-up and under moderately increased concentration. Accordingly, comparisons with previously reported protocols are best interpreted in terms of operational robustness rather than absolute yield under matched conditions. In the presence of TMSCl, the propargyl alcohol is activated via silyl ether formation. Under the reaction conditions, C–O bond cleavage provides access to a delocalised cationic manifold (Scheme 3), with the positive charge distributed over the propargyl/allenyl framework. While the propargyl cationic form is expected to be the more stabilised contributor, the allenyl form is likely more reactive toward nucleophilic capture, leading to preferential chloride trapping and formation of TPCA.18 This qualitative picture is consistent with the predominant formation of TPCA and with the cleaner one-pot behaviour observed under TMSCl-based conditions.
 | ||
| Scheme 3 Plausible pathway for the TMSCl-mediated conversion of TPPA into TPCA. | ||
In conclusion, this study examines and optimises the commonly employed TPPA–TPCA–rubrene sequence with the goal of establishing a truly one-pot process, where all steps proceed in a single operation without isolation of intermediates. The use of PCl5 proved instrumental in enabling a systematic analysis of the TPCA reactivity landscape, revealing both the robustness of the pathway and the intrinsic competition between productive and unproductive reaction channels. Building on these insights, the adoption of TMSCl provided a decisive improvement at the level of the one-pot process. Under TMSCl-mediated conditions, rubrene is obtained in 61% yield of isolated product after simple MeOH trituration of the crude reaction mixture and filtration, with effective suppression of cyclobutene by-products and limitation of oxidation to reversible endoperoxide formation. By eliminating the low-temperature step and a key purification bottleneck, this single-solvent, one-pot approach enables more practical and reproducible synthetic access to rubrene. More broadly, these results illustrate how subtle changes in chloride activation can bias the reactivity of the chloroallene manifold, providing a rational framework for controlling a complex cascade process in rubrene synthesis.
A. O. investigation and validation; M. B. investigation and validation; G. T. investigation; S. S. conceptualisation and investigation; F. R. methodology; L. V. investigation; A. P. conceptualisation, writing – original draft, and writing – review and editing; T. C. methodology, M. M. conceptualisation, writing – original draft, writing – review and editing, and supervision.
Conflicts of interest
There are no conflicts to declare.Data availability
All experimental and characterisation data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general experimental procedures, HPLC-MS traces, 1H and 13C NMR spectra of selected compounds and 13C NMR UDEFT spectra (PDF). See DOI: https://doi.org/10.1039/d6cc00341a.Acknowledgements
We thank L. Dordevic (University of Padova) for his kind support with MW experiments, and M. Parravicini (University of Milano Bicocca) and L. Mezzarobba (University of Padova) for the initial synthetic studies. A. O. gratefully acknowledges financial support from the Department of Chemical Sciences, University of Padova (P-DiSC project C93C22009260001 - MUR-DE 2023 Chemical Complexity - C2).References
- (a) C. Moureu, C. Dufraisse and P. M. Dean, Compt. Rend., 1926, 182, 1440 CAS; (b) C. Moureu, C. Dufraisse and L. Enderlin, Compt. Rend., 1928, 187, 406 CAS; (c) C. Dufraisse and L. Enderlin, Bull. Soc. Chim., 1932, 51, 132 Search PubMed; (d) C. Dufraisse, Bull. Soc. Chim., 1933, 53, 789 CAS; (e) B. Furniss, Vogel's Textbook of Practical Organic Chemistry, Longman Scientific & Technical, Essex, UK, 5th edn, 1989 Search PubMed; (f) R. A. Irgashev, A. S. Steparuk, A. E. Aleksandrov, G. L. Rusinov, I. R. Sayarov and A. R. Tameev, Molbank, 2026, M2122 CrossRef; (g) F. Henke, A. Kishonti, S. L. Woltering and K. Leo, J. Org. Chem., 2026, 91, 1969 CrossRef CAS PubMed.
- M. M. Richter, Chem. Rev., 2004, 104, 3003 CrossRef CAS PubMed.
