
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and reactivity of a strongly pyramidalized P(III)-compound embedded into a pyrrolide (ONO)3− pincer ligand
Malte Kaste†
a,
Tobias Heilmann†a,
Johannes Kircherb,
Christopher Golza,
Ricardo A. Matab and
Manuel Alcarazo *a
*a
aInstitut für Organische und Biomolekulare Chemie, Georg-August-Universität Göttingen. Tammannstr. 2, 37077, Göttingen, Germany. E-mail: manuel.alcarazo@chemie.uni-goettingen.de
bInstitut für Physikalische Chemie, Georg-August-Universität Göttingen. Tammannstr. 6, 37077, Göttingen, Germany
First published on 20th February 2026
Abstract
A Cs-symmetric P(III) complex bearing a pyrrolide-centered trianionic pincer ligand is reported and its reactivity explored. This compound reacts with alcohols and amines via oxidative addition of their respective O–H and N–H bonds at the P-center, delivering the corresponding P(V) species. DFT calculations, as well as kinetic and isotope labelling analyses suggest the assistance of one of the phenolate side arms in the process.
The embedment of a formal tricationic P-centre into the rigid environment created by pincer ligands is known to deliver geometrically distorted phosphabicyclic compounds that are characterized by unique electronic structure and reactivity.1–4 For instance, compound A is a competent catalyst for transfer hydrogenation reactions,5 while B undergoes E-H activation reactions (E = –OR, –NHR, BR2);6 mimicking in both cases the behavior of transition metal complexes in small molecule activation processes. The origins of this specific reactivity lie in the decreased HOMO–LUMO gap when compared with their undistorted C3v-symmetric congeners, which is caused by the imposed non-trigonal geometries and facilitates oxidative addition reactivity. Moreover, alternative activation pathways based on the cooperation of the P-center with the flanking ligand might also get involved.7 Considering the key role played by the pincer framework to finely tune and leverage these reactivity modes, it is not surprising that during the last years a number of tri- and dianionic scaffolds containing ONO,5,8,9 NNN,6,10–12 NPN,13 OCO,14 CNC15 or ONP16 donor sites have been evaluated among others.17–19 They afford neutral (A–E) or cationic species (F–I),20 respectively; each one depicting unique modes of reactivity (Fig. 1a).
 | ||
| Fig. 1 (a) Selection of previously studied constrained P(III)-species; (b) This work: a P(III)-compound embedded into a pyrrolide (ONO)3− pincer ligand. | ||
Some years ago, Veige described a new trianionic pyrrolide containing pincer ligand 2,21 which has been used for the synthesis of W, Ti and Zr complexes (Scheme 1).22 During these studies the ability of the pyrrolide moiety to undergo reversible remote protonation at the backbone was noticed. This unusual behaviour motivated us to study whether its P-derivative 1 might also undergo E–H activation processes (E = –OR, –NHR) through a cooperation mode that involves the P-center and the remote 3-position of the pyrrolide ligand.
 | ||
| Scheme 1 (a) Synthesis of 1; (b) structure of 1 in the solid state. | ||
Hence, phosphine 1 was prepared by treatment of 2 with PCl3 in the presence of slight excess of Et3N (3.5 equiv.). The 31P NMR spectrum of the reaction crude showed a well-defined signal at 114.7 ppm, which closely matches the values reported for highly pyramidalized F (117 ppm in CD2Cl2)9 or G (112 ppm in CDCl3).11 Subsequent evaporation of the solvent and crystallization from n-pentane afforded analytically pure 1 as pale-yellow needles (86% yield), which were used to determine its molecular structure by X-ray crystallography (Scheme 1b). In the solid state, 1 depicts strong pyramidalization at the P-centre (∑<P = 288°); comparable to that of F(∑<P = 286°) and G(∑<P = 293°). Both P–O bond lengths (1.635(1)Å and 1.631(1)Å) fall within the expected range for P(III)–O bonds, while the P–N bond length in 1 (1.704(1)Å) is significantly shorter than that in cationic F (1.809(2)Å) or even the neutral phosphoramidite D (1.757(1)Å).8
Willing to obtain some evidence about the impact that the distortion provoked by the pincer ligand generates on the electronic properties, we decided to assess the donor strength of 1 by IR spectroscopy, using the cyclopentadienyliron dicarbonyl cation [FeCp(CO)2]+ as probe. The IR spectrum of 3 shows two CO stretching vibrations (![[small nu, Greek, tilde]](https://www.rsc.org/images/entities/char_e0e1.gif) asym = 2030 cm−1 and sym = 2073 cm−1), which are higher in energy than that of the analogue phosphoramidite complex 6, and better align the donor properties of 1 with these of phosphites 4-5, (Fig. 2a).23–25 A substantial decrease of the donor ability upon distortion is observed by comparison of 7 and 8;26 an effect that probably also plays a role in 3 (when compared to 6). The qualitative trend is also corroborated by the JPSe coupling constant in 9 (JP–Se = 1018 Hz), which is significantly higher than that of 10 or 11, and indicates that 1 is a weaker donor than of P(OMe)3 or (Me2N)P(OMe)2 (Fig. 2b).27 Calculations at the B3LYP-D3(BJ)/def2-TZVP theory level support this conclusion.28 They reveal a LUMO substantially lower in energy than that for non-constrained analogues, which is expected to confer enhanced Lewis acidity at the P-atom. It also suggests its possible involvement in E–H bond activation processes (see Fig. S17 and S18 for details).
