
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Computational modeling of triplet energy transfer processes: progress and future challenges
Lee M.
Thompson
 *,
Megan J.
Mackintosh
,
Saptarshi
Saha
and
Pawel M.
Kozlowski
*
*,
Megan J.
Mackintosh
,
Saptarshi
Saha
and
Pawel M.
Kozlowski
*
Chemistry Department, University of Louisville, 2320 South Brook St, Louisville, 40208, Kentucky, USA. E-mail: lee.thompson1@louisville.edu; pawel.kozlowski@louisville.edu
First published on 10th February 2026
Abstract
In this feature article, we review progress and future challenges in the development of computational approaches for modelling triplet energy transfer processes. These processes are of fundamental importance for understanding biological systems, developing new synthetic chemistry tools, photochemistry and transition-metal based photocatalysis, as well as designing materials with improved properties. Describing these processes requires calculation of diabatic states where the character of the wavefunction is constant across the reaction coordinate. Diabatic states are often difficult to access directly and are poorly defined when trying to construct them from more easily obtained adiabatic states. Another challenge is determining how to properly include the charge transfer states in the theoretical framework. Here, we review the previous developments within the field, as well as our recent work on triplet energy transfer. Subsequently, we highlight the future directions and challenges that remain to provide reliable and computationally expedient predictions of triplet energy transfer kinetics.
 Lee M. Thompson | Lee M. Thompson joined the Department of Chemistry at the University of Louisville as Assistant Professor in August 2017, before being promoted to Associate Professor in 2023. Prior to his arrival in Louisville, he obtained a Master of Chemistry degree from University of Southampton (2004–2008). He carried out his doctoral research in the computational photochemistry group at Imperial College London (2009–2013), where he developed and applied computational methods for simulating the vibrational spectra of photoactive proteins. In 2014 he moved to the USA to take up a postdoctoral research position at the University of California, Merced (2014–2017) where he worked on approximate projection methods and their application to elucidation of transition metal oxide cluster photodetachment spectra. |
 Megan J. Mackintosh | Megan J. Mackintosh received her MS and PhD degrees in chemistry from the University of Louisville (UofL) in the group of Prof. Pawel M. Kozlowski. Her research at UofL involved studying the photochemistry of B12 dependent systems via multiscale methods with her main interest being the B12 photoreceptor CarH. After her graduation from UofL in December of 2021, she was awarded the Zuckerman STEM Leadership Program's two-year postdoctoral fellowship to conduct research in Israel in Prof. Igor Schapiro's group at the Hebrew University of Jerusalem (HUJI). She continued to study photoreceptors at HUJI including phytochromes and bacteriorhodopsins. In 2024 Megan returned to UofL to continue research in the area of computational chemistry, focusing her efforts on dynamic correlation of nonorthogonal CI methods. |
 Saptarshi Saha | Saptarshi Saha received his BSc degree in chemistry from Visva-Bharati University, Santiniketan, India. Afterwards, he pursued his MSc degree in chemistry from the Indian Institute of Technology Kharagpur (IITKGP), India. During his two-year master's course, he completed his MSc thesis work under the supervision of Prof. Sabyasachi Mishra, working on ultrafast luminescent decay of [Re(CO)3(phen)(im)]+. Since 2022, he has been pursuing his doctoral studies under the supervision of Prof. Pawel M. Kozlowski in the University of Louisville, Kentucky, USA. His primary research focus is to develop kinetic modelling of photocatalytic reaction pathways using non-orthogonal configuration-interaction (NOCI) methodologies. |
 Pawel M. Kozlowski | Pawel M. Kozlowski is a Professor of Chemistry in the Department of Chemistry, University of Louisville. He received his M.Sc. degree from Jagiellonian University, Poland, and his PhD from the University of Arizona. He did postdoctoral work at Indiana University with Professor Ernest R. Davidson, and the University of Arkansas with Professor Peter Pulay. He was a research staff member of Professor Thomas G. Spiro's group at Princeton University, before joining the University of Louisville. His research is concerned with application of methods of computational chemistry to solve problems of bioinorganic structure and function. |
1. Introduction
Energy transfer (EnT) is a fundamental phenomenon involved in a host of photophysical processes.1–6 EnT mechanisms have been implicated in light-harvesting complexes and photosynthesis,7–9 solar energy conversion and photovoltaics,10,11 materials science,12–18 photocatalysis and photochemistry.19–23 EnT involves the exchange of energy between molecules or molecular fragments. There are two types of EnT mechanisms which have been identified – Förster and Dexter EnT.24,25 One key distinction between these two mechanisms involves the distances between the molecules or molecular fragments. Förster EnT involves the transfer of energy over long distances, while Dexter EnT occurs over short distances.25 In Förster EnT, the electronic excitation of the acceptor results from dipole–dipole coupling between the transition dipole moment of the donor, associated with electronic deexcitation, and a resonant transition dipole moment in the acceptor.24 In contrast, Dexter EnT results from the transfer of electrons between donor–acceptor molecular species, which requires large overlap and so results in strong distance dependence. Triplet EnT (TEnT) is a subclass of Dexter EnT in which energy transfer from a triplet donor leads to a triplet acceptor. EnT remains an open area of research due to the lack of fundamental understanding concerning the mechanisms and rates in a theoretical framework. In this feature article, we describe progress towards the computational modeling of EnT processes with an emphasis on TEnT. We include a summary of some specific cases where Dexter TEnT plays an important role.TEnT belongs to the Dexter EnT category, and, unlike singlet EnT, the spin on each fragment is not conserved. Rather, TEnT involves a change in the electron spin and multiplicity of the acceptor from the initial singlet state and final triplet state. Ultimately the triplet donor will transfer energy to an acceptor molecule or molecular fragment leading to a triplet state on the acceptor. This TEnT process can be thought of as an electron transfer (ET) process where two electrons of different spin are transferred concertedly. As for ET, TEnT is most conveniently described in terms of diabatic states, which represent electronic configurations in which the frontier orbitals are localized to one of the two fragments. Use of two diabatic states, in which the triplet resides on either the donor or the acceptor fragment, results in the two state-model, while inclusion of the sequential pathways, where each fragment is a doublet, provides a four-state model (Fig. 1).26–31 The two-state model involves two locally-excited (LE) states: the reactants (D*A) in which the triplet resides on the donor, and the products (DA*) where the triplet is localized on the acceptor (A*). The pathway from D*A to DA*, called the direct path, is achieved by a simultaneous two-electron transfer. TEnT is more accurately described with a four-state model which, in addition to the LE states (D*A and DA*), also includes two charge transfer (CT) states. The two CT states can be described as electron (D+A−) and hole (D−A+) transfer intermediates (Fig. 1), respectively. In the four-state model, energy exchange can occur by a simultaneous two-electron transfer, i.e. the direct (D*A → DA*) pathway, or through two sequential one-electron transfers, through either electron (D*A → D+A− → DA*) or hole (D*A → D−A+ → DA*) transfer pathways. The one- and two-electron interactions that couple the donor and acceptor are known as Dexter coupling.28 Systems involving transition metal complexes or bridging moieties may require even more states in the model.28
 | ||
| Fig. 1 Four-state model of triplet energy transfer containing concerted two-electron, direct energy transfer mechanism, and two sequential one-electron energy transfer mechanisms involving either electron or hole transfer. The two-state model is also depicted (D*A → DA*, with the pathway labeled as direct) and involves a concerted two electron transfer. | ||
Having identified the relevant intermediate states, EnT rates can be determined from Fermi's Golden rule32
 | (1) |
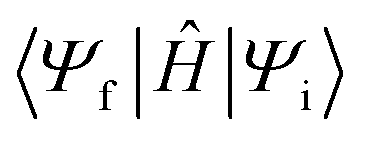 is the diabatic coupling element between initial and final states. For the two-state system described previously, the diabatic coupling element is
is the diabatic coupling element between initial and final states. For the two-state system described previously, the diabatic coupling element is  . The density of states per unit energy of the final state is ρ(E), which depends on the nuclear reaction coordinate, and for a harmonic oscillator is equal to 1/ℏω, where ω is the angular frequency.33 When EnT is more strongly coupled to nuclear motion or solvent rearrangement, Marcus theory provides an alternative formulation for kTEnT. Consequently, the diabatic states can be described as parabolas along this nuclear coordinate and ρ(E) obtained through geometric considerations (eqn (2)).34–38
. The density of states per unit energy of the final state is ρ(E), which depends on the nuclear reaction coordinate, and for a harmonic oscillator is equal to 1/ℏω, where ω is the angular frequency.33 When EnT is more strongly coupled to nuclear motion or solvent rearrangement, Marcus theory provides an alternative formulation for kTEnT. Consequently, the diabatic states can be described as parabolas along this nuclear coordinate and ρ(E) obtained through geometric considerations (eqn (2)).34–38 | (2) |
 | ||
Fig. 2 Schematic representation of coupling between diabatic potential energy surfaces used for determination of the rate constant according to Marcus theory. (a) Pictorial definition of the Gibbs activation energy ΔG‡, solvent reorganization energy λ, Gibbs reaction energy ΔG°, and diabatic electronic coupling  . (b) Four-state model showing the case in which direct coupling outcompetes both CT pathways. (c) Four-state model showing the case in which CT coupling outcompetes the direct pathway. (d) Four-state model showing the case in which both CT and direct pathways are competitive. Reprinted with permission from J. Chem. Theory Comput. 2023, 19, 7685–7694. Copyright 2023 American Chemical Society. . (b) Four-state model showing the case in which direct coupling outcompetes both CT pathways. (c) Four-state model showing the case in which CT coupling outcompetes the direct pathway. (d) Four-state model showing the case in which both CT and direct pathways are competitive. Reprinted with permission from J. Chem. Theory Comput. 2023, 19, 7685–7694. Copyright 2023 American Chemical Society. | ||
For a four-state model, additional diabatic coupling elements between the reactant/product and the CT states are required  . To calculate the overall TEnT reaction rate, it is necessary to consider all these coupling elements simultaneously. Marcus theory can be extended to the four-state model by considering CT and LE diabatic states as parabolas along the reaction coordinate (Fig. 2b–d).30 Both the relative energies of the CT states compared to LE states and the magnitude of the diabatic coupling element defines the extent to which CT states contribute to kTEnT. Although CT states are generally relatively high in energy, they show much larger coupling with reactant/product states than for the direct coupling. If the CT states are too high in energy, then even a large coupling element will not enable them to stabilize the reaction coordinate (Fig. 2b). However, when CT states are sufficiently low in energy, as shown in Fig. 2d, all states must be accounted for to determine the rate.
. To calculate the overall TEnT reaction rate, it is necessary to consider all these coupling elements simultaneously. Marcus theory can be extended to the four-state model by considering CT and LE diabatic states as parabolas along the reaction coordinate (Fig. 2b–d).30 Both the relative energies of the CT states compared to LE states and the magnitude of the diabatic coupling element defines the extent to which CT states contribute to kTEnT. Although CT states are generally relatively high in energy, they show much larger coupling with reactant/product states than for the direct coupling. If the CT states are too high in energy, then even a large coupling element will not enable them to stabilize the reaction coordinate (Fig. 2b). However, when CT states are sufficiently low in energy, as shown in Fig. 2d, all states must be accounted for to determine the rate.
