
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot asymmetric sulfoxidation using in situ generated H2O2 from H2 and O2 catalyzed by rhodium and vanadium complexes
Takeshi
Yatabe
 abc,
Naoto
Kanda
ab,
Mizuki
Tsuji
ab,
Sho
Sakogashira
ab,
Norifumi
Tashiro
d,
Hidetaka
Nakai
e and
Seiji
Ogo
*abc
abc,
Naoto
Kanda
ab,
Mizuki
Tsuji
ab,
Sho
Sakogashira
ab,
Norifumi
Tashiro
d,
Hidetaka
Nakai
e and
Seiji
Ogo
*abc
aDepartment of Chemistry and Biochemistry, Graduate School of Engineering, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan. E-mail: ogo.seiji.872@m.kyushu-u.ac.jp
bInternational Institute for Carbon-Neutral Energy Research (WPI Academy I2CNER), Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
cCenter for Small Molecule Energy, Kyushu University, 744 Moto-oka, Nishi-ku, Fukuoka 819-0395, Japan
dMitsubishi Gas Chemical Company Inc., Mitsubishi Building, 5-2, Marunouchi 2-chome, Chiyoda-ku, Tokyo 100-8324, Japan
eDepartment of Energy and Materials, Faculty of Science and Engineering, Kindai University, 3-4-1 Kowakae, Higashi-Osaka, Osaka 577-8502, Japan
First published on 11th February 2026
Abstract
This paper reports the first example of a one-pot asymmetric sulfoxidation using in situ generated H2O2. This reaction is catalyzed by two homogeneous catalysts: a Rh complex for the direct synthesis of H2O2 from H2 and O2, and a V complex for the asymmetric oxidation of sulfides.
In the chemical industry, one-pot oxidation reactions that employ in situ generated H2O2 from H2 and O2 are highly attractive because they eliminate the need to purify, transport, or store explosive H2O2 solutions, thereby reducing both capital and operational costs in large-scale processes.1–3 While one-pot oxidations of alkanes, benzene, propylene, and sulfides using in situ generated H2O2 have been extensively studied,1–8 no example of asymmetric oxidation using in situ formed H2O2 has, to the best of our knowledge, been achieved to date (Fig. 1(a)).
 | ||
| Fig. 1 (a) The target sequential reaction. (b) The strategy for the sequential reaction. | ||
This paper focuses on the asymmetric oxidation of sulfides, which affords chiral sulfoxides that are valuable for pharmaceuticals and asymmetric synthesis.9,10 To achieve this one-pot asymmetric sulfoxidation, we consider the following requirements: (1) the direct synthesis of H2O2 and the asymmetric sulfoxidation be carried out by different catalysts, and (2) each catalyst must retain its activity in the presence of the additives, reactants, products, and intermediates associated with the other reaction (Fig. 1(b)). Our strategies are therefore as follows. (1) We employed our recently developed homogeneous Rh catalyst [RhII2(L′)2(OH2)4](NO3)4 {[1](NO3)4, L′ = 2,6-bis(1-methylimidazol-2-ylidene)pyridine} for the direct synthesis of H2O211 and a novel homogeneous V catalyst [VV(O)(L)(OiPr)] {2, L = 5-chloro-3-(((3,3-dimethyl-1-oxidobutan-2-yl)imino)methyl)-[1,1′-biphenyl]-2-olate} for the asymmetric sulfoxidation. (2) To prevent deactivation of the V complex by sodium acetate used as an additive in the H2O2 synthesis, and to avoid poisoning of the Rh catalyst for H2O2 formation by either the sulfide substrate or the resulting sulfoxide, the one-pot sulfoxidation was carried out in a biphasic system, and a sequential reaction protocol was employed (Fig. 1(b)).
In the first step of this sequential reaction, the Rh catalyst 1 is used to generate a small amount of H2O2 in water. The second step requires the development of a highly active V catalyst capable of capturing the small amount of H2O2 in a solvent immiscible with water. After screening for enantioselectivity (Table S1, SI) and catalytic activity, the V complex 2, featuring a tridentate Schiff base ligand and soluble in CH2Cl2, was selected as the catalyst for the second step.12–21