asym = 2030 cm−1 and sym = 2073 cm−1), which are higher in energy than that of the analogue phosphoramidite complex 6, and better align the donor properties of 1 with these of phosphites 4-5, (Fig. 2a).23–25 A substantial decrease of the donor ability upon distortion is observed by comparison of 7 and 8;26 an effect that probably also plays a role in 3 (when compared to 6). The qualitative trend is also corroborated by the JPSe coupling constant in 9 (JP–Se = 1018 Hz), which is significantly higher than that of 10 or 11, and indicates that 1 is a weaker donor than of P(OMe)3 or (Me2N)P(OMe)2 (Fig. 2b).27 Calculations at the B3LYP-D3(BJ)/def2-TZVP theory level support this conclusion.28 They reveal a LUMO substantially lower in energy than that for non-constrained analogues, which is expected to confer enhanced Lewis acidity at the P-atom. It also suggests its possible involvement in E–H bond activation processes (see Fig. S17 and S18 for details).
 | ||
| Fig. 2 Evaluation of the donor properties of 1: (a) IR stretching frequencies of L[CpFe(CO)2] complexes; (b) JPSe of model phosphorous selenides; (c) synthesis of 1+˙; (d) CV of 1; (e) EPR spectrum of 1+˙; (f) SOMO of 1+˙. | ||
The redox chemistry of 1 was subsequently studied by cyclic voltammetry (CV). Its voltammogram shows a quasi-reversible wave with an oxidation potential of Eox = 0.69 V vs. Fc0/+. In agreement with that value, addition of magic blue to a CH2Cl2 solution of 1 at −78 °C resulted in the development of an intense green colour. The recorded X-band isotropic spectrum of the newly generated species, which we assume to be 1+˙, has been simulated with a g value of 2.003 and hyperfine constants of 1.66 G to the central 31P(I = 1/2), 0.98 G to the pyrrolic 14N(I = 1), and 4.32 G, 2.67 G and 2.13 G to three pairs of protons 1H(I = 1/2) (Fig. 2e). The calculated SOMO of 1+˙ is mainly delocalized over the pincer ligand, with Mulliken spin densities of 13.7% and 7.0% at the p-C and o-C of the phenolate units, respectively; 5.7% at each backbone carbon of the pyrrolide moiety, 5.5% at the N atom and only 0.3% at P (Fig. 2f).
Having synthesized and characterized 1, the screening of its ability to activate the N–H bond of amines started. Exposure of 1 to stoichiometric amounts of primary amines (butyl-, benzyl-, isobutyl- and neopentyl-) in chloroform at room temperature led to quantitative consumption of 1 and the clean formation of the corresponding hydridoamidophosphorane, which are characterized by high field resonances in the 31P NMR spectra (δ = −80 – (−90) ppm), and large 1JP–H coupling constants (1JP–H = 848–865 Hz), indicating pentacoordination of the P-atom and the formation of a P–H bond (For details see the SI). Bulkier amines such as isopropyl- and tert-butyl amines, or less nucleophilic anilines required longer reaction times and the reaction mixtures to be heated to 60 °C to ensure complete conversion (Scheme 2a). The formation of compounds of general formula 12 is a reversible process; for example, by heating 12b at 120 °C under vacuum (10−3 bar) 1 is recovered.19,29 In addition, heating 12c with an excess of iBuNH2 (5.0 equiv.) leads to amine exchange. Finally, reaction of 1 with deuterated isobutylamine afforded 12b-d2 (31P NMR δ = −84.7 ppm, 1JP–D = 129.4 Hz). No deuterium label was detected at the pyrrolide backbone via 2H NMR analysis, ruling out the involvement of that site during the N–H activation process (Scheme 2b).