As it is also difficult to assess the diabatic coupling element experimentally, computational models for its calculation are desirable.42 Approximating that the wavefunction can be described as a product of the nuclear and electronic components, the diabatic coupling element can be written as in eqn (3);
 | (3) |
In the remainder of this feature article, we will summarize challenges and approaches for obtaining diabatic states in the context of studying TEnT processes. Subsequently we will discuss approaches for the construction of the diabatic coupling elements. We then describe the use of these computational approaches to study TEnT in several technologically relevant applications. Finally, we conclude with an outlook of the challenges and future directions of research involving modelling of TEnT processes.
2. Challenges in construction of diabatic states
The challenge with constructing diabatic states from adiabatic states is that it is not possible to create a strictly diabatic basis from an adiabatic basis – except in rare cases.46 Recall that we previously described a qualitative picture of diabats as those which do not change character regardless of changes in the nuclear geometry. This qualitative picture holds true in mathematical sense as well, in that the derivative coupling between two diabatic states vanishes at every possible nuclear configuration (R). Thus, for a manifold of strict diabatic states ,
, | (4) |
To construct the diabatic states from the adiabats, it would be ideal if the diabatic states of interest could be completely described by the subset of adiabatic states of importance (span the same Hilbert subspace). Such a transformation could be made through use of an adiabatic-to-diabatic transformation matrix.44,71 However, in practice this transformation leads to conditions which cannot be satisfied.54,72 Instead, a set of diabatic states can be obtained that do not completely eliminate the nonadiabatic couplings, which are referred to as quasi-diabatic states.50,51,73–77
Owing to the fact that definitions of quasi-diabatic states are not unique, different descriptions of diabatic states can be obtained with different strengths and weaknesses. For the purposes of this feature article, these approaches can be broadly organized into those that optimize diabatic states variationally, and those that construct diabats from approximate transformation of adiabatic states. Here, we first give a brief outline of the different approaches to transform adiabatic states to the diabatic representation, before providing a more detailed discussion of how diabats can be directly optimized. The major challenge with transformation-based approaches is that they require a good quality description of a significant number of adiabatic states to provide a suitable basis in which to construct diabatic states. This often leads to situations where relatively expensive computations must be performed to generate many states, from which much information is discarded in constructing a few diabatic states of interest.30 Additionally, it is not always obvious how the adiabatic states must be constructed to correctly include all the required information of the diabatic states. However, these methods have the advantage of being more rigorously defined, at least in the limit of a complete basis of adiabatic states or when the subset of adiabatic states obey the curl condition.46,56 To transform from the adiabatic representation to the diabatic representation, variation of the parameters in the adiabatic-to-diabatic transformation matrix can be performed to minimize some measure of adiabaticity. Therefore, these methods are distinguished based on the objective function that is used in the variational minimization. Based on the definition of eqn (4), the most obvious choice is minimization of the nonadiabatic derivative coupling elements,78 although such a choice is not generally practical. Instead, the transformation can be carried out through ensuring “configurational uniformity” such that diabatic states change continuously along a given path.52,79–81 Alternatively, a similar outcome can be obtained through block diagonalization of the Hamiltonian.82,83 The Hamiltonian must be partitioned into active and external blocks, where the active block contains states of interest. Diabatic states are then obtained through minimization of the norm of the off-diagonal Hamiltonian blocks. The active block after the block-diagonalization is an effective Hamiltonian of diabatic states.
Rather than trying to ensure configurational uniformity directly, properties can be used to identify diabatic states. Minimizing the average transition dipole matrix elements results in a form of Boys localization applied to states rather than orbitals and ensures all states have as localized a dipole as possible, which is particularly useful in studies of exciton transfer.84,85 Generalized Mulliken–Hush (GMH) follows a similar approach, but in its most common form it ensures the transition dipole matrix element between two states vanishes.86 GMH is tailored for two-state charge transfer problems, and so is popular for ET studies where the relevant diabatic states are charge localized so that the dipole of each state does not significantly change with nuclear coordinate. Use of an Edmiston–Ruedenberg localization approach instead of Boys localization, in which the off-diagonal Coulomb interaction terms are minimized, leads to diabatic states with localized charge density.38 Similarly, the GMH scheme can be modified to use properties other than the two-state transition dipole matrix element. In the fragment charge difference model (FCD) the interfragment charge density is minimized, which has the effect of localizing charge on a particular fragment in each diabat.87 More useful for TEnT, the fragment spin density (FSD) model uses the spin density instead to obtain spin-localized diabatic states.42 For singlet EnT (SEnT), charge or spin density are less useful, and instead the excitation number (number of electron–hole pairs) can be used, as in the fragment excitation number difference scheme.88,89 While property based approaches to defining diabats are useful, they are often difficult to generalize beyond two states and cannot easily describe diabats with different character, such as those in the four-state TEnT model (Fig. 1).
Rather than determine adiabatic-to-diabatic transformations, an alternative approach is the direct construction of quasi-diabatic states. This approach avoids the need to construct adiabatic states and circumvents the two-state limitation of approaches like FSD. A particularly suitable framework for such an approach is valence bond theory due to the use of localized molecular orbitals. Using valence bond self-consistent field (VBSCF), an effective two-state model permits variational optimization of diabatic states.77,90 However, in general valence bond theory is used in combination with molecular orbitals theory to provide a more computationally tractable description of diabatic states. The block-localized wavefunction (BLW) method optimizes orbitals such that they have zero contribution from atomic orbitals that reside on different fragments.91,92 Similarly motivated, constrained density functional theory (CDFT) uses density-based constraints, such as number of electrons, charge, or spin, to define diabats.93,94 Rather than constraining orbital localization, Beratan constructed diabatic configuration interaction singles (CIS) states through transformation of orbitals to a natural localized molecular orbital (NLMO) basis.28 Finally, it has been observed for some time that solutions to the SCF equations can have quasi-diabatic character, particularly in the context of ET.95–99 However, only recently have developments in SCF optimization techniques100–106 been able to circumvent variational collapse and allow reliable convergence of SCF solutions with quasi-diabatic character.30,31 In our work, as we discuss below, these quasi-diabatic SCF (QD-SCF) solutions feature orbitals that are localized to different fragments and so can be used to represent diabatic states. Deficiencies in the single-reference framework of QD-SCF states can be overcome using orbital expansions based on UNOCAS,107 or projection (including half-projection).108
3. Methods for calculating the diabatic coupling
Having obtained a set of diabatic states, it follows that the next goal in calculation of eqn (1) is the determination of the diabatic coupling. The diabatic coupling is the off-diagonal matrix element of the Hamiltonian in the diabatic representation. When using adiabatic-to-diabatic transformation approaches for determining the diabatic states, the diabatic coupling elements arise naturally from the calculation. Nonetheless, as we will discuss in more detail below, an analytical form for the diabatic coupling may be available for two-state methods that avoids the requirement to explicitly determine the diabatic states (e.g. GMH, FCD etc.) However, where diabatic states are directly variationally constructed, the matrix elements between these states must be calculated from the diabats (e.g. CDFT, QD-SCF etc.).For models that are limited to describing two-states, the diabatic coupling element can be determined analytically from parameters of the two adiabatic states, without having to explicitly compute the diabatic states. The formulas are derived from analytical formulas for the characteristic roots of the Hamiltonian matrix, and so in principle can be obtained for up to four states (Abel-Ruffini theorem), although in practice it is more straightforward to build and diagonalize the operator matrix, which is equivalent to explicitly determining the diabatic states. In two-state GMH, the vertical excitation energy (E2 − E1), the difference between the adiabatic dipole moments (μ1–μ2), and the transition dipole moment μ12 is used to determine the diabatic coupling
 | (5) |
The methods that are derived from GMH (FCD, FSD, FED) all have formulas for the diabatic coupling that are analogous to eqn (5), where a different property is used in place of the dipole. For example, in the FCD model, the diabatic coupling element is computed using the fragment localized charge difference (Δq1, Δq2 and Δq12)
 | (6) |
While the above methods construct diabatic states implicitly to determine the diabatic coupling element, the connection between the diabatic coupling strength and the fragment orbital overlap permits the diabatic coupling to be assessed without ever constructing the diabatic states themselves. These approaches are closely based on energy gap formulation in GMH and related approaches (eqn (5)) but motivated by electronic structure rather than state properties. You, et al., developed an approach based on CIS to compute the strengths of the electronic coupling of TEnT.109 Their analysis showed that the energy gap between the two lowest triplet CIS states is proportional to the TEnT diabatic coupling. An extension of the CIS energy gap method to more than two diabatic states was developed so that transfer between a bridge-linked donor and acceptor could be performed.28 In this case the CIS was performed in a NLMO basis and the CIS energies obtained by removing different components of the CIS Hamiltonian depending on which diabatic coupling element was being computed. Rather that approximate orbital overlaps based on the energy gap method, empirical scaling of the frontier molecular orbital overlap was also found to provide reliable diabatic coupling elements for a four-state TEnT model.29
In cases where the diabats are calculated directly, the diabatic coupling element must be computed explicitly. Unlike in methods that transform the adiabatic Hamiltonian to a diabatic representation, the computed diabats are generally nonorthogonal, requiring special consideration of how to compute matrix elements and orthogonalize states. In the case of QD-SCF diabats, there are several different approaches for computing the nonorthogonal matrix element.110–124 A particularly exciting development is that of the GRONOR program, which is designed for deployment on leadership-class computing facilities, enabling accurate descriptions of diabatic wavefunctions on large systems.119,124 Where states are all expressed in a different orbital basis, a suitable approach is to construct transition density matrices and contract them with the Hamiltonian operator
 | (7) |
 where, as before, the prime indicates these are nonorthogonal diabats, the following expression is obtained
where, as before, the prime indicates these are nonorthogonal diabats, the following expression is obtained | (8) |
In the above equation, Fi is the free energy and ![[V with combining circumflex]](https://www.rsc.org/images/entities/i_char_0056_0302.gif) i is the constraining potential operator of the ith diabatic state, and Nfi is the diabat overlap.129–132 Care must be taken when using CDFT in cases of near-degeneracies in frontier orbitals that allow for fractional electron transfer between fragments.131 Having computed the couplings in the nonorthogonal diabatic representation, the remaining task is orthogonalization of the diabats to determine the final diabatic couplings. While there are numerous different orthogonalization methods, Löwdin symmetric orthogonalization133 is best suited to obtaining orthogonal states that retain their diabatic character as the transformation rotates all states equally, minimizing the angle that any one state rotates from the nonorthogonal diabatic state.30,31,131 Where there are only two states of interest, it is possible to construct an analytic formulation of the orthogonalized diabatic coupling element
i is the constraining potential operator of the ith diabatic state, and Nfi is the diabat overlap.129–132 Care must be taken when using CDFT in cases of near-degeneracies in frontier orbitals that allow for fractional electron transfer between fragments.131 Having computed the couplings in the nonorthogonal diabatic representation, the remaining task is orthogonalization of the diabats to determine the final diabatic couplings. While there are numerous different orthogonalization methods, Löwdin symmetric orthogonalization133 is best suited to obtaining orthogonal states that retain their diabatic character as the transformation rotates all states equally, minimizing the angle that any one state rotates from the nonorthogonal diabatic state.30,31,131 Where there are only two states of interest, it is possible to construct an analytic formulation of the orthogonalized diabatic coupling element
 | (9) |
As eqn (9) orthogonalizes only two states at a time, it provides an approximation of the true orthogonal diabatic states. However, the results of the two-state approximation can be compared to the results from full orthogonalization to establish the extent to which the two-states of interest are coupled to the remaining states. As highlighted below, we have found that the two-state approximation underestimates direct diabatic couplings, giving around half the value of four-state models.30
4. Modeling TEnT
Having explored how various research groups have tackled the technical challenges in computing diabatic couplings to determine TEnT kinetics, we now provide several examples of areas where studies have made use of these computational techniques.4.1. Bridge-mediated Dexter energy transfer
There are many practical examples of bridge-mediated TEnT chemical systems, where the donor–acceptor architecture is connected by a bridging moiety, including solar-energy harvesting,10 OLEDs,134 and light-harvesting complexes.7Skourtis, et al. presented a theory of bridge-mediated TEnT mechanisms using the CIS approach.28 The TEnT pathways including the contributions of the bridge, in addition to CT states, were investigated using alkyl-bridged and norbornyl-bridged dienes as example systems. A key distinction between this theory and earlier ones27,135 is that vertical excited states of the bridge are considered in the assessment of the diabatic coupling. It was demonstrated that the CT states with an electron or hole on the donor and the other on acceptor were the most significant contribution to the bridge-mediated TEnT coupling involving systems with short bridges and high tunneling-energy gaps. On the other hand, for systems with longer bridges or a lower bridge energy gap the excitations localized on the bridge dominate the coupling. In the case of the bridge excitations, the electron and hole are both found on the bridging unit. In the context of bridge-mediated TEnT, the Fermi golden rule for the rate can also be used but the coupling term takes on a different form (eqn (10)) than that shown in eqn (1) .