We first describe the synthesis of the V complex 2, followed by the optimization of an asymmetric sulfoxidation using 2 and authentic H2O2. Finally, we demonstrate the asymmetric sulfoxidation using in situ generated H2O2 from H2 and O2, catalyzed by the Rh and V complexes, and discuss the whole reaction cycle.
Complex 2 was synthesized by the reaction of [VV(O)(OiPr)3] with H2L in iPrOH and characterized by using 1H NMR (Fig. S1, SI), UV-vis absorption (Fig. 2), and IR spectroscopy (Fig. 3), electron-spray ionization mass spectrometry (Fig. S2, SI), and elemental analysis.
 | ||
| Fig. 2 UV-vis-NIR absorption spectra of (a) 2 (36.7 µM) in methanol and (b) the reaction solution of 2 (36.7 µM) with 10 equivalents of H2O2 in methanol. The light path length is 1.0 cm. | ||
 | ||
| Fig. 3 IR spectra of (a) 2, (b) 3 obtained from the reaction of 2 with H216O2 and (c) 18O-labled 3 obtained from the reaction of 2 with H218O2. | ||
A 1H NMR spectrum of 2 in the diamagnetic region shows the signals derived from L and iPrO− (Fig. S1). An UV-vis absorption spectrum of 2 exhibits the absorption bands at 338 nm and 370–500 nm (Fig. 2a), which are assigned as intraligand π → π* transition and phenolate oxygen atom or alcoholate oxygen atom → d orbital transition each other.22–24 An IR spectrum of 2 shows a typical IR band of ν(V![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) at 966 cm−1 (Fig. 3a).13–18,24
O) at 966 cm−1 (Fig. 3a).13–18,24
We have examined the reactivity of 2 toward H2O2. Complex 2 reacts with H2O2 in methanol at room temperature to form a V peroxo complex [VV(O)(L)(O2)]− (3) (eqn (1)). This reaction was monitored by UV-vis absorption (Fig. 2b) and IR spectroscopy (Fig. 3). Upon addition of 10 equivalents of H2O2 into 2 in methanol, characteristic bands of 2 disappeared together with the appearance of the absorption bands derived from 3 with broad bands at 338 and 434 nm (Fig. 2b). The latter absorption band is assigned as the peroxo-to-V charge transfer band, which is typical for the V(V) peroxo complex.25,26
 | (1) |
An IR spectrum in solid state of 3 shows an isotope-sensitive band at 897 cm−1, which is assigned as the O–O bond stretching vibration (Fig. 3). Isotopic substitution of 16O2 by 18O2 in the peroxido ligand of 3 resulted in a band shift to 868 cm−1. The wavenumber of the O–O bond stretching vibration is similar to that found in other V peroxide complexes.25–29 For example, V peroxo complex having Schiff base ligand shows the corresponding vibration at 878 cm−1.29
Since complex 2 forms the peroxo complex 3 by the reaction with H2O2, we initiated our study by examining the asymmetric sulfoxidation of thioanisole as a model substrate by complex 2 in the presence of authentic H2O2. The reaction conditions were designed taking into account those used for the direct H2O2 synthesis catalyzed by 1. The produced sulfoxide was characterized and quantified by 1H NMR spectroscopy and its enantiomeric excess was determined by chiral HPLC after purification.
The reaction solvent was explored by using several biphasic system (entries 2–7 in Table S2, SI) in the presence of sodium acetate to meet the requirement 2 because the direct H2O2 synthesis using 1 proceeded in the aqueous solution with sodium acetate that acts as a Lewis base to remove protons of H2 in the reaction of 1 with H2.11 An CH3CN inhibits the direct H2O2 synthesis and was therefore excluded as a candidate solvent. The concentration of H2O2 was adjusted to 55 mM in the aqueous phase, which corresponds to the predicted amount formed by the direct H2O2 synthesis by 1. A significant yield of 61% was observed in H2O/CH2Cl2 (1/1) (entry 2 in Table S2) compared to the other biphasic system (18–58%) (entries 3–7 in Table S2). The highest ee of 60% was observed in H2O/CH2Cl2, whereas the ee values obtained with other solvents ranged from 29% to 53% ee. In contrast to the biphasic system, the ee value of the asymmetric sulfoxidation decreased drastically in methanol (2% ee, entry 8 in Table S2). This decrease is attributed to the decomposition of 2 in the presence of sodium acetate which dissociates L from V center (Fig. S3, SI).
