
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Cu(OTf)2-catalyzed access to 2,4,5-trisubstituted imidazoles from acyclic Reissert compounds
Swetha
Sathyendran
a,
Vikraman Ganesh
Moorthi
a,
Sharmila
Nokku
a,
Aron Manick
Joel
a,
Suryanarayanan
Chandrasekaran
a,
Wei-Yu
Lin
 b and
Gopal Chandru
Senadi
*ab
b and
Gopal Chandru
Senadi
*ab
aGreen and Sustainable Synthesis Laboratory, Department of Chemistry, College of Engineering and Technology, SRM Institute of Science and Technology, SRM Nagar, Kattankulathur - 603 203, Chengalpattu District, Tamil Nadu, India. E-mail: chandrug@srmist.edu.in
bDepartment of Medicinal and Applied Chemistry, Kaohsiung Medical University, Kaohsiung, Taiwan
First published on 21st April 2026
Abstract
A Cu(OTf)2-catalyzed cascade self-coupling of N-acyl-α-aminonitriles (acyclic Reissert compounds) has been developed to afford 2,4,5-trisubstituted imidazoles in moderate to good yields. Single-crystal X-ray diffraction confirmed the structure of a representative 1H-2,4-disubstituted-5-acylimidazole, while NMR studies revealed N1/N3 tautomerism in selected cases. The synthetic utility of this protocol was further demonstrated by gram-scale synthesis and carbonyl reduction.
Imidazole, a privileged core structure, finds diverse applications in medicinal chemistry,1 natural products,2 and drug discovery.3 Substitution on the imidazole ring further extends its utility to functional materials,4a,b polymers,4c,d and coordination chemistry,4e,f spurring interest in 2,4,5-trisubstituted imidazoles.5 Traditional syntheses rely on the Radziszewski multicomponent cyclocondensation of aldehydes, ammonia, and 1,2-diketones,4g,h with numerous refinements enabling substituted analogues.5 However, methods for 5(4)-acylimidazoles bearing ketones or esters remain underexplored.6,7 For instance, Muthusubramanian and coworkers (2014) reported an Er(OTf)3-catalyzed multicomponent reaction of α-azido chalcones, aryl aldehydes, and anilines (Scheme 1a).7a Li and coworkers (2015) developed an FeCl3/I2-catalyzed aerobic oxidative coupling of amidines and chalcones for regioselective acylimidazoles (Scheme 1b).7e Guchhait and coworkers (2017) devised a tandem aza-Michael addition/SN2 redox-neutral process from α-bromo enones (Scheme 1c).7e With such limited precedents, substantial opportunities remain for new approaches to C-acylimidazoles.
 | ||
| Scheme 1 Prior art on acylimidazoles and N-acyl-α-aminonitriles, and this work. | ||
More than 160 years after their discovery,8 α-aminonitriles remain powerful synthetic linchpins, undergoing transformations9via deprotonation or decyanation to iminium ions, as demonstrated in our amide and 2,4-diarylquinoline syntheses.10a–c These findings prompted us to explore N-acyl-α-aminonitriles,10d an open-chain class of Reissert analogues. The 1,4- and 1,2-addition reactions of deprotonated Reissert compounds and their open-chain analogues have been widely exploited for the construction of diverse molecular frameworks.11 The synergistic electrophilicity of the nitrile group and nucleophilicity of the amine functionality has enabled versatile access to N-heterocycles from these bifunctional scaffolds. In this context, cyclization reactions incorporating the nitrile moiety,12a–c as well as the use of electron-deficient arenes in place of classical electrophiles,12d further expanded the synthetic potential of N-acyl-α-aminonitriles.
In a seminal contribution, Mcewen and co-workers developed a condensation of the conjugate base of N-acyl-α-aminonitriles with vinyltriphenylphosphonium bromide for the synthesis of substituted pyrroles (Scheme 1d).11e,f Later, Zhong et al. reported the synthesis of 2,4-disubstituted 5-halo-1H-imidazoles from N-acyl-α-aminonitriles, which were subsequently converted into 2,4,5-trisubstituted imidazoles via Pd-catalyzed cross-coupling (Scheme 1e).12e Nearly two decades later, we describe a simple transformation of the same N-acyl-α-aminonitriles into C-acylimidazoles in a single step using catalytic Cu(OTf)2 (Scheme 1f). Our initial hypothesis targeted the synthesis of 5-amino oxazole via a Lewis acid–promoted 5-exo-dig cyclization; however, the reaction selectively delivered imidazoles through a self-coupling cascade. Given the limited availability of efficient methods for synthesizing 5(4)-acyl-substituted imidazoles, this conversion provides a useful route and highlights a different reactivity pathway of N-acyl-α-aminonitriles.