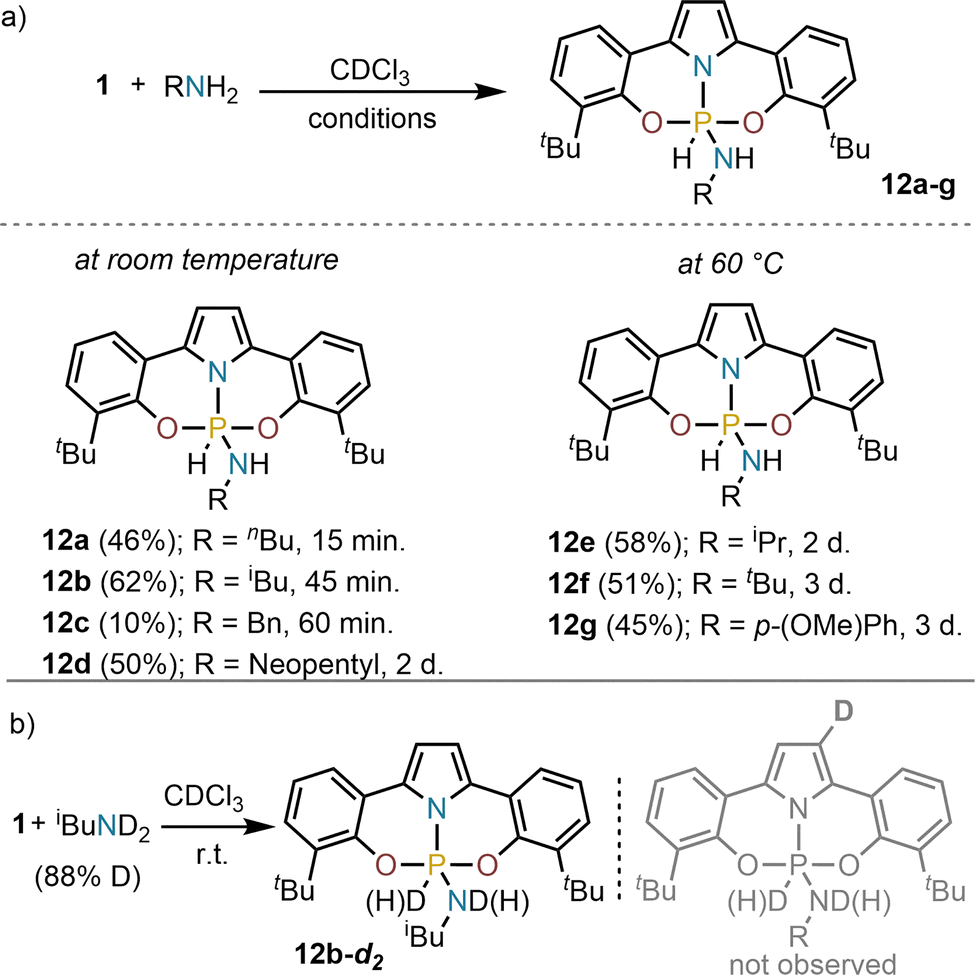 | ||
| Scheme 2 (a) N–H oxidative addition products 12a–g; (b) preparation of deuterium labelled 12b-d2. Complete conversions were achieved. Yields are of the isolated products. | ||
Compound 12d has been crystallized from n-pentane and its structure is depicted in Fig. S14. When compared with 1, the structure of 12d exhibits slight elongation of the P–O bond lengths (P1–O1, 1.685(1)Å and P1–O2, 1.713(1)Å), while the P1–N1 (1.700(1)Å) remains unchanged within the error range. Moreover, the pentacoordinate phosphorus atom adopts a nearly undistorted trigonal bipyramidal geometry (τ5 = 0.97).30
Kinetic studies have been undertaken by monitoring the formation of phosphorane 12d via 1H NMR spectroscopy during 12 hours. Tetrakis(trimethylsilyl)silane was employed as standard and the temperature was kept constant at 25 °C. Under these conditions the orders in 1 and amine have been determined to be 1.0 in both cases using a variable time normalization analysis (VTNA) (Fig. S7–S11).31–33
Next, the reactivity of 1 with alcohols was evaluated. No reaction was observed when MeOH, EtOH or iPrOH were added to solutions of 1 in CHCl3; yet, the activation of the O–H bond slowly occurs if an acid catalyst ([HOEt2][B(C6H3(CF3)2)4]; 10 mol%) is present in the reaction mixture delivering compounds 13a–e (Scheme 3a). This observation suggests that an initial protonation of 1 is necessary to boost reactivity. To evaluate this hypothesis 1 was exposed to CD3OD under acidic conditions, and the incorporation of the isotope label determined by multinuclear NMR. In 13a-d4 deuterium was found statistically distributed among the two positions of the pyrrolide backbone and the P-centre (Scheme 3b). This observation suggests an active participation of the pyrrolide moiety in the activation process. Most probably, the protonation of 1 at the pyrrole backbone generates a phosphenium cation of enhanced Lewis acidity, which is more capable of coordinating the alcohol substrate at P; thus, initiating the activation process.9,11,13 Yet, protonation at phosphorus followed by attack of the alcohol cannot be excluded. In that case, the H/D exchange at C3 of the pyrrole should occur after the formal oxidative addition step.21,22