.
 | (10) |
Beratan and coworkers computed diabatic couplings based on the adiabatic energy-gap approach.136 Their study focused on noncovalently stacked naphthalene dimers and naphthalene-ethylene-naphthalene (Nap-Et-Nap) bridge-mediated assemblies. The minimum energy splitting approach was first used to study the dependence of diabatic couplings on the donor–acceptor orientation of an asymmetric naphthalene dimer system (i.e. without the bridge). It was demonstrated that the direct energy splitting method is unable to provide diabatic couplings consistent with the reference results from the FSD method.42,88 In contrast, the couplings from the minimum energy splitting method were in-line with the FSD method. They also computed diabatic couplings as a function of z-distance in a nonsymmetrically placed naphthalene dimer. When compared to the results using the FSD approach, the direct energy splitting method overestimated the couplings. On the other hand, the minimum energy splitting results exhibited close agreement with the FSD results. Their final investigation was analyzing how the diabatic couplings varied with the distance between the ethylene bridge and one naphthalene monomer along the z-axis considering a nonsymmetric Nap-Et-Nap structure. In agreement with the previous case, the minimum energy splitting method provided accurate outcomes compared to overestimated results of the direct energy splitting method when contrasted with FSD results. They also tested various quantum chemical levels of theory in this study for determining the energy splitting and diabatic couplings. For the basis set, it was concluded that diffuse functions are more important than basis-set size. Regarding the method, use of the Tamm–Dancoff approximation was found to be an improvement over full TD-DFT and TD-HF approaches. Finally, the accuracy of the diabatic couplings was dependent upon the quality of the CT state energies, and so functionals such as CAM-B3LYP were recommended.
In another investigation by Zhao and coworkers, the TEnT processes involving a benzophenone donor to a naphthalene acceptor via fluorene bridge, namely, the Bp-F-Nap system, was studied using CDFT.137 For this kind of donor-bridge-acceptor (DBA) system, two transfer pathways for TEnT are possible - tunneling (superexchange) and sequential hopping. In superexchange, the electron tunnels from the donor to the acceptor through the bridges (3DBA → DB3A), whereas the sequential hopping mechanism involves temporary localization of energy on the bridges and production of intermediate states (3DBA → D3BA → DB3A). Discussing the Bp-F system first, at the CDFT optimized 3Bp-F and Bp-3F diabatic state geometries, diabatic couplings of 2.4 and 5.7 meV were obtained, respectively. The variation in the diabatic potential energies of the 3Bp-F and Bp-3F states was also considered along the linear reaction coordinate (LRC), along with the change in the diabatic coupling. The LRC was taken as the elongation of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O in benzophenone. The diabatic couplings showed near-constant values of 3.9 meV at the crossing point of the two diabatic potential energy surfaces. Next, the naphthalene molecule was attached to the bridge to form the Bp-F-Nap system. At the point along the LRC where the diabatic energies of donor (3Bp-F-Nap) and acceptor (Bp-F-3Nap) states are equal, the superexchange coupling is found to be two orders of magnitude smaller than the direct coupling. Lastly, TEnT rates were calculated using both Fermi's Golden Rule and Marcus formulas, where it was found that the rates for the hopping pathways are significantly higher than superexchange pathways.
O in benzophenone. The diabatic couplings showed near-constant values of 3.9 meV at the crossing point of the two diabatic potential energy surfaces. Next, the naphthalene molecule was attached to the bridge to form the Bp-F-Nap system. At the point along the LRC where the diabatic energies of donor (3Bp-F-Nap) and acceptor (Bp-F-3Nap) states are equal, the superexchange coupling is found to be two orders of magnitude smaller than the direct coupling. Lastly, TEnT rates were calculated using both Fermi's Golden Rule and Marcus formulas, where it was found that the rates for the hopping pathways are significantly higher than superexchange pathways.
4.2. Excitation energy transfer and TEnT
Chen and coworkers focused on developing a scheme to account for the excitation energy transfer (EET) coupling with the generalization of the FCD scheme used for electron-transfer coupling.138 The short-range coupling, described by the contribution from diabatic coupling and the overlap effect, was obtained as the difference of EET coupling and the Coulomb coupling. For a pair of stacked naphthalenes, the short-range coupling was found to be very similar to the TEnT coupling in terms of both magnitudes and distance dependence. In EET, the electronic coupling consists of three terms, the Coulomb coupling (Coulomb interaction between electronic transitions), Dexter exchange coupling, and a third term arising from the overlap of electron densities of donor and acceptor moieties. In their proposed scheme, namely, fragment excitation difference (FED), the EET coupling is formulated as a function of the difference in the excitation population and the energy difference between the diabatic states. Their new formulation of Coulomb coupling involved the evaluation of two-electron integrals in atomic basis functions. They first tested their FED and Coulomb scheme with a pair of stacked naphthalene molecules. It was seen that with increase in inter-monomer distance, both the FED and Coulomb coupling showed decay consistent with d−3 dipole–dipole interaction. For small basis sets, a Gaussian decay was seen in the difference of FED and Coulomb couplings. The basis set dependence of this decay was observed to be very similar to that of TEnT coupling. This result indicated that the difference of FED and Coulomb couplings behaves almost like an “exchange” component such as the TEnT coupling. In order to investigate through bond effects, they considered truncated models of polynorbornyl-linked dimers. Although the FED scheme is applicable to all of the models regardless of their molecular symmetry, the Coulomb coupling can only be obtained for systems composed of separated fragments as individual calculation of donor and acceptor excitations is needed. When all of the three low-energy excited states of naphthalene, namely, Lb, La, and Bb, were considered for linked naphthalene dimers, the coupling values increased when more bridge fragments were restored in the truncated models. This confirmed the existence of through-bond EET coupling. As seen by the increase of Coulomb coupling with larger bridge fragments, the presence of significant Coulomb effect in the through-bond EET coupling was concluded. The contribution of short-range couplings was found to be very large, especially for La and Lb states. Compared to experimental results, their calculated FED couplings were found to be 20–40% higher.4.3. Singlet fission and the involvement of TEnT
Singlet fission is a photophysical process in which an excited singlet state on one fragment, produced by photon absorption, forms a correlated triplet excited state over two fragments which decoheres to generate two distinct triplet excited states (eqn (11)).139,140 While this section will primarily focus on the involvement of Dexter energy transfer in the context of singlet fission, a more comprehensive review of theoretical approaches for modeling singlet fission can be found in the review from Casanova.141The mechanism for singlet fission can be represented with the following stepwise kinetic model:139–142
| S0 + S1 ↔ 1(T1T1) ↔ T1 + T1 | (11) |
| S0 + S1 → [1(T1T1)] ↔ 1(T⋯T)] ↔ T1 + T1 | (12) |
In a combined experimental and computational study, Korovina and coworkers investigated the photophysics of two types of covalently linked 5-ethynyl-tetracence dimers. The first dimer had large cofacial overlap of the tetracene π-orbitals (called BET-X), while the second dimer exhibited a twisted geometry with less overlap of the π-orbitals (called BET-B).151 BET-B exhibited a long-lived 1(T1T1) state, while BET-X, relaxed to the ground state very rapidly. The possibility of TEnT in the mechanism of 1(T1T1) dissociation was explored by doping BET-B into a film of 5,12-diphenyltetracene (DPT). The transient absorption spectrum of BET-B doped in DPA showed a ground state bleach which suggests that TEnT occurs from the BET-B 1(T1T1) state to the film thus generating two independent triplet states – one on BET-B and the other on DPA. Thus, it was concluded that for systems with endothermic singlet fission, TEnT is essential to generate independent triplet excitons. The experimental work was supported by computational assessment of diabatic couplings, excitation energies and rates. The calculations indicated that the 1(T1T1) state of BET-X will be short-lived compared to that of BET-B as the coupling of the 1(T1T1) state to the S0 state of BET-X is much larger than in BET-B. Ultimately, the spectroscopy and calculations were used to demonstrate that the relative orientation of the tetracence analogues affects the energetics associated with the singlet, triplet, and 1(T1T1) states, as well as the couplings between the states. The relaxation pathways are also distinct depending on the degree of π-orbital overlap. In BET-X, with a large amount of overlap, the singlet excited state decays rapidly to either the excimer pathway or to a trapped 1(T1T1) that does not result in triplet excitons. In contrast, in BET-B, where the π-orbital overlap was lower than in BET-X, dissociation to the excimer configuration was not observed, and instead the triplet was generated through a singlet fission mechanism.
Scholes and coworkers also investigated the separation of the correlated triplet pair using temperature-dependent transient absorption spectroscopy complemented by calculations.152 Their model system was 6,3-bis(triisopropylsilylethynyl)pentacene (TIPS-Pn) films which exhibited exothermic singlet fission. They found that the correlated triplet pair evolves into triplet excitons by a TEnT mechanism on the picosecond timescale. While other possibilities were considered, this analysis was used to corroborate the conclusion that the dissociation of the correlated triplet pair in TIPS-Pn was governed by TEnT.