The optimization began with N-(cyano(phenyl)methyl)benzamide 1a as the model substrate. Screening of common metal halides in 1,4-dioxane at 90 °C (Table 1, entries 1–5) delivered the desired product 2a in modest yields (17–63%). Subsequent evaluation of various metal triflates (Table 1, entries 6–10) identified Cu(OTf)2 as the most effective catalyst, providing 2a in 73% yield at 20 mol% loading (Table 1, entry 7). With copper triflate selected, solvent screening was carried out next. Replacing 1,4-dioxane with ethanol, isopropanol (IPA), ethylene glycol, ethyl acetate, or acetonitrile (Table 1, entries 11–15) failed to improve the reaction efficiency. Catalyst loading studies showed that reducing Cu(OTf)2 to 15 mol% lowered the yield (Table 1, entry 16), while increasing it to 25 mol% offered no further enhancement (Table 1, entry 17). Temperature variation (Table 1, entries 18 and 19) confirmed that 90 °C was optimal for the formation of 2a.
|
|
||||
|---|---|---|---|---|
| S. no | Catalyst (x mol%) | Solvent | Temp. (°C)/time (h) | Yield (%) |
| a Reaction conditions: all reactions were carried out using 1a (0.5 mmol), solvent (0.15 M) and catalyst (x mol%) in a sealed vial at indicated temperature and time unless otherwise noted. b Isolated yield. c See the SI (Tables S1–S4) for the detailed optimization studies. | ||||
| 1 | FeCl3 (20) | 1,4-dioxane | 90/12 | 63 |
| 2 | CoCl2.6H2O (20) | 1,4-dioxane | 90/24 | 17 |
| 3 | CuCl2 (20) | 1,4-dioxane | 90/24 | 44 |
| 4 | CuBr2 (20) | 1,4-dioxane | 90/16 | 56 |
| 5 | ZnCl2 (20) | 1,4-dioxane | 90/24 | 31 |
| 6 | Fe(OTf)2 (20) | 1,4-dioxane | 90/12 | 59 |
| 7 | Cu(OTf) 2 (20) | 1,4-dioxane | 90/08 | 73 |
| 8 | Sm(OTf)3 (20) | 1,4-dioxane | 90/24 | 48 |
| 9 | AgOTf (20) | 1,4-dioxane | 90/24 | 54 |
| 10 | Yb (OTf)3 (20) | 1,4-dioxane | 90/24 | 51 |
| 11 | Cu(OTf)2 (20) | Ethanol | 80/24 | 42 |
| 12 | Cu(OTf)2 (20) | IPA | 80/24 | 38 |
| 13 | Cu(OTf)2 (20) | Ethylene glycol | 100/12 | 61 |
| 14 | Cu(OTf)2 (20) | Ethyl acetate | 80/24 | 34 |
| 15 | Cu(OTf)2 (20) | Acetonitrile | 80/24 | 19 |
| 16 | Cu(OTf)2 (15) | 1,4-dioxane | 90/24 | 64 |
| 17 | Cu(OTf)2 (25) | 1,4-dioxane | 90/8 | 72 |
| 18 | Cu(OTf)2 (20) | 1,4-dioxane | 70/24 | 58 |
| 19 | Cu(OTf)2 (20) | 1,4-dioxane | 110/8 | 74 |

Control experiments under N2 and O2 were performed to probe the reaction pathway (SI, Table S4). Under N2, the reaction gave only 29% yield of 2a without water, which increased to 56% and 68% upon addition of 5 and 10 equiv. of H2O, respectively, indicating the role of water (Scheme 2a). Under O2, the reaction afforded 2a in 58% yield with 30% of decyanative product 2a′ in the absence of water, while similar yields of 2a (57%) and reduced 2a′ (24%) were observed in the presence of water, indicating competing oxygen-mediated pathways (Scheme 2b). Next, the reaction was performed13 in H218O using dry 1,4-dioxane, and LC–MS analysis showed a mixture of isotopic and non-isotopic mass peaks (SI, Fig. S10). The non-isotopic mass suggests that oxygen may also arise from water generated in situ during the course of the reaction and from residual moisture (Scheme 2c). Further, cross-over experiments with 1a/1b (electron-donating) and 1a/1f (halogen-substituted) under standard conditions afforded only the corresponding products 2a/2b and 2a/2f, respectively (Scheme 2d and e). No heterocoupled products were detected, indicating exclusive homocoupling (SI, Fig. S8 and S9). In the presence of TEMPO, the yield of 2a decreased to 33%, along with formation of hydrolysed product 2″ in 41% yield likely due to the Cu/TEMPO system. In contrast, BHT afforded 2a in 70% yield (cf. 73% standard). These results suggest that the reaction does not proceed via a radical pathway (Scheme 2f).