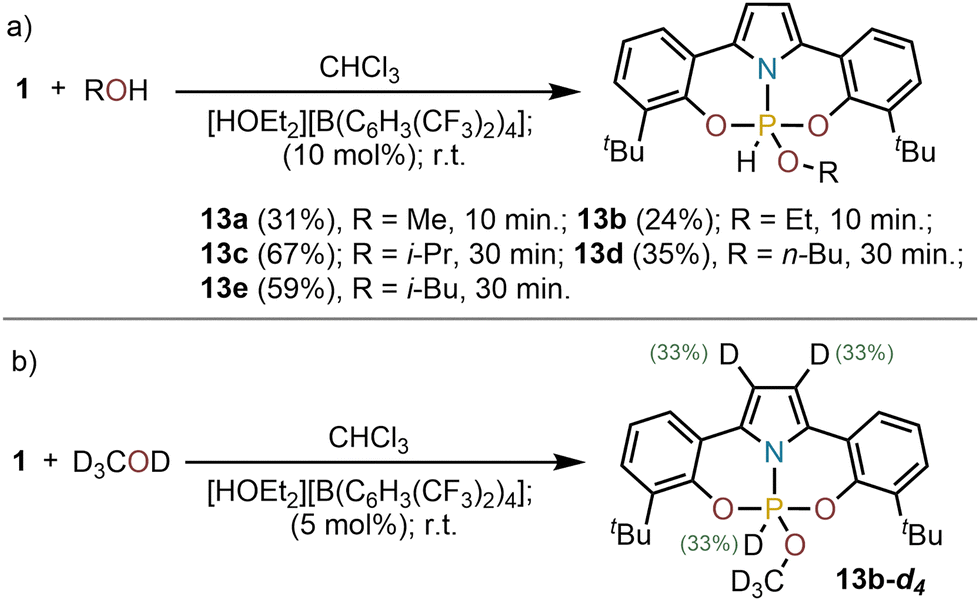 | ||
| Scheme 3 (a) O–H oxidative addition products 13a–e; (b) incorporation of the deuterium label in the ONO supporting ligand; Complete conversions were achieved. Yields are of the isolated products. | ||
To get deeper insight into the mechanism leading to the formation of hydridoamidophosphoranes 12, the reaction pathway was evaluated through computational methods. The reaction intermediates and selected transition states were optimized at the B3LYP D3(BJ)/def2-TZVP level of theory, making use of the D3 Grimme dispersion correction with Becke–Johnson damping.34 Solvation effects were accounted through single point corrections using the SMD model and chloroform as solvent at the B3LYP-D3(BJ)/def2-TZVP level.35 The structure of transition state TS4 was obtained via the nudged-elastic band method.36
Our calculations indicate that at the very first step the amine coordinates the phosphorous atom through TS1. Subsequent shuttle of a proton from the amine moiety in Int1 to the oxygen atom of the neighbouring phenolate ligand with concomitant P–O bond cleavage delivers Int2 (Fig. 3). From this point, the system evolves to Int3 via rotation of the phenolate ligand, and finally, the proton is transferred to the P-atom via TS4. Considering the uncertainty associated with the DFT computed barriers and the relatively small energetic difference between TS1 and TS4 (22.0 and 25.1 kcal mol−1, respectively), both steps are potentially rate-determining. However, independent of where the bottleneck is, the reaction rate should still be first-order with respect to the reacting phosphine and amine. If TS1 is the rate-limiting step, this follows immediately. If TS4 is rate-limiting, an effective first order rate constant with respect to both reagents can be deduced assuming 1 and the amine to be in chemical equilibrium with Int3 (See the SI for the analysis). Finally, we have also considered that proton transfer from Int2 to 12 could be assisted by amine through a hydrogen bond network. This pathway requires a series of elementary steps but proceeds through a much lower barrier (16.0 kcal mol−1; Fig. S20). In this scenario, which might be operative at an initial stage of the reaction, TS1 is again rate determining.