- (a) K. Saxena, D. S. Mehta, V. K. Rai, R. Srivastava, G. Chauhan, M. N. Kamalasanan and V. K. Jain, Phys. Status Solidi A, 2009, 206, 1660 Search PubMed; (b) H.-H. Huang, S.-Y. Chu, P.-C. Kao and Y.-C. Chena, Thin Solid Films, 2008, 516, 5669 CrossRef CAS; (c) D.-H. Lee, J. S. Choi, H. Chae, C.-H. Chung and S. M. Cho, Curr. Appl. Phys., 2009, 9, 161 CrossRef; (d) Z. Wang, Y. Lou, S. Naka and H. Okada, J. Lumin., 2010, 130, 1198 CrossRef CAS; (e) B. Ding, W. Zhu, X. Jiang and Z. Zhang, Solid State Commun., 2008, 148, 226 CrossRef CAS; (f) A. M. C. Ng, A. B. Djurišic, W.-K. Chan and J.-M. Nunzi, Chem. Phys. Lett., 2009, 474, 141 CrossRef CAS.
- (a) R. W. I. de Boer, M. E. Gershenson, A. F. Morpurgo and V. Podzorov, Phys. Status Solidi A, 2004, 201, 1302 Search PubMed; (b) J. Takeya, M. Yamagishi, Y. Tominari, R. Hirahara, Y. Nakazawa, T. Nishikawa, T. Kawase, T. Shimoda and S. Ogawa, Appl. Phys. Lett., 2007, 90, 102120 CrossRef; (c) K. K. Zhang, K. Tan, C. Zou, M. Wikberg, L. E. McNeil, S. G. Mhaisalkar and C. Kloc, Org. Electron., 2010, 11, 1928 Search PubMed.
- (a) V. C. Sundar, J. Zaumseil, V. Podzorov, E. Menard, R. L. Willett, T. Someya, M. E. Gershenson and J. A. Rogers, Science, 2004, 303, 1644 Search PubMed; (b) D. Braga and G. Horowitz, Adv. Mater., 2009, 21, 1473 CrossRef CAS; (c) A. Molinari, I. Gutiérrez, I. N. Hulea, S. Russo and A. F. Morpurgo, Appl. Phys. Lett., 2007, 90, 212103 CrossRef; (d) V. Podzorov, E. Menard, A. Borissov, V. Kiryukhin, J. A. Rogers and M. E. Gershenson, Phys. Rev. Lett., 2004, 93, 086602 CrossRef CAS PubMed; (e) J. E. Anthony, Chem. Rev., 2006, 106, 5028 CrossRef CAS PubMed; (f) D. A. F. da Silva, E.-G. Kim and J.-L. Brédas, Adv. Mater., 2005, 17, 1072 CrossRef; (g) M. L. Clapham, E. C. Murphy and C. J. Douglas, Synthesis, 2021, 461 CAS.
- (a) A. S. Paraskar, A. R. Reddy, A. Patra, Y. H. Wijsboom, O. Gidron, L. J. W. Shimon, G. Leitus and M. Bendikov, Chem. – Eur. J., 2008, 14, 10639 CrossRef CAS PubMed; (b) E. V. Banide, B. C. Molloy, Y. Ortin, H. Müller-Bunz and M. J. McGlinchey, Eur. J. Org. Chem., 2007, 2611 CrossRef CAS; (c) L. E. Harrington, J. F. Britten and M. J. McGlinchey, Tetrahedron Lett., 2003, 44, 8057 Search PubMed; (d) L. E. Harrington, J. F. Britten and M. J. McGlinchey, Org. Lett., 2004, 6, 787 CrossRef CAS PubMed; (e) E. V. Banide, Y. Ortin, C. M. Seward, L. E. Harrington, H. Müller-Bunz and M. J. McGlinchey, Chem. – Eur. J., 2006, 12, 3275 CrossRef CAS; (f) J. A. Dodge, J. D. Bain and A. R. Chamberlin, J. Org. Chem., 1990, 55, 4190 CrossRef CAS; (g) Z. Chen, P. Müller and T. M. Swager, Org. Lett., 2006, 8, 273 CrossRef CAS PubMed; (h) Y.-C. Lin and C.-H. Lin, Org. Lett., 2007, 9, 205 Search PubMed; (i) E. Yagodkin and C. J. Douglas, Tetrahedron Lett., 2010, 51, 3037 CrossRef CAS; (j) Y. Hou, X. Chi, X. Wan and Y. Chen, J. Mol. Struct., 2008, 889, 265 Search PubMed.