 | ||
| Scheme 2 Control experiments to study the reaction pathway. | ||
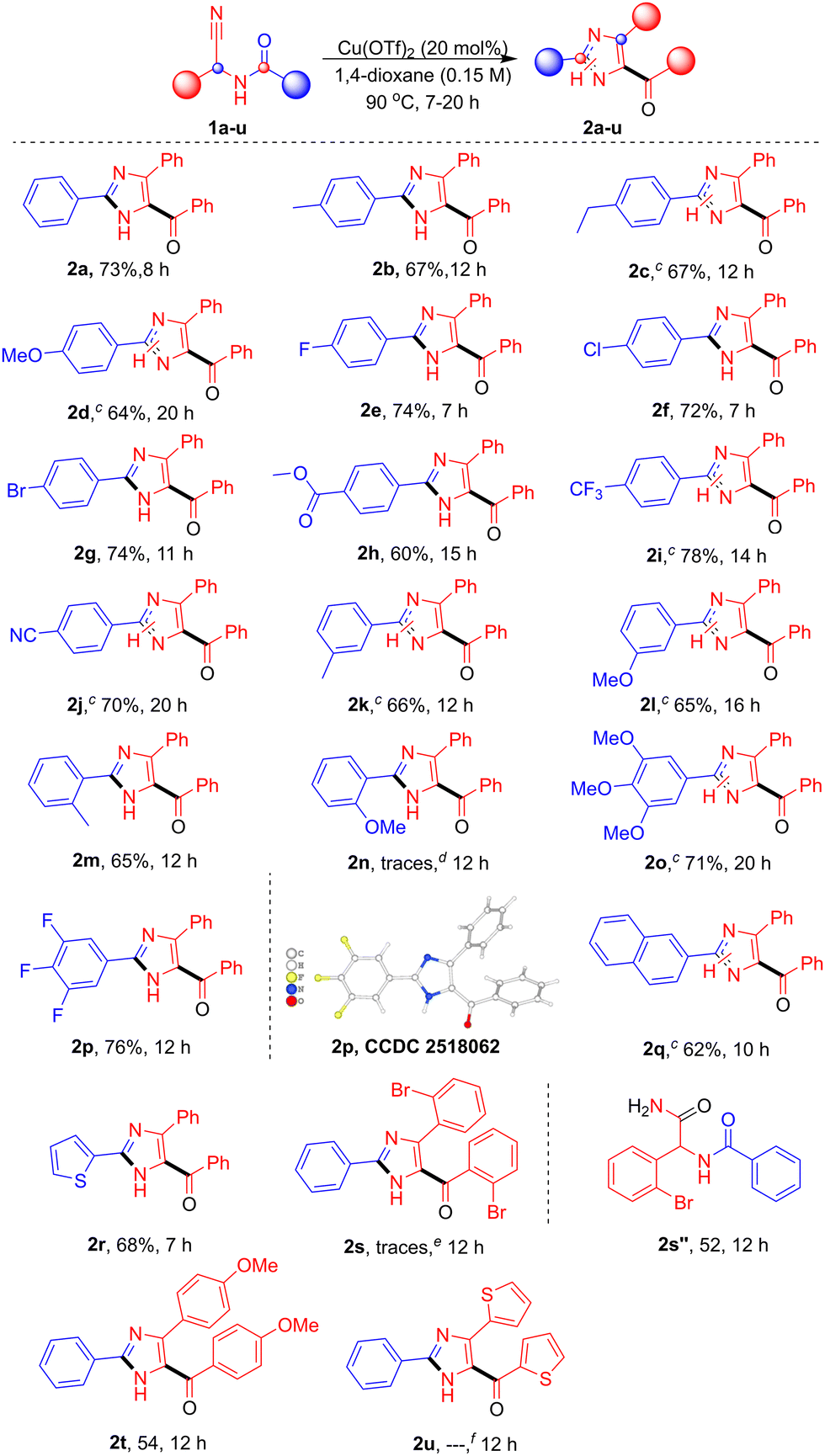
With the optimized conditions and control studies, the substrate scope of N-acyl-α-aminonitriles was examined (Table 2). A range of para-substituted phenyl derivatives, including p-Me (1b), p-Et (1c), p-OMe (1d), p-F (1e), p-Cl (1f), p-Br (1g), p-COOMe (1h), p-CF3 (1i), and p-CN (1j), were well tolerated, affording the corresponding products 2b–2j in 60–78% yield. Meta- and ortho-substituted substrates such as m-Me (1k), m-OMe (1l), and o-Me (1m) also reacted smoothly, affording 2k–2n in 65–66% yield. Whereas o-OMe (2n) was observed only in trace amounts and could not be isolated. Polysubstituted aromatics, including 3,4,5-tri-OMe (1o) and 3,4,5-trifluoro (1p), were compatible, producing 2o and 2p in 71% and 76% yield, respectively, with the structure of 2p confirmed by single-crystal X-ray analysis.14 Fused aromatics and heteroaryl substrates, such as naphthyl (1q) and 2-thienyl (1r), furnished 2q and 2r in 62–68% yields. Substrates with aryl groups attached to the α-carbon, including o-Br (1s), p-OMe (1t), and 2-thienyl (1u), gave 2t in 54% yield. For 1s, only traces of 2s were observed by TLC, and the major isolated product was the corresponding hydrolysed amide 2s″ in 52% yield. Similarly, 2u was confirmed by HRMS analysis, while its isolation was unsuccessful. Overall, para-substituted substrates generally gave higher yields and shorter reaction times, meta-substituents gave moderate results, and ortho-substitution lowered yields and required longer reactions, likely due to steric effects.
| a Reaction conditions: Compound 1a (0.5 mmol), Cu(OTf)2 (20 mol %), and 1,4-dioxane (0.15 M) were stirred at 90 °C for 7–20 h. b Isolated yields. c The presence of N1–N3 tautomeric forms was confirmed by NMR analysis. d Detected in trace amounts by TLC; not isolated due to multiple spots. e 2s was detected in trace amounts and not isolated; the corresponding amide was obtained. f The mass was confirmed by HRMS analysis; however, isolation for pure NMR characterization was unsuccessful. |
|---|
|