4.4. TEnT in molecular materials
TEnT has been observed in the family of azobenzene based photoswitches.155–159 Photoisomerization of azobenzene is notably fast in solution but not in the solid state. A challenge to overcome for efficient solid-state photoswitching of azobenzene is the low penetration depth of UV-vis light especially in thick materials. It is also known that there can be competing absorption between the isomers which complicates tunability. For various applications of azobenzene-based photoswitches, there is a need to red-shift the absorption as well as improve the spectral resolution between the E- and Z-isomers. Triplet-sensitized photoisomerization is one way to improve the efficiency and control of molecular photoswitches. Isokuortti et al., demonstrated near-unity photoconversion efficiency of Z → E isomerization of two ortho-functionalized bistable azobenzenes.160 TEnT was the driving force for the rapid photoisomerization following red and near-infrared excitation of porphyrin photosensitizers. It was further demonstrated that the process of triplet-sensitized photoisomerization is efficient despite cases where the photosensitizer's triplet energy is substantially lower than the azobenzene molecule.TEnT has also been implicated in azobenzene photoswitching mechanism within a host-photosensitizer complex.161 Gemen et al. tackled the challenge of obtaining the thermodynamically disfavored Z-isomer by placing the E-isomer into a supramolecular host along with a photosensitizer. They performed the E → Z isomerization of azobenzene via various wavelengths including red light, starting from stable E isomer of azobenzene and a dye that acts as an antenna placed in the supramolecular host. The dye was excited from the ground state to the singlet excited state and confinement inside of the host increased the dye's ability to undergo intersystem crossing, allowing for population of the dye triplet state for subsequent triplet sensitization. Interestingly, it was demonstrated by simulations that a small C–N–N–C dihedral angle torsion in the azobenzene reduced the azobenzene triplet energy, while increasing the triplet energy of the coencapsulated dye, such that the two triplet energies became quasi-degenerate. As a result, the dihedral twist was suggested as a contributing factor to the TEnT mechanism from the dye (donor) to the azobenzene (acceptor). One fate of the triplet azobenzene is conversion to the Z-isomer. Subsequently, the Z-isomer is expelled from the host molecule and cannot be resensitized.
While the azobenzene examples clearly highlight the beneficial nature of TEnT for some applications, in other instances, energy transfer is not a desired pathway. One such example is in the context of organic light emitting diodes (OLEDs). Improving the efficiency of these devices, which is a measure of the power required for a given amount of light produced, is a key goal.162–169 One approach for improving the efficiency of OLEDs is through thermally activated delayed fluorescence (TADF), in which population of the T1 state, produced through either intersystem crossing from the S1 state to the T1 state or direct excitation in the triplet manifold, undergoes reverse intersystem crossing (RISC) to the S1 state and subsequently fluoresces.170–175 As a result, OLED efficiency is improved because excitations in the triplet manifold can also contribute to OLED fluorescence. The rate of RISC depends on both the singlet–triplet energy gap (ΔEST) and the extent of spin–orbit coupling.176 However, energy transfer to a nearby phosphorescent emitter is an additional possibility for the fate of the triplet exciton besides RISC, as observed for some blue TADF emitters.177 An emerging area of research is developing TADF materials which can suppress the energy transfer pathway in order to ensure efficient OLEDs which do not suffer from trapping in a triplet exciton state.174 Reduction in the energy transfer pathway has been demonstrated already with one strategy being to reduce the triplet exciton population by fast kRISC and another being to spatially separate the TADF host and the emitter molecules.178,179
4.5. TEnT mediated photocatalysis
TEnT has been used in photocatalysis to access otherwise inaccessible triplet states of organic substrates in order to control reactivity in a variety of chemical processes such as cycloadditions,180–185 isomerizations23,159,186–188 and polymerizations.189–193 Computational studies center on (i) calculating triplet energies of catalysts and substrates,194 (ii) modeling how coordination or environment shifts those energies20,185 and (iii) analyzing donor–acceptor encounter complexes and orbital overlaps that govern TEnT rates.29,185TEnT photocatalytic reactions have been summarized in recent review articles,23 including cycloadditions,180–185E → Z isomerizations,23,159,186–188 deracemizations,6,195 strain-release reactions23,196–199 and C–C bond formations.23,159,196,200–202 Many of these reactions are driven by visible-light induced TEnT from an iridium, ruthenium based or organic PS to an organic substrate. [2+2]-photocycloadditions using direct excitation give poor quantum yield due to low extinction coefficients and requirement of higher frequency radiation. Through TEnT excitation of the olefinic substrate to its triplet state, radical addition to a second olefin partner in its singlet ground state readily occurs. [2+2] photocycloadditions have been reported using TEnT using chiral Lewis acids, in which Lewis acid coordination both reduces the triplet energy to allow exergonic triplet energy transfer from the photosensitizer and formation of enantioselective product.20 Thermodynamically disfavored E → Z isomerization of alkenes can be made feasible through the generation of high-energy twisted triplet species via TEnT, where differences in photosensitization rates of E- and Z- isomers controls stereoselectivity. Recent work203 shows that nitroarenes can function as PSs, where the triplet configuration of nitroarenes (3n,π* vs. 3π,π*) rather than the triplet energy alone was found to govern TEnT efficiency. High E → Z isomerization and [2+2] cycloaddition efficiency was associated with 3π,π* nitroarenes, while 3n,π* nitroarenes exhibited little or no reactivity. In deracemization reactions mediated by TEnT, a chiral PS binds to the substrate (cyclic204 or acyclic195 allenes and alkylidenecycloalkanes6 as well as centrally chiral 3-cyclopropylquinolin-2-one205 derivatives and spirocyclopropyl oxindoles206) stereospecifically due to steric differences in steric repulsion between enantiomers, leading to the depletion of the favored enantiomer. TEnT-induced energy-transfer polymerization189 uses generated triplet olefin species to perform polymerization without added radical initiators. Aldehyde activation and various cycloaddition reactions have been found to proceed in the presence of organic PSs, such as thioxanthone, in which mechanistic work has revealed TEnT as the operative pathway.207
To determine feasibility of proposed TEnT mechanisms, studies have computed vertical and adiabatic triplet energies of PSs and substrates to ensure that TEnT is thermodynamically feasible or to rationalize endergonic transfer. For example, in the Lewis acid catalyzed [2+2] cycloadditions of chalcone,20 the triplet energy of free chalcone was computed (∼51 kcal mol−1) and compared to a much lower triplet energy (∼32 kcal mol−1) upon coordination to Sc(III), explaining why TEnT from [Ru(bpy)3]2+ becomes favorable only in the complex. Computation of energy gaps has also been used to propose new structures to expand the range of available PSs. These studies have shown that Lewis acid,20,180,181 Brønsted acid,182 iminium catalysis,183 and H-bond donor184 species can lower the triplet energy of substrate, promising feasibility of TEnT in photocatalytic reactions. Recent work194 has used dynamic vertical triplet energies sampled from molecular dynamics to obtain distributions of triplet energies from conformational ensembles. This work demonstrated how conformational flexibility affects TEnT feasibility. Computational modelling has also been used to provide evidence of TEnT in order to distinguish the observed behavior from electron transfer photocatalyzed processes. Calculations that showed Lewis acid coordination mainly impacts triplet energy in the chalcone system was consistent with TEnT rather than photoredox activation.20,23 Several TEnT mechanism studies29,31,185 have been reported that build explicit donor–acceptor encounter complexes (PS + substrate) and compute electronic couplings and orbital overlaps to observe TEnT rates and selectivity. These analyses show how orientation and distance between π-systems impact Dexter-type coupling. Work on mechanisms of endothermic and irreversible TEnT208 has been used to investigate suppression of TEnT back-transfer and how multi-step or irreversible chemical follow-up reactions can allow effective uphill TEnT.
4.6. Nonorthogonal approach to TEnT
The suitability of the NOCI framework for the calculation of the electronic coupling element was recently demonstrated for the four systems shown in Fig. 3. The systems included the ethylene dimer, ethylene/methaniminium cation,30 as well as naphthalene dimer and 2,2′-bifluorene molecule.31 For the first three cases, the diabatic coupling elements were calculated as a function of the distance between the two optimized molecules, while for the 2,2′-bifluorene molecule, the diabatic coupling was calculated as a function of the rotation around the C2–C2′ covalent bond. Geometries were optimized using density functional theory with the ωB97xD functional, which incorporates empirical dispersion, as well as constraints on the point group symmetry.209 For the ethylene and ethylene/methaniminium cation the 6-311+G(d) basis set was used while 6-31G(d) was used for the naphthalene dimer and 2,2′-bifluorene. A modified version of Gaussian 16 was employed to locally optimize the quasi-diabatic SCF solutions. Subsequently, the nonorthogonal matrix elements were computed with a stand-alone in-house code that requires the MQCPack library interfaced with a modified version of Gaussian 16.210,211 | ||
| Fig. 3 The model systems which were used in the calculation of TEnT coupling elements using the nonorthogonal framework including (a) ethylene dimer, (b) ethylene/methaniminium cation, (c) the naphthalene dimer, and (d) 2,2′-bifluorene. | ||
The ethylene dimer model system contains two identical ethylene molecules and as a result the initially excited triplet state (D*A) and the final triplet state (DA*) resulting from the two-electron transfer are equivalent. Similarly, the CT states (D+A− and D−A+) which result from one-electron/hole transfer are also indistinguishable. Several conclusions can be drawn from the resulting orthogonal diabatic Hamiltonian (eqn (13)) which contains the relative diabatic coupling elements in meV where the lowest diabatic coupling element is set as 0.0 meV with other matrix elements shifted accordingly.
 | (13) |
There is an order of magnitude difference between the coupling elements which involve one-electron transfer and those which involve two-electron transfer. The two-electron coupling elements, either along the direct pathway (D*A and DA*, ∼44 meV), or between CT states (D+A− and D−A+, ∼46 meV), were similar in magnitude. Table 1 summarizes how the diabatic coupling elements change as a function of intermolecular distance (3.5–5.0 Å). For comparison, results from the two-state model using eqn (9) are also included in Table 1, along with the direct coupling and CIS results from You et al.109
| R (Å) | Diabatic coupling elements, direct pathway | Diabatic coupling elements, CT pathways | Z.-Q. You, et al. J. Chem. Phys. 2006, 124, 044506. | |||||
|---|---|---|---|---|---|---|---|---|
|
|
|
|
||||||
| SOH | Two-state model (VDA) | SOH | Two-state model (VDA) | SOH | Two-state model (VDA) | DC109 | CIS109 | |
| 3.5 | 43.5 | 98.4 | 654.1 | 655.5 | 539.3 | 540.8 | 99.1 | 94.2 |
| 4.0 | 26.3 | 26.2 | 443.2 | 443.3 | 311.8 | 311.6 | 28.5 | 27.2 |
| 4.5 | 11.6 | 5.7 | 292.2 | 292.1 | 173.3 | 172.2 | 8.2 | 8.0 |
| 5.0 | 4.7 | 1.3 | 277.3 | 275.7 | 75.6 | 71.7 | 2.3 | 2.2 |



With increasing intermolecular distance, the coupling elements decrease with distance for both the direct and CT pathways. The coupling elements for the direct pathway are consistently an order of magnitude smaller than those involving CT states at all distances. The results from the the four-state model are in good agreement with those from the two-state model. One distinction is that the coupling elements from the two-state model for the direct pathway decay faster than for those in the four-state model computed using the symmetric orthogonalized Hamiltonian. This suggests that at long distances, the two-state model underestimates the involvement of the direct pathway. Conversely, at short distances, the two-state model overestimates the involvement of the direct TEnT pathway.