Catalyst concentration was investigated the range from 1 to 20 mol% (entries 2, 9–11 in Table S2). Catalyst loading of 10 mol% resulted in the highest yield with 61% and 60% ee (entry 2 in Table S2). Lowering the reaction temperature to 0 °C decreased the yield from 61% to 36% and ee from 60 to 32% ee (entry 12 in Table S2). Reaction time of 2 h was enough to obtain the sulfoxide (entries 1, 2, and 13 in Table S2). A series of control experiments confirmed that the methyl phenyl sulfoxide was not formed in the absence of 2 or H2O2 (entries 14 and 15 in Table S2). These results indicate that complex 2 catalyzes the asymmetric sulfoxidation in the biphasic H2O/CH2Cl2 (1/1) system, even in the presence of sodium acetate. Thus, the conditions of entry 1 were used as standard conditions.
The obtained ee values were moderate compared to those of the reported asymmetric sulfoxidation catalyzed by the other V complex (up to 98% ee).19,20 The yield (< 64%) is slightly lower than that of previously reported systems, in which the sulfoxidation of thioanisole catalyzed by V complexes was carried out in dry CH2Cl2. This may be due to the low concentration of H2O2 used for the sulfoxidation by 2 or the decomposition of 2 in the presence of water.
With the optimized reaction conditions of the sulfoxidation, we began to investigate the one-pot asymmetric sulfoxidation of various sulfides using in situ generated H2O2 from H2 and O2. The one-pot asymmetric sulfoxidation was carried out at room temperature using 1 and 2 as catalysts for direct H2O2 synthesis and asymmetric sulfoxidation, respectively. This is our strategy 1 and meets the requirements 1 as mentioned above. Catalyst 1 was synthesized according to the literature method.11 The one-pot sulfoxidation, carried out in a sequential manner, was performed through the initial formation of H2O2 from H2 and O2 using catalyst 1, followed by the addition of 2 and sulfides to the reaction solution to initiate the sulfoxidation reaction. The H2/O2 (95/5) pressure was adjusted to avoid the explosion and maximize the yield of H2O2.11 The formed sulfoxides were identified and quantified by 1H NMR spectroscopy after purification (Fig. S4–14, SI).
The yield of the one-pot sulfoxidation of thioanisole (entry 1 in Table 1) was comparable to that of the sulfoxidation using authentic H2O2 (entry 1 in Table S2), suggesting the one-pot sulfoxidation was not suppressed by Rh complexes present in the reaction mixture. In other words, this demonstrates that the requirement 2 was successfully fulfilled by the strategy 2. With the thioanisole derivatives, electron-donating substituents increase the yield of sulfoxide (entries 1–5 in Table 1). This trend of yield is similar to those of the reported asymmetric sulfoxidation by Ru catalyst.30 The highest yield of 88% was obtained with benzyl methyl sulfide (entry 6 in Table 1), while the moderate yields of 33% and 38% were observed in naphthyl methyl sulfide and benzyl phenyl sulfide, respectively (entries 7 and 8 in Table 1).
|
|
||||
|---|---|---|---|---|
| Entry | Sulfide | Sulfoxide | Yieldb (%) (TON)c | eed (%) |
a Reaction conditions for direct synthesis of H2O2: complex 1 (0.4 µmol), sodium acetate (2 mmol), H2O (4 mL), H2 (1.9 MPa), O2 (0.09 MPa), r.t., 12 h. Reaction conditions for asymmetric sulfoxidation: complex 2 (20 µmol), sulfide (200 µmol), H2O/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v, 8 mL), r.t., 2 h.
b Determined by 1H NMR analysis. The values are isolated yields.
c Turnover numbers (TONs, mol of sulfoxide/mol of 2) based on complex 2.
d Determined by chiral HPLC analysis. :1, v/v, 8 mL), r.t., 2 h.
b Determined by 1H NMR analysis. The values are isolated yields.
c Turnover numbers (TONs, mol of sulfoxide/mol of 2) based on complex 2.
d Determined by chiral HPLC analysis.
|
||||
| 1 |