We further evaluated the scalability of the methodology on a gram-scale using the standard optimized conditions. Representative substrates bearing an electron-donating substituent (1d) and an electron-withdrawing substituent (1h) furnished the corresponding products 2d and 2h in moderate yields of 61% and 65%, respectively (Scheme 3a & b). In addition, reduction of carbonyl functional group in 2a with NaBH4 proceeded smoothly to afford the corresponding secondary alcohol 3a in 84% yield (Scheme 3c).
 | ||
| Scheme 3 Synthetic applications and control studies. | ||
A plausible mechanism, supported by control experiments and literature precedent,11,12,15 is outlined in Scheme 4. Coordination of Cu(OTf)2 to the nitrile group of α-aminonitrile 1 generates intermediate A, increasing the electrophilicity of the nitrile carbon and enabling intramolecular 5-exo-dig cyclization to form imino-oxazolone B. DFT calculations using UB3LYP/def2-TZVP with a 1,4-dioxane solvent model (see SI for computational details) support preferential nitrogen coordination (ΔG = −8.04 kcal mol−1) over oxygen (ΔG = −3.35 kcal mol−1), with HOMO–LUMO overlap and a smaller band gap (2.13 eV) favouring the nitrile binding pathway (SI, Fig. S13). Further, to assess the ring-closure mechanism and stability of cyclized intermediate B, transition state free energy calculations were performed (see SI, Fig. S15). The process proceeds via transition state A with an activation free energy of ΔG‡ = 23.4 kcal mol−1, leading to intermediate B, which is thermodynamically stabilized by −4.46 kcal mol−1 relative to the initial reactant 1.16 Next, nucleophilic addition of a second molecule of 1 to B, followed by ring opening, affords intermediate C. Owing to the lability of Cu(II) coordination, intramolecular exchange between imine and amide carbonyl binding facilitates cyclization to imidazoline D. Dehydration of D regenerates Cu(OTf)2 and furnishes intermediate E, which upon isomerization and HCN elimination gives F. Proton transfer affords aromatic intermediate G, and final hydrolysis yields the 2,4,5-trisubstituted imidazole 2 along with amide 4, confirmed by 1H NMR spectroscopy (SI, Fig. S57).