- (a) A. J. Brattesani, E. Maverick, O. J. Jr Muscio and T. L. Jacobs, J. Org. Chem., 1992, 57, 7346 CrossRef CAS; (b) E. V. Dehmlow and G. C. Ezimora, Tetrahedron Lett., 1972, 13, 1265 CrossRef; (c) E. V. Dehmlow, Angew. Chem., Int. Ed. Engl., 1972, 11, 322 CAS; (d) B. Alcaide, P. Almendros and C. Aragoncillo, Chem. Soc. Rev., 2010, 39, 783 RSC.
- (a) J. Rigaudy and P. Capdevielle, Tetrahedron, 1977, 33, 767 CrossRef CAS; (b) T. L. Jacobs and D. M. Fenton, J. Org. Chem., 1965, 30, 1808 CrossRef CAS.
- J. D. Roberts and C. M. Sharts, Org. React., 1962, 12, 1 CAS.
- (a) J. J. Gajewski and C. N. Shih, J. Am. Chem. Soc., 1967, 89, 4532 CrossRef CAS; (b) J. J. Gajewski and C. N. Shih, J. Am. Chem. Soc., 1969, 91, 5900 CrossRef CAS; (c) J. J. Gajewski and C. N. Shih, J. Org. Chem., 1972, 37, 64 CrossRef CAS; (d) J. J. Gajewski and C. N. Shih, J. Am. Chem. Soc., 1972, 94, 1675 CrossRef CAS.
- M. D. Schiavelli, S. C. Hixon, H. W. Moran and C. J. Boswell, J. Am. Chem. Soc., 1971, 93, 6989 CrossRef CAS.
- (a) D. Braga, A. Jaafari, L. Miozzo, M. Moret, S. Rizzato, A. Papagni and A. Yassar, Eur. J. Org. Chem., 2011, 4160 CrossRef CAS; (b) L. M. Uttiya, E. M. Fumagalli, S. Bergantin, R. Ruffo, M. Parravicini, A. Papagni, M. Moret and A. Sassella, J. Mater. Chem. C, 2014, 2, 4147 RSC.
- (a) W. E. Truce and R. W. Campbell, J. Am. Chem. Soc., 1966, 88, 3599 Search PubMed; (b) J. F. King and J. R. Manoir, J. Am. Chem. Soc., 1975, 97, 2566 CrossRef CAS.
- (a) T. Katsuhira, T. Harada and A. Oku, J. Org. Chem., 1994, 59, 4010 CrossRef CAS; (b) W. E. Truce, R. W. Campbell and J. R. Norell, J. Am. Chem. Soc., 1964, 86, 288 CrossRef CAS.
- G. V. Karunakar and M. Periasamy, J. Org. Chem., 2006, 71, 7463 Search PubMed.
- J. T. Ly, S. A. Lopez, J. B. Lin, J. J. Kim, H. Lee, E. K. Burnett, L. Zhang, A. Aspuru-Guzik, K. N. Houk and A. L. Briseno, J. Mater. Chem. C, 2018, 6, 3757 Search PubMed.
- Z. Sun, K. Xiang, H. Tao, L. Guo and Y. Li, Org. Biomol. Chem., 2018, 16, 6133 RSC.
- Although theoretical calculations (B3LYP/6-31G(d,p) level) indicate, based on the electrostatic potential map, a higher positive charge density at the diphenyl-substituted propargylic carbon, the allenyl contributor presents a more accessible sp-hybridised electrophilic centre, leading to chloride capture and TPCA formation. It is worth noting that access to TPCA may also be envisaged via chloride attack at the β-alkynyl carbon of the silyl ether, followed by elimination of the trimethylsilyl alkoxide, in a pathway analogous to mechanism A in Scheme 2.
Footnotes |
| † These authors contributed equally. |
| ‡ S.S. present address: Procter and Gamble, Brussels Innovation Centre, Temselaan 100, Strombeek-bever, B-1853, Belgium. |
| This journal is © The Royal Society of Chemistry 2026 |