|
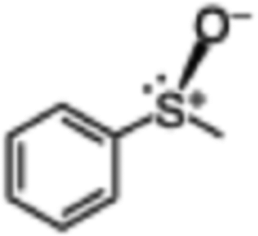
|
56 (5.6) | 59 |
| 2 |

|

|
50 (5.0) | 58 |
| 3 |
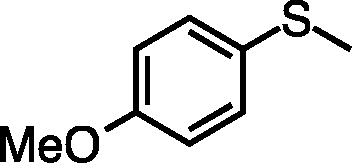
|

|
58 (5.8) | 38 |
| 4 |

|

|
77 (7.7) | 63 |
| 5 |

|

|
67 (6.7) | 39 |
| 6 |

|

|
88 (8.8) | 12 |
| 7 |

|

|
33 (3.3) | 42 |
| 8 |

|

|
38 (3.8) | 69 |
| 9 |

|

|
53 (5.3) | 57 |
| 10 |

|

|
59 (5.9) | 14 |
| 11 |

|

|
48 (4.8) | 13 |
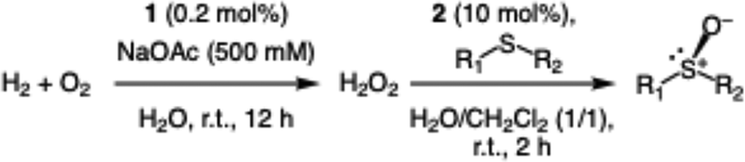
The one-pot sulfoxidation of sulfides bearing one aryl substituent afforded moderate ee values (38–69% ee; entries 1–5 and 7–9 in Table 1). Notably, benzyl phenyl sulfide gave the highest enantioselectivity (69% ee, entry 8 in Table 1), which may be attributed to steric congestion around the sulfur center. In contrast, sulfides bearing two aliphatic substituents exhibited low ee value (<14% ee) (entries 6, 10, and 11 in Table 1), presumably due to enhanced conformational flexibility arising from free rotation about the C–S bonds, which diminishes steric congestion at the sulfur center. Because no overoxidation of the sulfoxide was observed, the enantioselectivity is attributed to asymmetric induction in the sulfoxidation step, not to kinetic resolution.30 The sulfoxides were formed predominantly as the S enantiomer, attributable to the substrate aryl group orienting away from the biphenyl motif of L, as proposed for related chiral vanadium complexes with tridentate Schiff base ligands.20 The maximum yield and enantioselectivity of the one-pot asymmetric sulfoxidation are comparable to those of the reported asymmetric sulfoxidation that used O2 and reductant.31
The asymmetric sulfoxidation of thioanisole using 18O2 instead of 16O2 gives the 18O-incorporating methyl phenyl sulfoxide, confirmed by GC-MS (Fig. S15, SI). This means that the O atom of sulfoxide is derived from in situ generated H2O2. There is no sulfoxide from the reaction without H2, O2, or 1 (entries 1–3 in Table S3, SI). The trace amount of product with 0% ee was formed in the sulfoxidation without 2 (entry 4 in Table S3), which derived from the background reaction of thioanisole with the in situ generated H2O2. This result corresponds to the sulfoxidation without 2 as shown in entry 15 of Table S2 and indicates that complex 1 does not catalyze the sulfoxidation. This circumvention of the undesired sulfoxidation promoted by 1, which produces racemic sulfoxides, is essential for constructing the one-pot asymmetric sulfoxidation using in situ generated H2O2.
Based on above results, we have proposed the reaction mechanism as shown in Fig. 4. The Rh complex 1 gives H2O2 from H2/O2 mixture as reported previously. Complex 2 reacts with in situ generated H2O2 to form the V peroxo complex 3. 3 oxidizes sulfide to sulfoxide and converts to complex 2. This is the first example not only of a one-pot asymmetric sulfoxidation, but also of any one-pot asymmetric oxidation that employs H2O2 generated in situ from H2 and O2.