 | ||
| Scheme 4 Plausible reaction mechanism. | ||
In conclusion, a copper-catalyzed synthesis of C-acylimidazoles from open-chain analogues of Reissert compound is disclosed. The reaction is proposed to proceed through an imino-oxazolone pathway, enabling access to 5-acylimidazole scaffolds. Broad aryl-substrate tolerance, gram-scale feasibility, and reduction of the carbonyl to the corresponding alcohol demonstrate the practicality of the method. A plausible mechanism was proposed based on control studies and DFT calculations. This work expands the synthetic toolbox for N-acyl-α-aminonitriles and provides a useful entry to acylimidazoles.
Conflicts of interest
There are no conflicts to declare.Data availability
The supporting data has been provided as part of the supplementary information (SI). Supplementary information: Tables S1–S4 and Fig. S1 and S2. NMR spectra and further experimental details. See DOI: https://doi.org/10.1039/d6cc00157b.CCDC 2518062 contains the supplementary crystallographic data for this paper.14
Acknowledgements
S. S., V. G. M., S. N and A. M. J., thank the SRM Institute of Science and Technology for the PhD fellowship. Dr G.C.S acknowledges Anusandhan National Research Foundation (ANRF) for Core Research Grant (File No: CRG/2022/006963). The authors wish to thank department of chemistry and Interdisciplinary Institute of Indian System of Medicine (IIISM), SRM Institute of Science and Technology for providing HRMS, NMR and computational HPCC facilities. The authors also thank Prof. Jeh-Jeng Wang, Kaohsiung Medical University, Kaohsiung, Taiwan for providing support for SCXRD and HRMS analysis.References
-
(a) G. Yadav and R. Jain, Eur. J. Med. Chem., 2025, 290, 117524 CrossRef CAS PubMed
; (b) L. Zhang, X.-M. Peng, G. L. Damu, R.-X. Geng and C.-H. Zhou, Med. Res. Rev, 2014, 34, 340–437 CrossRef CAS PubMed
-
(a) Z. Jin, Nat. Prod. Rep., 2016, 33, 1268–1317 RSC
-
(a) S. Gupta, M. A. Babu, R. Kumar, T. G. Singh, A. Goel, S. Rastogi, P. Sharma, Y. Tyagi, K. K. Goel and B. Kumar, Chem. Biodivers., 2025, 22, e202403020 CrossRef CAS PubMed
-
(a) Y. Yao, Z. Li, Y. Zhang, K. Xie, Y. Song and Y. Tang, ACS Sens., 2025, 10, 8489–8497 CrossRef CAS PubMed
-
(a) D. A. Shabalin and J. E. Camp, Org. Biomol. Chem., 2020, 18, 3950–3964 RSC
-
(a) B. Hu, Z. Wang, N. Ai, J. Zheng, X.-H. Liu, S. Shan and Z. Wang, Org. Lett., 2011, 13, 6362–6365 CrossRef CAS PubMed
-
(a) K. Rajaguru, R. Suresh, A. Mariappan, S. Muthusubramanian and N. Bhuvanesh, Org. Lett., 2014, 16, 744–747 CrossRef CAS PubMed
- A. Strecker, Liebigs Ann. Chem., 1850, 75, 27–45 CrossRef
-
(a) C. Grundke, N. Vierengel and T. Opatz, Chem. Rec., 2020, 20, 989–1016 CrossRef CAS PubMed
-
(a) S. Swetha and G. C. Senadi, Adv. Synth. Catal., 2022, 364, 2872–2882 CrossRef CAS
-
(a) V. Boekelheide and J. C. Godfrey, J. Am. Chem. Soc., 1953, 75, 3679–3685 CrossRef CAS
-
(a) M. Uchibayashi, Yakugaku Zasshi, 1958, 78, 845–849 CrossRef CAS
- For detailed isotopic studies with H218O refer supporting information Fig. S10 to Fig. S12.
-
CCDC 2518062: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qj7tb
-
(a) W. E. McEwen, A. V. Grossi, R. J. MacDonald and A. P. Stamegna, J. Org. Chem., 1980, 45, 1301–1308 CrossRef CAS
- N. Dhama, M. K. Tiwari, V. K. Vishvakarma, R. Yadav, M. Kumar and D. T. Masram, Inorg. Chim. Acta, 2026, 589, 122928 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2026 |