The TEnT diabatic coupling elements were also determined for the ethylene/methaniminium cation system.30 Due to the asymmetric monomers, the energies of the CT diabatic states will be different, where the hole transfer path is much higher in energy than the electron transfer path. Consequently, the higher energy hole CT state can be neglected from the four-state model. Like the ethylene dimer case, the coupling elements were determined as a function of the distance between the face-to-face ethylene and methaniminium cation. Diabatic coupling elements were determined for the direct and electron CT pathways using symmetric orthogonalization of the Hamiltonian (Table 2).
| R (Å) | Diabatic coupling elements, direct pathway | Diabatic coupling elements, CT (electron) pathway | Z.-Q. You, et al. J. Chem. Phys. 2006, 124, 044506 | |||
|---|---|---|---|---|---|---|
|
|
|
|||||
| SOH | Two-state model (VIJ) | SOH | Two-state model (VIJ) | DC109 | CIS109 | |
| 3.5 | 5.6 | 209.3 | 497.1 | 476.9 | 212.3 | 171.4 |
| 4.0 | 4.3 | 60.2 | 278.9 | 275.0 | 78.9 | 56.8 |
| 4.5 | 0.4 | 19.2 | 158.9 | 158.3 | 21.2 | 16.1 |
| 5.0 | 1.6 | 7.6 | 87.8 | 87.8 | 5.7 | 4.5 |


As shown in Table 2, there are two orders of magnitude difference between the diabatic coupling elements of the direct and electron CT. The three-state model is in close agreement with the two-state model for coupling along the CT pathway, while along the direct pathway, there is a significant difference, especially at short-distance (5.6 meV vs. 209.3 meV). Therefore, while the two-state model suggests that both CT and direct pathways are competitive, especially at short distance, the three-state model, suggests that the rate only depends on the CT pathway.
For the naphthalene dimer in D2h symmetry, using the two-state model, the electronic couplings were evaluated for intermolecular z distances 3.6 Å–5.2 Å, as well as for x- and y-displacements 0.0 Å–4.0 Å of one naphthalene monomer with respect to other.31 The electronic couplings obtained from the four-state model are shown in Fig. 4. With increasing intermolecular distance in the z direction, the electronic coupling of the direct and CT pathways decrease monotonically, which is consistent with the decreasing frontier molecular orbital (MO) overlap. The coupling element of the CT pathways is an order of magnitude larger than the coupling element of the direct pathway. With increasing x-displacement, the electronic couplings decay and approach zero where the orbital overlap is minimal. In the direct pathway, the electronic couplings vanish at 0.7 Å, 1.25 Å, and 3.75 Å x-displacements, due to nodal structure in the π and π* orbitals of the donor and acceptor naphthalene molecules at those geometries. Similarly, the CT coupling elements show a similar trend, but with fewer nodes, at 1.6 Å and 3.3 Å. Conversely, maxima in the electronic coupling elements occur along the direct pathway at 0.0 Å, 0.9 Å, 1.4 Å and 2.6 Å, and at 2.25 Å along the CT pathways, which indicate geometries where the frontier π and π* orbitals have maximal overlap. For displacement in the y direction, the direct and CT electronic couplings decay from 0.0 Å to 4.0 Å due to the decreased overlap between the frontier π and π* orbitals as y-displacement increases. Along the direct coupling pathway, certain values of y-displacement (i.e.1.1 Å) show vanishing electronic coupling values due to nodal positions in the frontier π and π* MOs. However, along the CT pathways the electronic coupling is always non-zero. As was seen along the z-direction, the direct coupling for displacements along x- and y-directions is also significantly smaller compared to the CT couplings.
 | ||
| Fig. 4 The TEnT coupling elements (absolute values) for naphthalene dimer using the four-state model for direct (a) and (d) and (g), electron (b), (e) and (h) and hole (c) and (f) and (i) transfer mechanisms along intermolecular z distance (a)–(c), x-distance (d)–(f) and y-distance (g)–(i). Adopted with permission from Ref. 31. Copyright 2025 American Chemical Society. | ||
The four-state model was also utilized to compute the electronic couplings for 2,2′-bifluorene for both the direct and CT pathways.31 Unlike the dispersion-bound naphthalene dimer, in 2,2′-bifluorene, two fluorene rings are connected through a C2–C2′ covalent bond. The direct and CT couplings along the torsional angle Φ for 2,2′-bifluorene are shown in Fig. 5, where both types of couplings show a maximum at Φ = 0° and Φ = 180° due to coplanar geometry of 2,2′-bifluorene where the frontier MO overlap is maximized. The couplings have a minimum at Φ = 90° where the planes of the two fluorene rings are perpendicular to one another. Compared to the naphthalene dimer, where CT couplings are an order of magnitude larger than direct coupling, in 2,2′-bifluorene, CT and direct couplings are similar in magnitude. Therefore, the direct coupling plays a greater role in the triplet energy transfer mechanism in 2,2′-bifluorene than in the naphthalene dimer. The change in the relative magnitude of couplings in 2,2′-bifluorene results from both a decrease in the magnitude of CT couplings compared to the naphthalene dimer, and an increase in magnitude of direct coupling, rather than just a change in the magnitude of any one type of coupling element. The reason for the larger magnitude of the direct coupling in 2,2′-bifluorene is the conductive pathway that is created between the two fluorene rings through the C2–C2′ covalent bond, resulting from π-conjugation between the fluorene rings at a quasi-planar geometry. The decrease in the CT coupling magnitude is a consequence of reduced frontier π and π* MO overlap in 2,2′-bifluorene, where naphthalene dimer can form a parallel orientation of the monomers that is not possible in 2,2′-bifluorene.
 | ||
| Fig. 5 TEnT coupling elements (absolute values) for (a) direct, (b) electron, and (c) hole pathways for 2,2′-bifluorene evaluated using the four-state model along the torsional angle (degrees) between the two fluorene rings. Adopted with permission from Ref. 31. Copyright 2025 American Chemical Society. | ||
The main challenge of using quasi-diabatic SCF solutions is ensuring that the correct quasi-diabatic states have been identified. The spin densities and biorthogonal orbitals can be analyzed to verify that the correct electronic structure has been obtained through the calculations. The spin densities for the LE states (D*A and DA*) as well as for the CT states (D+A− and D−A+) of the naphthalene dimer at the D2h symmetry with the intermolecular distance of 3.6 Å are shown in Fig. 6. It can be seen, as expected from the four-state model, for the LE states (D*A and DA*) there is net localization of spin density only on one fragment of the dimer, whereas for CT states (D+A− and D−A+) there is an equal distribution of spin density over both fragments. Although spin densities provide a useful way to verify that correct diabatic states have been obtained, the specific information about the nature of the frontier orbitals cannot be determined just from the spin densities, especially for CT states. As a result, it is necessary to perform a biorthogonal orbital transformation in order to determine on which monomer the unpaired electrons reside.
 | ||
| Fig. 6 (a) Spin density plots for the initial or product state (D*A or DA*, top panel) and the CT state (D+A− or D−A+, bottom panel) and (b) biorthogonal orbitals of the D+A− state for the naphthalene dimer. The first number in the parentheses indicates the molecular orbital number and the second number indicates the overlap between corresponding alpha and beta orbitals. Reprinted with permission from J. Phys. Chem. A https://doi.org/10.1021/acs.jpca.4c06478. Copyright 2025 American Chemical Society. | ||
The biorthogonal orbitals for the D+A− state are shown in Fig. 6b. For the CT states, in contrast to the LE states, there is a distribution of the two unpaired alpha electrons over both the donor and acceptor moieties of the naphthalene dimer. The biorthogonal orbitals can distinguish between the delocalization of singly occupied orbitals over both monomers and the localization of singly occupied orbitals on each monomer. Similar analysis of spin density and biorthogonal orbitals can be done for 2,2′-bifluorene. As was demonstrated for the naphthalene dimer, the excess alpha spin density is localized only on one fragment for the LE states (D*A and DA*) for 2,2′-bifluorene (Fig. 7a) whereas it is distributed equally over the two fluorene rings for the CT states (D+A− and D−A+) (Fig. 7b). Additional analysis of biorthogonal orbitals allows for the correct identification of the localization of singly occupied orbitals in the LE and CT states for this molecule.
 | ||
| Fig. 7 Spin density plots for the (a) initial or product state (D*A or DA*) and (b) CT state (D+A− or D−A+) of the 2,2′-bifluorene. Reprinted with permission from J. Phys. Chem. A https://doi.org/10.1021/acs.jpca.4c06478. Copyright 2025 American Chemical Society. | ||
Outlook, challenges and future directions
While significant advancements have been made in the computational modelling of TEnT processes, there are several fronts that we can identify as areas for continued research. We can divide these into two categories, the first being primarily theoretical developments related to improving computational models of energy transfer processes. The second category broadly encompasses applications, in which new methodologies will be applied to understand larger and more experimentally-relevant systems.The first challenge in improving the theoretical framework is proper inclusion of the vibronic contribution to the rate equation. While it is generally accepted that the TEnT rate depends most strongly on the electronic coupling element, the Franck–Condon weighted density of states modifies the rate associated with each pathway.30,31 A proper computational approach requires calculation of the Frank–Condon weighted density of states, which necessitates correctly identifying the nuclear motion along which the states are connected nonadiabatically. The second challenge is the development of a multistate formulation of the Fermi Golden rule. Our current approach is to use the quasidiabatic states as a basis in which diabatic coupling matrix elements are constructed using the two-state Fermi Golden Rule formulation. Orthogonalization of the diabatic coupling matrix generates effective diabatic coupling elements in which multistate effects incorporating contributions from intermediate charge-transfer states are introduced. Proper consideration of intermediate states is of particular value to the study of TEnT involving transition metals, which may involve additional states than in the four-state model for organic systems, such as ligand-to-metal charge transfer states. The third computational challenge is the case of strongly correlated diabatic states. For TEnT, the diabats are generally high spin, which are well described by a single determinant. However, in singlet fission, photoinduced electron transfer, or singlet EnT, the diabats are not high spin and so not well-described by a single determinant. Additionally, in the case of transition metals, there may be a low-spin open-shell core that must be correctly symmetry adapted. Finally, the fourth computational challenge is the inclusion of dynamic correlation energy which can potentially be accomplished using multireference perturbation theory.
The above developments in the computational tools for the study of TEnT will permit investigation of systems that are larger and of greater relevance to developing technologies and chemical applications. As illustrated in this article, TEnT mechanisms play a role in molecular materials and photocatalysts, and yet these systems are often challenging to model computationally due to the presence of transition metals or complex environments. Through continued development of computational models, it will be possible to study of systems containing transition metals, as well as reactions with singlet oxygen. Although we focus our discussion on TEnT, the general scheme is transferable to singlet EnT, photoinduced ET, singlet fission, long-range ET, and proton-coupled ET. In all these cases, the challenge is forming the correct effective Hamiltonian, which involves identifying a suitable set of diabatic basis states.
Conflicts of interest
There are no conflicts of interest to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
This material is based on the work supported by the National Science Foundation under Grant 2144905. This work was conducted in part using the resources of the University of Louisville's Research Computing group and the Cardinal Research Cluster.References
- R. Silbey, Annu. Rev. Phys. Chem., 1976, 27, 203–223 CrossRef CAS.
- D. S. Tyson and F. N. Castellano, J. Phys. Chem. A, 1999, 103, 10955–10960 CrossRef CAS.
- C. B. Murphy, Y. Zhang, T. Troxler, V. Ferry, J. J. Martin and W. E. Jones, J. Phys. Chem. B, 2004, 108, 1537–1543 CrossRef CAS.