 | ||
| Fig. 4 Reaction cycle of a one-pot asymmetric sulfoxidation using in situ generated H2O2 from H2 and O2 by means of a Rh and V homogeneous catalysts. | ||
In conclusion, we have demonstrated the principle that one-pot asymmetric sulfoxidation is achievable. This was accomplished by developing a highly active novel V catalyst capable of capturing the small amount of H2O2 generated by the Rh catalyst in the first step, despite chiral V complexes for asymmetric sulfoxidation having been known for approximately 30 years. The combination of a biphasic H2O/CH2Cl2 system and a sequential reaction protocol was essential to prevent deactivation of both catalysts and enable selective operation of each catalytic cycle. This work provides a general strategy for integrating incompatible H2O2-forming and asymmetric catalytic reactions in a single vessel, offering a foundation for the development of future one-pot asymmetric oxidation processes based solely on homogeneous catalysts. We anticipate that these principles will enable the development of a broader range of asymmetric oxidation processes driven by in situ H2O2 generated directly from H2 and O2.
This work was supported by JST CREST (grant number JPMJCR18R2), Japan, JSPS KAKENHI (grant numbers JP26000008 (specially promoted research) and JP22K05130), the Iwatani Naoji Foundation, and TANAKA Precious Metal Memorial Foundation.
Conflicts of interest
There are no conflicts to declare.Data availability
The supporting data has been provided as part of the supplementary information (SI). Supplementary information: Tables S1–S3, Fig. S1–S15, and further experimental details. See DOI: https://doi.org/10.1039/d6cc00169f.References
- S. Zhang, D. Zeng, H. Wang, X. Tang, Y. Jiang and C. Yu, Chem. – Eur. J., 2024, 30, e202402767, DOI:10.1002/chem.202402767.
- J. R. Lewis and G. J. Hutchings, ChemCatChem, 2019, 11, 298–308, DOI:10.1002/cctc.201801435.
- B. Puértolas, A. K. Hill, T. García, B. Solsona and L. Torrente-Murciano, Catal. Today, 2015, 248, 115–127, DOI:10.1016/j.cattod.2014.03.054.
- M. H. Ab Rahim, M. M. Forde, R. L. Jenkins, C. Hammond, Q. He, N. Dimitratos, J. A. Lopez-Sanchez, A. F. Carley, S. H. Taylor, D. J. Willock, D. M. Murphy, C. J. Kiely and G. J. Hutchings, Angew. Chem., Int. Ed., 2013, 52, 1280–1284, DOI:10.1002/anie.201207717.
- T. Miyake, M. Hamada, Y. Sasaki and M. Oguri, Appl. Catal., A, 1995, 131, 33–42, DOI:10.1016/0926-860X(95)00134-4.
- W. Li, L. Chen, M. Qiu, W. Li, Y. Zhang, Y. Zhu, J. Li and X. Chen, ACS Catal., 2023, 13, 10487–10499, DOI:10.1021/acscatal.3c02206.