 | ||
| Fig. 3 Free energy profile for the reaction of 1 with methylamine calculated at the B3LYP-D3(BJ)/def2-TZVP(SMD)//B3LYP-D3(BJ)/def2-TZVP level of theory. tert-Bu groups were replaced by methyl groups for these calculations. | ||
In conclusion, we describe herein the synthesis of a structurally distorted P(III)-complex stabilized by a pyrrolide (ONO)3− pincer ligand and preliminarily discuss its oxidation and coordination chemistry. Compound 1 readily reacts with amines to deliver the corresponding hydridoamidophosphoranes, and theoretical calculations indicate that the operative pathway involves the cooperation between the P-centre and a phenolate arm. Contrarily, the oxidative addition of alcohols requires the intervention of a Brønsted acid catalyst to proceed. Deuterium labelling experiments suggest that in such reactions a phosphenium cation of enhanced Lewis's acidity is probably formed by protonation of the pyrrolide backbone. This cationic species is the one that presumably initiates the O–H oxidative addition by coordination of the alcohol substrate to the electrophilic P-centre.
M. A. conceived and coordinated the project. M. K. and T. H. performed the experiments. M. K, J. K and R. A. M. carried out the computational studies. C. G. did the X-ray crystallographic studies. M. K., R. A. M. and M. A. prepared the manuscript. All authors discussed the results.
This work was supported by the Deutsche Forschungs-gemeinschaft (DFG) through the BENCh Research Training Group 389479699/GRK2455 and projects INST 186/1318-1 and INST 186/1324-1.
Conflicts of interest
There are no conflicts to declare.Data availability
Further details of the experimental procedures, NMR spectra, and DFT data are available in the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc07240a.CCDC 2515996 (1) and 2515997 (12d) contain the supplementary crystallographic data for this paper.37a,b
References
- S. Kundu, Chem. – Asian J., 2020, 15, 3209–3224 CrossRef CAS PubMed.
- J. Abbenseth and J. M. Goicoechea, Chem. Sci., 2020, 11, 9728–9740 RSC.
- A. Maiti, R. Yadav and L. Greb, Structural constraint effects on p-block elements: Recent advances, Adv. Inorg. Chem., 2023, 82, 261–299 CrossRef CAS.
- T. J. Hannaha and S. S. Chitnis, Chem. Soc. Rev., 2024, 53, 764–792 Search PubMed.
- N. L. Dunn, M. Ha and A. T. Radosevich, J. Am. Chem. Soc., 2012, 134, 11330–11333 Search PubMed.
- (a) W. Zhao, S. M. McCarthy, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2014, 136, 17634–17644 CrossRef CAS PubMed; (b) Y.-C. Lin, E. Hatzakis, S. M. McCarthy, K. D. Reichl, T.-Y. Lai, H. P. Yennawar and A. T. Radosevich, J. Am. Chem. Soc., 2017, 139, 6008–6016 CrossRef CAS PubMed.
- J. M. Lipshultz, G. Li and A. T. Radosevich, J. Am. Chem. Soc., 2021, 143, 1699–1721 CrossRef CAS PubMed.