- T. Lazarides, D. Sykes, S. Faulkner, A. Barbieri and M. D. Ward, Chem. – Eur. J., 2008, 14, 9389–9399 Search PubMed.
- M. E. Daub, H. Jung, B. J. Lee, J. Won, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2019, 141, 9543–9547 Search PubMed.
- T. Kratz, P. Steinbach, S. Breitenlechner, G. Storch, C. Bannwarth and T. Bach, J. Am. Chem. Soc., 2022, 144, 10133–10138 CrossRef CAS PubMed.
- R. J. Cogdell and H. A. Frank, Biochim. Biophys. Acta, Bioenerg., 1987, 895, 63–79 Search PubMed.
- T. Mirkovic, E. E. Ostroumov, J. M. Anna, R. van Grondelle, Govindjee and G. D. Scholes, Chem. Rev., 2017, 117, 249–293 Search PubMed.
- C. Curutchet and B. Mennucci, Chem. Rev., 2017, 117, 294–343 CrossRef CAS PubMed.
- J. Lin, X. Hu, P. Zhang, A. Van Rynbach, D. N. Beratan, C. A. Kent, B. P. Mehl, J. M. Papanikolas, T. J. Meyer, W. Lin, S. S. Skourtis and M. Constantinou, J. Phys. Chem. C, 2013, 117, 22250–22259 CrossRef CAS.
- G. J. Hedley, A. Ruseckas and I. D. W. Samuel, Chem. Rev., 2017, 117, 796–837 CrossRef CAS PubMed.
- F.-C. Chen, S.-C. Chang, G. He, S. Pyo, Y. Yang, M. Kurotaki and J. Kido, J. Polym. Sci., Part B:Polym. Phys., 2003, 41, 2681–2690 CrossRef CAS.
- A. Köhler and H. Bässler, J. Mater. Chem., 2011, 21, 4003–4011 RSC.
- C. Mongin, S. Garakyaraghi, N. Razgoniaeva, M. Zamkov and F. N. Castellano, Science, 2016, 351, 369–372 CrossRef CAS PubMed.
- N. Hildebrandt, C. M. Spillmann, W. R. Algar, T. Pons, M. H. Stewart, E. Oh, K. Susumu, S. A. Díaz, J. B. Delehanty and I. L. Medintz, Chem. Rev., 2017, 117, 536–711 CrossRef CAS PubMed.
- Y. Jiang and J. McNeill, Chem. Rev., 2017, 117, 838–859 CrossRef CAS PubMed.
- J. A. Bender, E. K. Raulerson, X. Li, T. Goldzak, P. Xia, T. Van Voorhis, M. L. Tang and S. T. Roberts, J. Am. Chem. Soc., 2018, 140, 7543–7553 CrossRef CAS PubMed.
- X. Luo, R. Lai, Y. Li, Y. Han, G. Liang, X. Liu, T. Ding, J. Wang and K. Wu, J. Am. Chem. Soc., 2019, 141, 4186–4190 CrossRef CAS.
- D. C. Fabry, M. A. Ronge and M. Rueping, Chem. – Eur. J., 2015, 21, 5350–5354 CrossRef CAS.
- T. R. Blum, Z. D. Miller, D. M. Bates, I. A. Guzei and T. P. Yoon, Science, 2016, 354, 1391–1395 CrossRef CAS.
- K. L. Skubi, J. B. Kidd, H. Jung, I. A. Guzei, M.-H. Baik and T. P. Yoon, J. Am. Chem. Soc., 2017, 139, 17186–17192 CrossRef CAS PubMed.
- A. Acharya, A. M. Bogdanov, B. L. Grigorenko, K. B. Bravaya, A. V. Nemukhin, K. A. Lukyanov and A. I. Krylov, Chem. Rev., 2017, 117, 758–795 CrossRef CAS PubMed.
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068–1089 RSC.
- T. Förster, Ann. Phys., 1948, 437, 55–75 Search PubMed.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836–850 Search PubMed.
- G. D. Scholes and K. P. Ghiggino, J. Chem. Phys., 1994, 101, 1251–1261 CrossRef CAS.
- R. D. Harcourt, G. D. Scholes and K. P. Ghiggino, J. Chem. Phys., 1994, 101, 10521–10525 CrossRef CAS.
- S. S. Skourtis, C. Liu, P. Antoniou, A. M. Virshup and D. N. Beratan, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8115–8120 CrossRef CAS.
- S. Bai, P. Zhang and D. N. Beratan, J. Phys. Chem. C, 2020, 124, 18956–18960 CrossRef CAS.
- L. M. Thompson, E. M. Kempfer-Robertson, S. Saha, S. Parmar and P. M. Kozlowski, J. Chem. Theory Comput., 2023, 19, 7685–7694 CrossRef CAS PubMed.
- S. Saha, M. J. Mackintosh, L. M. Thompson and P. M. Kozlowski, J. Phys. Chem. A, 2024, 129, 967–977 CrossRef PubMed.
- E. Fermi, Nuclear Physics, University of Chicago Press, Chicago, 1974 Search PubMed.
- C. Serpa, L. G. Arnaut, S. J. Formosinho and K. R. Naqvi, Photochem. Photobiol. Sci., 2003, 2, 616–623 CrossRef CAS PubMed.
- R. A. Marcus, J. Phys. Chem., 1963, 67, 853–857 CrossRef CAS.
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, Rev. Bioenerg., 1985, 811, 265–322 CrossRef CAS.
- A. Nitzan, Chemical Dynamics in Condensed Phases: Relaxation, Transfer and Reactions in Condensed Molecular Systems, Oxford University Press, 2006 Search PubMed.
- G. C. Schatz and M. A. Ratner, Quantum mechanics in chemistry, Courier Corporation, 2002 Search PubMed.
- J. E. Subotnik, J. Vura-Weis, A. J. Sodt and M. A. Ratner, J. Phys. Chem. A, 2010, 114, 8665–8675 CrossRef CAS PubMed.
- L. M. Frutos, O. Castaño, J. L. Andrés, M. Merchán and A. U. Acuña, J. Chem. Phys., 2004, 120, 1208–1216 CrossRef CAS PubMed.
- L. M. Frutos and O. Castaño, J. Chem. Phys., 2005, 123, 104108 CrossRef PubMed.
- F. Zapata, M. Marazzi, O. Castaño, A. U. Acuña and L. M. Frutos, J. Chem. Phys., 2014, 140, 034102 CrossRef PubMed.
- Z.-Q. You and C.-P. Hsu, J. Chem. Phys., 2010, 133, 074105 Search PubMed.
- F. T. Smith, Phys. Rev., 1969, 179, 111–123 CrossRef.
- M. Baer, Chem. Phys. Lett., 1975, 35, 112–118 Search PubMed.
- M. Baer, Chem. Phys., 1976, 15, 49–57 CrossRef CAS.
- C. A. Mead and D. G. Truhlar, J. Chem. Phys., 1982, 77, 6090–6098 CrossRef CAS.
- C. A. Mead, Rev. Mod. Phys., 1992, 64, 51–85 CrossRef CAS.
- A. Macias and A. Riera, J. Phys. B:At., Mol. Opt. Phys., 1978, 11, L489 CrossRef CAS.
- A. Macias and A. Riera, J. Phys. B:At., Mol. Opt. Phys., 1978, 11, 1077 CrossRef CAS.
- H. Werner and W. Meyer, J. Chem. Phys., 1981, 74, 5802–5807 Search PubMed.
- E. S. Kryachko and D. R. Yarkony, Int. J. Quantum Chem., 2000, 76, 235–243 CrossRef CAS.
- H. Nakamura and D. G. Truhlar, J. Chem. Phys., 2001, 115, 10353–10372 Search PubMed.
- Y. Shu, Z. Varga, S. Kanchanakungwankul, L. Zhang and D. G. Truhlar, J. Phys. Chem. A, 2022, 126, 992–1018 Search PubMed.
- T. Van Voorhis, T. Kowalczyk, B. Kaduk, L.-P. Wang, C.-L. Cheng and Q. Wu, Annu. Rev. Phys. Chem., 2010, 61, 149–170 CrossRef CAS PubMed.
- V. Sidis, Adv. Chem. Phys., 1992, 73–134 Search PubMed.
- H. Köuppel, W. Domcke and L. S. Cederbaum, Adv. Chem. Phys., 1984, 59–246 Search PubMed.
- U. Manthe and H. Köppel, J. Chem. Phys., 1990, 93, 1658–1669 CrossRef CAS.
- Y. Kim, J. C. Corchado, J. Villà, J. Xing and D. G. Truhlar, J. Chem. Phys., 2000, 112, 2718–2735 CrossRef CAS.
- R. Liyanage, R. J. Gordon and R. W. Field, J. Chem. Phys., 1998, 109, 8374–8387 CrossRef CAS.
- J. A. Mueller, M. L. Morton, S. L. Curry, J. P. D. Abbatt and L. J. Butler, J. Phys. Chem. A, 2000, 104, 4825–4832 Search PubMed.
- T. Ichino, A. J. Gianola, W. C. Lineberger and J. F. Stanton, J. Chem. Phys., 2006, 125, 084312 Search PubMed.
- J. M. Cohen and D. A. Micha, J. Chem. Phys., 1992, 97, 1038–1052 CrossRef CAS.
- J. C. Tully, Ann. Rev. Phys. Chem., 2000, 51, 153–178 Search PubMed.
- S. Mahapatra, H. Köppel and L. S. Cederbaum, J. Phys. Chem. A, 2001, 105, 2321–2329 Search PubMed.
- L. Pauling, The Nature of the Chemical Bond, Cornell University, Ithaca, NY, 3rd edn, 1960 Search PubMed.
- A. Warshel and R. M. Weiss, J. Am. Chem. Soc., 1980, 102, 6218–6226 CrossRef CAS.
- J. Åqvist and A. Warshel, Chem. Rev., 1993, 93, 2523–2544 Search PubMed.
- J. Juanos Timoneda and J. T. Hynes, J. Phys. Chem., 1991, 95, 10431–10442 CrossRef.
- S. Hammes-Schiffer and J. C. Tully, J. Chem. Phys., 1994, 101, 4657–4667 CrossRef CAS.
- A. M. Kuznetsov and J. Ulstrup, J. Chem. Phys., 1999, 77, 1085–1096 CAS.
- M. Baer, Mol. Phys., 1980, 40, 1011–1013 CrossRef CAS.
- M. Baer and R. Englman, Mol. Phys., 1992, 75, 293–303 CrossRef CAS.
- G. Hirsch, R. J. Buenker and C. Petrongolo, Mol. Phys., 1990, 70, 835–848 CrossRef CAS.
- M. Perić, S. D. Peyerimhoff and R. J. Buenker, Mol. Phys., 1990, 71, 693–719 CrossRef.
- X. Zhu and D. R. Yarkony, J. Chem. Phys., 2010, 132, 104101 CrossRef PubMed.
- C. L. Malbon and D. R. Yarkony, J. Phys. Chem. A, 2015, 119, 7498–7509 CrossRef CAS PubMed.
- J. Gao, M. Garcia-Viloca, T. D. Poulsen and Y. Mo, Adv. Phys. Org. Chem., 2003, 38, 161–181 CrossRef CAS.
- Z. Varga, K. A. Parker and D. G. Truhlar, Phys. Chem. Chem. Phys., 2018, 20, 26643–26659 RSC.