- C. Shi, B. Zhu, M. Lin and J. Long, Catal. Today, 2010, 149, 132–137, DOI:10.1016/j.cattod.2009.11.004.
- S. Ma, G. Li and X. Wang, Chem. Eng. J., 2010, 156, 532–539, DOI:10.1016/j.cej.2009.04.004.
- R. Bentley, Chem. Soc. Rev., 2005, 34, 609–624, 10.1039/b418284g.
- I. Fernández and N. Khiar, Chem. Rev., 2003, 103, 3651–3705, DOI:10.1021/cr990372u.
- S. Ogo, T. Yatabe, T. Tome, R. Takenaka, Y. Shiota and K. Kato, J. Am. Chem. Soc., 2023, 145, 4384–4388, DOI:10.1021/jacs.2c13149.
- A. G. J. Ligtenbarg, R. Hage and B. L. Feringa, Coord. Chem. Rev., 2003, 237, 89–101, DOI:10.1016/S0010-8545(02)00308-9.
- P. Plitt, H. Pritzkow and R. Krämer, Dalton Trans., 2004, 2314–2320, 10.1039/b403600j.
- M. R. Maurya, S. Agarwal, C. Bader and D. Rehder, Eur. J. Inorg. Chem., 2005, 147–157, DOI:10.1002/ejic.200400211.
- P. Wu, C. Celik, G. Santoni, J. Dallery and D. Rehder, Eur. J. Inorg. Chem., 2008, 5203–5213, DOI:10.1002/ejic.200800772.
- M. Maurya, B. Sarkar, F. Avecilla and I. Correia, Eur. J. Inorg. Chem., 2016, 4028–4044, DOI:10.1002/ejic.201600427.
- M. Debnath, M. Dolai, K. Pal, S. Bhunya, A. Paul, H. M. Lee and M. Ali, Dalton Trans., 2018, 47, 2799–2809, 10.1039/c7dt04718e.
- Z. Chen, Coord. Chem. Rev., 2022, 457, 214404, DOI:10.1016/j.ccr.2021.214404.
- C. Bolm and F. Beinewald, Angew. Chem., Int. Ed. Engl., 1995, 34, 2640–2642, DOI:10.1002/anie.199526401.
- S.-H. Hsieh, Y.-P. Kuo and H.-M. Gau, Dalton Trans., 2007, 97–106, 10.1039/b613212j.
- C. Ohta, H. Shimizu, A. Kondo and T. Katsuki, Synlett, 2002, 161–163, DOI:10.1055/s-2002-19361.
- D. C. Crans and P. K. Shin, J. Am. Chem. Soc., 1994, 116, 1305–1315, DOI:10.1021/ja00083a016.
- S. Battacharya and T. Gosh, Trans. Met. Chem., 2002, 27, 89–94, DOI:10.1023/A:1013499802610.
- J. Hartung, S. Drees, M. Greb, P. Schmidt, I. Svoboda, H. Fuess, A. Murso and D. Stalke, Eur. J. Org. Chem., 2003, 2388–2408, DOI:10.1002/ejoc.200200644.
- M. Sivák, M. Mad’arová, J. Tatiersky and J. Marek, Eur. J. Inorg. Chem., 2003, 2075–2081, DOI:10.1002/ejic.200200380.
- M. R. Maurya, U. Kumar and P. Manikandan, Dalton Trans., 2006, 3561–3575, 10.1039/b600822d.
- M. Časný and D. Rehder, Dalton Trans., 2004, 839–846, 10.1039/b315291j.
- M. R. Maurya, S. Agarwal, C. Bader and D. Rehder, Eur. J. Inorg. Chem., 2005, 147–157, DOI:10.1002/ejic.200400211.
- M. R. Maurya, A. Kumar, M. Ebel and D. Rehder, Inorg. Chem., 2006, 45, 5924–5937, DOI:10.1021/ic0604922.
- H. Tanaka, H. Nishikawa, T. Uchida and T. Katsuki, J. Am. Chem. Soc., 2010, 132, 12034–12041, DOI:10.1021/ja104184r.
- K. Imagawa, T. Nagata, T. Yamada and T. Mukaiyama, Chem. Lett., 1995, 335–336, DOI:10.1246/cl.1995.335.
| This journal is © The Royal Society of Chemistry 2026 |