- T. P. Robinson, D. M. de Rosa, S. Aldridge and J. M. Goicoechea, Angew. Chem., Int. Ed., 2015, 54, 13758–13763 CrossRef CAS PubMed.
- S. Volodarsky and R. Dobrovetsky, Chem. Commun., 2018, 54, 6931–6934 RSC.
- H. W. Moon, A. Maity and A. T. Radosevich, Organometallics, 2021, 40, 2785–2791 CrossRef CAS.
- S. Volodarsky, D. Bawari and R. Dobrovetsky, Angew. Chem., Int. Ed., 2022, 61, e202208401 CrossRef CAS PubMed.
- A. J. King, J. Abbenseth and J. M. Goicoechea, Chem. – Eur. J., 2023, 29, e202300818 CrossRef CAS PubMed.
- J. Cui, Y. Li, R. Ganguly, A. Inthirarajah, H. Hirao and R. Kinjo, J. Am. Chem. Soc., 2014, 136, 16764–16767 CrossRef CAS PubMed.
- D. Bawari, I. Malahov and R. Dobrovetsky, Angew. Chem., Int. Ed., 2025, 64, e202419772 CrossRef CAS PubMed.
- D. Bawari, D. Toami, K. Jaiswal and R. Dobrovetsky, Nat. Chem., 2024, 16, 1261–1266 CrossRef CAS PubMed.
- L. You, D. Roth and L. Greb, Chem. Sci., 2025, 16, 1716–1721 RSC.
- A. Brand, A. Hentschel, A. Hepp and W. Uhl, Eur. J. Inorg. Chem., 2020, 361–369 Search PubMed.
- D. Roth, A. T. Radosevich and L. Greb, J. Am. Chem. Soc., 2023, 145, 24184–24190 CrossRef CAS PubMed.
- J. Abbenseth, O. P. E. Townrow and J. M. Goicoechea, Angew. Chem., Int. Ed., 2021, 60, 23625–23629 CrossRef CAS PubMed.
- For a recent review see: D. Bawari, D. Toami and R. Dobrovetsky, Chem. Commun., 2025, 61, 5871–5882 RSC.
- M. E. O’Reilly, S. S. Nadif, I. Ghiviriga, K. A. Abboud and A. S. Veige, Organometallics, 2014, 33, 836–839 CrossRef.
- S. S. Nadif, M. E. O’Reilly, I. Ghiviriga, K. A. Abboud and A. S. Veige, Angew. Chem., Int. Ed., 2015, 54, 15138–15142 CrossRef CAS PubMed.
- H. Schumann, J. Organomet. Chem., 1985, 293, 75–91 CrossRef CAS.
- H. Nakazawa, Y. Kadoi and K. Miyoshi, Organometallics, 1989, 8, 2851–2856 CrossRef CAS.
- N. Nakazawa, K. Kubo, K. Tanisaki, K. Kawamura and K. Miyoshi, Inorg. Chim. Acta, 1994, 222, 123–129 CrossRef.
- G. T. Cleveland and A. T. Radosevich, Angew. Chem., Int. Ed., 2019, 58, 15005–15009 CrossRef CAS PubMed.
- D. E. Schiff, J. W. Richardson, R. A. Jacobson, A. H. Cowley, L. Lasch and J. G. Verkade, Inorg. Chem., 1984, 23, 3373–3377 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- N. Beims, T. Greven, M. Schmidtmann and J. I. van der Vlugt, Chem. – Eur. J., 2023, 29, e202302463 CrossRef CAS PubMed.
- A. W. Addison, N. T. Rao, J. Reedijk, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 2028–2031 CrossRef PubMed.
- J. Burés, Angew. Chem., Int. Ed., 2016, 55, 16084–16087 CrossRef PubMed.
- C. D.-T. Nielsen and J. Burés, Chem. Sci., 2019, 10, 348–353 RSC.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- O. Weser, B. Hein-Janke and R. A. Mata, J. Comput. Chem., 2023, 44, 710–726 CrossRef CAS PubMed.
- (a) CCDC 2515996: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qg35h; (b) CCDC 2515997: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2qg36j.
Footnote |
| † Equal contribution from both authors. |
| This journal is © The Royal Society of Chemistry 2026 |