- K. Ruedenberg and G. J. Atchity, J. Chem. Phys., 1993, 99, 3799–3803 CrossRef CAS.
- G. J. Atchity and K. Ruedenberg, Theor. Chem. Acc., 1997, 97, 47–58 Search PubMed.
- H. Nakamura and D. G. Truhlar, J. Chem. Phys., 2002, 117, 5576–5593 CrossRef CAS.
- L. S. Cederbaum, H. Köppel and W. Domcke, Int. J. Quantum Chem., 1981, 20, 251–267 CrossRef.
- T. Pacher, C. A. Mead, L. S. Cederbaum and H. Köppel, J. Chem. Phys., 1989, 91, 7057–7062 CrossRef CAS.
- R. J. Cave and M. D. Newton, Chem. Phys. Lett., 1996, 249, 15–19 CrossRef CAS.
- J. E. Subotnik, S. Yeganeh, R. J. Cave and M. A. Ratner, J. Chem. Phys., 2008, 129, 244101 CrossRef PubMed.
- R. J. Cave and M. D. Newton, J. Chem. Phys., 1997, 106, 9213–9226 CrossRef CAS.
- A. A. Voityuk and N. Rösch, J. Chem. Phys., 2002, 117, 5607–5616 CrossRef CAS.
- C.-P. Hsu, Acc. Chem. Res., 2009, 42, 509–518 CrossRef CAS PubMed.
- Z.-Q. You and C.-P. Hsu, Int. J. Quantum Chem., 2014, 114, 102–115 CrossRef CAS.
- L. Song and J. Gao, J. Phys. Chem. A, 2008, 112, 12925–12935 CrossRef CAS PubMed.
- Y. Mo and J. Gao, J. Phys. Chem. A, 2000, 104, 3012–3020 CrossRef CAS.
- Y. Mo and J. Gao, J. Comput. Chem., 2000, 21, 1458–1469 CrossRef CAS.
- N. Davari, P.-O. Åstrand and T. Van Voorhis, Mol. Phys., 2013, 111, 1456–1461 CrossRef CAS.
- B. Kaduk, T. Kowalczyk and T. Van Voorhis, Chem. Rev., 2012, 112, 321–370 CrossRef CAS PubMed.
- K. Ohta, G. L. Closs, K. Morokuma and N. J. Green, J. Am. Chem. Soc., 1986, 108, 1319–1320 Search PubMed.
- A. Broo and S. Larsson, Chem. Phys., 1990, 148, 103–115 Search PubMed.
- A. Farazdel, M. Dupuis, E. Clementi and A. Aviram, J. Am. Chem. Soc., 1990, 112, 4206–4214 CrossRef CAS.
- L. Y. Zhang, R. A. Friesner and R. B. Murphy, J. Chem. Phys., 1997, 107, 450–459 CrossRef CAS.
- M. D. Newton, Int. J. Quantum Chem., 1980, 18, 363–391 CrossRef.
- A. J. W. Thom and M. Head-Gordon, Phys. Rev. Lett., 2008, 101, 193001 CrossRef PubMed.
- A. T. B. Gilbert, N. A. Besley and P. M. W. Gill, J. Phys. Chem. A, 2008, 112, 13164–13171 CrossRef CAS PubMed.
- H.-Z. Ye, M. Welborn, N. D. Ricke and T. Van Voorhis, J. Chem. Phys., 2017, 147, 214104 CrossRef PubMed.
- L. M. Thompson, J. Chem. Phys., 2018, 149, 194106 CrossRef PubMed.
- G. M. J. Barca, A. T. B. Gilbert and P. M. W. Gill, J. Chem. Theory Comput., 2018, 14, 1501–1509 CrossRef CAS PubMed.
- K. Carter-Fenk and J. M. Herbert, J. Chem. Theory Comput., 2020, 16, 5067–5082 Search PubMed.
- X. Dong, A. D. Mahler, E. M. Kempfer-Robertson and L. M. Thompson, J. Chem. Theory Comput., 2020, 16, 5635–5644 Search PubMed.
- J. M. Bofill and P. Pulay, J. Chem. Phys., 1989, 90, 3637–3646 CrossRef CAS.
- G. E. Scuseria, C. A. Jiménez-Hoyos, T. M. Henderson, K. Samanta and J. K. Ellis, J. Chem. Phys., 2011, 135, 124108 CrossRef PubMed.
- Z.-Q. You, C.-P. Hsu and G. R. Fleming, J. Chem. Phys., 2006, 124, 044506 Search PubMed.
- P.-O. Löwdin, Phys. Rev., 1955, 97, 1490–1508 CrossRef.
- H. Fukutome, Prog. Theor. Phys., 1988, 80, 417–432 Search PubMed.
- J. Verbeek and J. H. Van Lenthe, J. Mol. Struct. THEOCHEM, 1991, 229, 115–137 Search PubMed.
- J. Verbeek and J. H. Van Lenthe, Int. J. Quantum Chem., 1991, 40, 201–210 Search PubMed.
- F. Dijkstra and J. H. Van Lenthe, Int. J. Quantum Chem., 1998, 67, 77–83 CrossRef CAS.
- R. Broer, L. Hozoi and W. C. Nieuwpoort, Mol. Phys., 2003, 101, 233–240 CrossRef CAS.
- I. Mayer, Simple theorems, proofs, and derivations in quantum chemistry, Springer, New York, 2003 Search PubMed.
- A. J. W. Thom and M. Head-Gordon, J. Chem. Phys., 2009, 131, 124113 Search PubMed.
- H. G. A. Burton and A. J. W. Thom, J. Chem. Theory Comput., 2019, 15, 4851–4861 Search PubMed.
- T. P. Straatsma, R. Broer, S. Faraji, R. W. A. Havenith, L. E. A. Suarez, R. K. Kathir, M. Wibowo and C. de Graaf, J. Chem. Phys., 2020, 152, 064111 CrossRef CAS PubMed.
- A. D. Mahler and L. M. Thompson, J. Chem. Phys., 2021, 154, 244101 CrossRef CAS PubMed.
- H. G. A. Burton, J. Chem. Phys., 2021, 154, 144109 Search PubMed.
- H. G. A. Burton, J. Chem. Phys., 2022, 157, 204109 Search PubMed.
- E. M. Kempfer-Robertson, A. D. Mahler, M. N. Haase, P. Roe and L. M. Thompson, J. Phys. Chem. Lett., 2022, 13, 12041–12048 Search PubMed.
- T. P. Straatsma, R. Broer, A. Sánchez-Mansilla, C. Sousa and C. de Graaf, J. Chem. Theory Comput., 2022, 18, 3549–3565 Search PubMed.
- M. Pavanello, T. Van Voorhis, L. Visscher and J. Neugebauer, J. Chem. Phys., 2013, 138, 054101 CrossRef PubMed.
- W.-L. Chan, T. C. Berkelbach, M. R. Provorse, N. R. Monahan, J. R. Tritsch, M. S. Hybertsen, D. R. Reichman, J. Gao and X.-Y. Zhu, Acc. Chem. Res., 2013, 46, 1321–1329 Search PubMed.
- H. Ren, M. R. Provorse, P. Bao, Z. Qu and J. Gao, J. Phys. Chem. Lett., 2016, 7, 2286–2293 Search PubMed.
- Y. Lu and J. Gao, J. Chem. Theory Comput., 2024, 20, 8474–8481 Search PubMed.
- Q. Wu, C.-L. Cheng and T. Van Voorhis, J. Chem. Phys., 2007, 127, 164119 Search PubMed.
- Q. Wu, B. Kaduk and T. Van Voorhis, J. Chem. Phys., 2009, 130, 034109 Search PubMed.
- M. G. Mavros and T. Van Voorhis, J. Chem. Phys., 2015, 143, 231102 CrossRef PubMed.
- S. Yeganeh and T. V. Voorhis, J. Phys. Chem. C, 2010, 114, 20756–20763 Search PubMed.
- P.-O. Löwdin, in Advances in Quantum Chemistry, ed. P.-O. Löwdin, Academic Press, 1970, vol. 5, pp. 185–199 Search PubMed.
- M. A. Baldo, D. F. O’Brien, Y. You, A. Shoustikov, S. Sibley, M. E. Thompson and S. R. Forrest, Nature, 1998, 395, 151–154 CrossRef CAS.
- G. L. Closs, M. D. Johnson, J. R. Miller and P. Piotrowiak, J. Am. Chem. Soc., 1989, 111, 3751–3753 CrossRef CAS.
- S. Bai, P. Zhang and D. N. Beratan, J. Phys. Chem. A, 2024, 128, 5721–5729 Search PubMed.
- Y. Si, W. Liang and Y. Zhao, J. Phys. Chem. C, 2012, 116, 12499–12507 CrossRef CAS.
- C.-P. Hsu, Z.-Q. You and H.-C. Chen, J. Phys. Chem. C, 2008, 112, 1204–1212 Search PubMed.
- M. B. Smith and J. Michl, Chem. Rev., 2010, 110, 6891–6936 CrossRef CAS PubMed.
- M. B. Smith and J. Michl, Ann. Rev. Phys. Chem., 2013, 64, 361–386 CrossRef CAS PubMed.
- D. Casanova, Chem. Rev., 2018, 118, 7164–7207 Search PubMed.
- C. E. Swenberg and N. E. Geacintov, In Organic Molecular Photophysics, Wiley-Interscience, 1973 Search PubMed.
- E. C. Greyson, B. R. Stepp, X. Chen, A. F. Schwerin, I. Paci, M. B. Smith, A. Akdag, J. C. Johnson, A. J. Nozik, J. Michl and M. A. Ratner, J. Phys. Chem. B, 2010, 114, 14223–14232 CrossRef CAS PubMed.
- L. Sebastian, G. Weiser and H. Bässler, Chem. Phys., 1981, 61, 125–135 CrossRef CAS.
- K. O. Lee and T. T. Gan, Chem. Phys. Lett., 1977, 51, 120–124 CrossRef CAS.
- C. Zeiser, L. Moretti, D. Lepple, G. Cerullo, M. Maiuri and K. Broch, Angew. Chem., Int. Ed., 2020, 59, 19966–19973 CrossRef CAS PubMed.
- G. B. Piland and C. J. Bardeen, J. Phys. Chem. Lett., 2015, 6, 1841–1846 CrossRef CAS PubMed.
- R. D. Pensack, E. E. Ostroumov, A. J. Tilley, S. Mazza, C. Grieco, K. J. Thorley, J. B. Asbury, D. S. Seferos, J. E. Anthony and G. D. Scholes, J. Phys. Chem. Lett., 2016, 7, 2370–2375 CrossRef CAS PubMed.
- G. D. Scholes, J. Phys. Chem. A, 2015, 119, 12699–12705 Search PubMed.
- A. R. Srimath Kandada, A. Petrozza and G. Lanzani, Phys. Rev. B:Condens. Matter Mater. Phys., 2014, 90, 075310 CrossRef.
- N. V. Korovina, S. Das, Z. Nett, X. Feng, J. Joy, R. Haiges, A. I. Krylov, S. E. Bradforth and M. E. Thompson, J. Am. Chem. Soc., 2016, 138, 617–627 CrossRef CAS PubMed.
- T. S. Lee, Y. L. Lin, H. Kim, R. D. Pensack, B. P. Rand and G. D. Scholes, J. Phys. Chem. Lett., 2018, 9, 4087–4095 Search PubMed.
- V. Abraham and N. J. Mayhall, J. Phys. Chem. Lett., 2021, 12, 10505–10514 CrossRef CAS PubMed.
- N. J. Mayhall, J. Chem. Theory Comput., 2016, 12, 4263–4273 CrossRef CAS PubMed.
- J. Moreno, M. Gerecke, L. Grubert, S. A. Kovalenko and S. Hecht, Angew. Chem., Int. Ed., 2016, 55, 1544–1547 CrossRef CAS PubMed.
- P. Bharmoria, S. Ghasemi, F. Edhborg, R. Losantos, Z. Wang, A. Mårtensson, M. Morikawa, N. Kimizuka, Ü. İşci, F. Dumoulin, B. Albinsson and K. Moth-Poulsen, Chem. Sci., 2022, 13, 11904–11911 RSC.
- L. B. Jones and G. S. Hammond, J. Am. Chem. Soc., 1965, 87, 4219–4220 CrossRef CAS.
- E. Fischer, J. Am. Chem. Soc., 1968, 90, 796–797 Search PubMed.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190–7202 Search PubMed.
- J. Isokuortti, K. Kuntze, M. Virkki, Z. Ahmed, E. Vuorimaa-Laukkanen, M. A. Filatov, A. Turshatov, T. Laaksonen, A. Priimagi and N. A. Durandin, Chem. Sci., 2021, 12, 7504–7509 Search PubMed.
- J. Gemen, J. R. Church, T.-P. Ruoko, N. Durandin, M. J. Białek, M. Weißenfels, M. Feller, M. Kazes, M. Odaybat, V. A. Borin, R. Kalepu, Y. Diskin-Posner, D. Oron, M. J. Fuchter, A. Priimagi, I. Schapiro and R. Klajn, Science, 2023, 381, 1357–1363 Search PubMed.
- A. Endo, M. Ogasawara, A. Takahashi, D. Yokoyama, Y. Kato and C. Adachi, Adv. Mater., 2009, 21, 4802–4806 Search PubMed.
- A. Endo, K. Sato, K. Yoshimura, T. Kai, A. Kawada, H. Miyazaki and C. Adachi, Appl. Phys. Lett., 2011, 98, 083302 Search PubMed.
- K. Goushi, K. Yoshida, K. Sato and C. Adachi, Nat. Photonics, 2012, 6, 253–258 CrossRef CAS.
- H. Tanaka, K. Shizu, H. Miyazaki and C. Adachi, Chem. Commun., 2012, 48, 11392–11394 Search PubMed.
- H. Uoyama, K. Goushi, K. Shizu, H. Nomura and C. Adachi, Nature, 2012, 492, 234–238 Search PubMed.
- Q. Zhang, J. Li, K. Shizu, S. Huang, S. Hirata, H. Miyazaki and C. Adachi, J. Am. Chem. Soc., 2012, 134, 14706–14709 Search PubMed.
- F. B. Dias, K. N. Bourdakos, V. Jankus, K. C. Moss, K. T. Kamtekar, V. Bhalla, J. Santos, M. R. Bryce and A. P. Monkman, Adv. Mater., 2013, 25, 3707–3714 Search PubMed.
- Y. Tao, K. Yuan, T. Chen, P. Xu, H. Li, R. Chen, C. Zheng, L. Zhang and W. Huang, Adv. Mater., 2014, 26, 7931–7958 Search PubMed.
- Y. Olivier, J.-C. Sancho-Garcia, L. Muccioli, G. D’Avino and D. Beljonne, J. Phys. Chem. Lett., 2018, 9, 6149–6163 Search PubMed.
- X.-K. Chen, D. Kim and J.-L. Brédas, Acc. Chem. Res., 2018, 51, 2215–2224 Search PubMed.
- T. J. Penfold, F. B. Dias and A. P. Monkman, Chem. Commun., 2018, 54, 3926–3935 Search PubMed.
- F. B. Dias, T. J. Penfold and A. P. Monkman, Methods Appl. Fluoresc., 2017, 5, 012001 Search PubMed.
- J. M. Dos Santos, D. Hall, B. Basumatary, M. Bryden, D. Chen, P. Choudhary, T. Comerford, E. Crovini, A. Danos, J. De, S. Diesing, M. Fatahi, M. Griffin, A. K. Gupta, H. Hafeez, L. Hämmerling, E. Hanover, J. Haug, T. Heil, D. Karthik, S. Kumar, O. Lee, H. Li, F. Lucas, C. F. R. Mackenzie, A. Mariko, T. Matulaitis, F. Millward, Y. Olivier, Q. Qi, I. D. W. Samuel, N. Sharma, C. Si, L. Spierling, P. Sudhakar, D. Sun, E. Tankelevičiūtė, M. Duarte Tonet, J. Wang, T. Wang, S. Wu, Y. Xu, L. Zhang and E. Zysman-Colman, Chem. Rev., 2024, 124, 13736–14110 CrossRef CAS PubMed.
- P. de Silva, C. A. Kim, T. Zhu and T. Van Voorhis, Chem. Mater., 2019, 31, 6995–7006 Search PubMed.
- C. A. Kim and T. Van Voorhis, J. Phys. Chem. A, 2021, 125, 7644–7654 Search PubMed.
- N. Haase, A. Danos, C. Pflumm, P. Stachelek, W. Brütting and A. P. Monkman, Mater. Horiz., 2021, 8, 1805–1815 Search PubMed.
- C. Duan, Y. Xin, Z. Wang, J. Zhang, C. Han and H. Xu, Chem. Sci., 2022, 13, 159–169 RSC.
- R. Dong, D. Liu, J. Li, M. Ma, Y. Mei, D. Li and J. Jiang, Mater. Chem. Front., 2022, 6, 40–51 RSC.
- X. Huang, T. R. Quinn, K. Harms, R. D. Webster, L. Zhang, O. Wiest and E. Meggers, J. Am. Chem. Soc., 2017, 139, 9120–9123 Search PubMed.
- R. Kleinmans, S. Dutta, K. Ozols, H. Shao, F. Schäfer, R. E. Thielemann, H. T. Chan, C. G. Daniliuc, K. N. Houk and F. Glorius, J. Am. Chem. Soc., 2023, 145, 12324–12332 Search PubMed.
- E. M. Sherbrook, H. Jung, D. Cho, M.-H. Baik and T. P. Yoon, Chem. Sci., 2020, 11, 856–861 Search PubMed.
- F. M. Hörmann, C. Kerzig, T. S. Chung, A. Bauer, O. S. Wenger and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 9659–9668 Search PubMed.
- J. Ma, S. Chen, P. Bellotti, R. Guo, F. Schäfer, A. Heusler, X. Zhang, C. Daniliuc, M. K. Brown, K. N. Houk and F. Glorius, Science, 2021, 371, 1338–1345 Search PubMed.
- E. Lee, H. Moon, J. Park and M.-H. Baik, J. Comput. Chem., 2025, 46, e70155 CrossRef CAS PubMed.
- T. Neveselý, M. Wienhold, J. J. Molloy and R. Gilmour, Chem. Rev., 2022, 122, 2650–2694 CrossRef PubMed.
- S. I. Faßbender, J. J. Molloy, C. Mück-Lichtenfeld and R. Gilmour, Angew. Chem., Int. Ed., 2019, 58, 18619–18626 CrossRef PubMed.
- J. J. Molloy, J. B. Metternich, C. G. Daniliuc, A. J. B. Watson and R. Gilmour, Angew. Chem., Int. Ed., 2018, 57, 3168–3172 CrossRef CAS PubMed.
- J. Liu, Y. Wei, L. Ma, X. Jin, S. Sun, S. Zhou, X. Xu, H. Tian and X. Ma, Natl. Sci. Rev., 2025, 12, nwaf381 Search PubMed.
- C. Wu, N. Corrigan, C.-H. Lim, W. Liu, G. Miyake and C. Boyer, Chem. Rev., 2022, 122, 5476–5518 Search PubMed.
- Y. Kwon, W. Jeon, J. Gierschner and M. S. Kwon, Acc. Chem. Res., 2025, 58, 1581–1595 CrossRef CAS PubMed.
- Y. Lyu, H. Nie, J. Du and K. Wu, Nat. Commun., 2025, 16, 10013 CrossRef CAS PubMed.
- X. Tang, L.-Q. Meng, R. Li, S. Zhu, S. Lian, B. Jia and Z. An, Angew. Chem., Int. Ed., 2026, 65, e22611 CrossRef CAS PubMed.
- M. V. Popescu and R. S. Paton, Chem, 2024, 10, 3428–3443 CAS.
- M. Plaza, J. Großkopf, S. Breitenlechner, C. Bannwarth and T. Bach, J. Am. Chem. Soc., 2021, 143, 11209–11217 CrossRef CAS PubMed.
- F. Strieth-Kalthoff and F. Glorius, Chem, 2020, 6, 1888–1903 CAS.
- R. Guo, Y.-C. Chang, L. Herter, C. Salome, S. E. Braley, T. C. Fessard and M. K. Brown, J. Am. Chem. Soc., 2022, 144, 7988–7994 CrossRef CAS PubMed.
- R. I. Rodríguez, V. Corti, L. Rizzo, S. Visentini, M. Bortolus, A. Amati, M. Natali, G. Pelosi, P. Costa and L. Dell’Amico, Nat. Catal., 2024, 7, 1223–1231 CrossRef.
- V. P. Demertzidou, E. Skolia and C. G. Kokotos, ChemCatChem, 2025, 17, e00760 CrossRef CAS.
- J. D. Bell and J. A. Murphy, Chem. Soc. Rev., 2021, 50, 9540–9685 RSC.
- A. Solé-Daura and F. Maseras, Chem. Sci., 2024, 15, 13650–13658 RSC.
- R. Kancherla, K. Muralirajan, B. Maity, S. Karuthedath, G. S. Kumar, F. Laquai, L. Cavallo and M. Rueping, Nat. Commun., 2022, 13, 2737 CrossRef CAS PubMed.
- M. Rihtaršič, B. Kweon, P. T. Błyszczyk, A. Ruffoni, E. M. Arpa and D. Leonori, Nat. Catal., 2025, 8, 1361–1369 CrossRef PubMed.
- A. Hölzl-Hobmeier, A. Bauer, A. V. Silva, S. M. Huber, C. Bannwarth and T. Bach, Nature, 2018, 564, 240–243 CrossRef PubMed.
- A. Tröster, A. Bauer, C. Jandl and T. Bach, Angew. Chem., Int. Ed., 2019, 58, 3538–3541 CrossRef PubMed.
- X. Li, R. J. Kutta, C. Jandl, A. Bauer, P. Nuernberger and T. Bach, Angew. Chem., Int. Ed., 2020, 59, 21640–21647 CrossRef CAS PubMed.
- N. F. Nikitas, P. L. Gkizis and C. G. Kokotos, Org. Biomol. Chem., 2021, 19, 5237–5253 RSC.
- A. Kalpattu and J. T. Fourkas, Chem. Phys. Rev., 2025, 6, 031305 CrossRef CAS.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- L. M. Thompson, X. Sheng, A. D. Mahler, D. Mullally and H. P. Hratchian, MQCPack, 2022, 22, 6 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |