
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Selective nucleophilic addition of aryllithium reagents to α-ketonitriles giving ketones under microflow conditions
Mohmmad S.
Qenawy
 ,
Kazuhiro
Okamoto
and
Aiichiro
Nagaki
*
,
Kazuhiro
Okamoto
and
Aiichiro
Nagaki
*
Department of Chemistry, Faculty of Science, Hokkaido University, Kita 10-jo, Nishi 8-chome, Kita-ku, Sapporo 060-0810, Japan
First published on 18th March 2026
Abstract
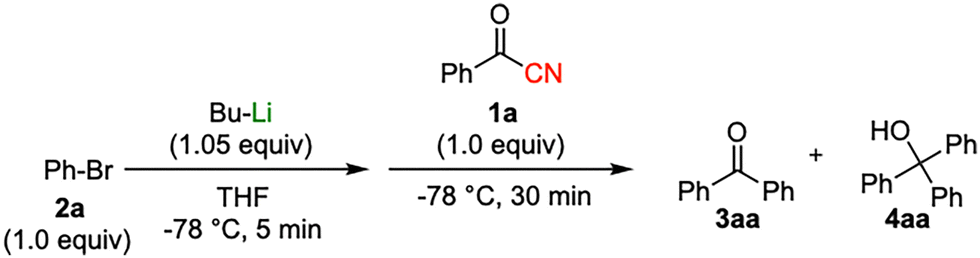
We report the selective nucleophilic addition of aryllithium species to acyl cyanides—electrophiles bearing both carbonyl and cyano functional groups—under catalyst-free continuous flow conditions. Using benzoyl cyanide as a model substrate, we demonstrate that the flow microreactor system enables highly selective addition to the carbonyl moiety while eliminating the cyano group as a leaving group, affording polyfunctional ketones in significantly improved yields compared to traditional batch processes.
The acylation of organometallic intermediates with acid chlorides represents a classical and widely utilized strategy for the synthesis of polyfunctional ketones, which are key structural motifs in numerous pharmaceuticals and materials science applications.1–3 A broad spectrum of organometallic reagents—including organomanganese, organozinc, and organomagnesium compounds—have been developed and applied in acylation reactions.4–8 While organomanganese reagents have demonstrated particular utility in such transformations, polyfunctional organozinc reagents, especially when combined with stoichiometric copper reagent9–11 or palladium catalysis, have also enabled efficient access to functionalized ketone frameworks.12,13 The preparation of functionalized arylzinc species remains synthetically challenging,14 often necessitating cobalt catalysis15–17 or the use of highly reactive activated zinc.18–20 Arylmagnesium halides can be readily generated via halogen–magnesium exchange methods and have been successfully employed in the synthesis of multifunctional diarylketones through acylation reactions.21–23 Previous studies have demonstrated moderate success in these transformations, with yields typically ranging from 50% to 58% when employing benzoyl chloride as the electrophile, despite optimization efforts and catalyst screening.24–39 These results align with broader literature observations indicating that acylation of organometallics with acid chlorides often proceeds with only moderate efficiency.3 A significant improvement in acylation efficiency was achieved by replacing acid chlorides with acyl cyanides.40–42 Acyl cyanides exhibit enhanced electrophilicity due to the strong electron-withdrawing nature of the cyano group.
Unlike the chlorine substituent in acid chlorides, the cyano group withdraws electron density from the adjacent carbonyl, thereby increasing its susceptibility to nucleophilic attack (Scheme 1).43 Given the high reactivity of organolithium species and their propensity for over-addition or decomposition under conventional batch conditions, we hypothesized that flow microreactor technology could overcome these limitations by offering rapid mixing, precise temperature control, and minimized residence times.44–65 Here we report the unique reactivity of acyl cyanides towards aryllithium reagents under microflow conditions. While prior studies predominantly focused on organomagnesium species using batch processes, the chemoselectivity of highly reactive aryllithium intermediates toward bifunctional electrophiles bearing both carbonyl and cyano groups remains underexplored under flow microreactor systems.66
 | ||
| Scheme 1 Reaction pathways for nucleophilic addition of Ar1Li to acyl cyanide Ar2COCN. | ||
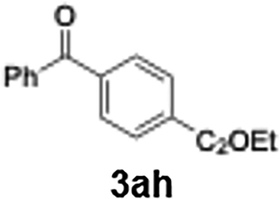
Initial study begun with the investigation of the chemoselectivity of aryllithium reagents toward bifunctional electrophiles bearing both carbonyl and cyano groups under flow conditions, employing benzoyl cyanide as a model electrophile (Scheme 2). In a batch reactor system, aryllithium intermediates were generated in situ via halogen–lithium exchange between bromobenzene and n-butyllithium (n-BuLi) at −78 °C, followed by the reaction with 1 equiv. of benzoyl cyanide (Scheme 1). The desired benzophenone (3aa) was obtained in 15% yield because of nucleophilic addition of the aryllithium species to the carbonyl group of benzoyl cyanide, accompanied by the formation of triphenylmethanol (4aa) in 42% yield. Given the high reactivity of organolithium species, any unreacted aryllithium species could readily undergo secondary addition to the ketone intermediate.
 | ||
| Scheme 2 Reaction of benzoyl cyanide (1a) and PhLi generated from bromobenzene (2a). | ||
The same transformation was subsequently examined under continuous flow conditions utilizing a microreactor system composed of two T-shaped micromixers and two microtube reactors (Scheme 3). A systematic investigation of the reaction temperature was conducted wherein the temperature at the second micromixer was varied between 0 °C and −78 °C. A pronounced temperature dependence was observed, whereby both the yield of the desired product 3aa and the overall chemoselectivity were markedly enhanced under flow conditions relative to the corresponding batch reaction. The reaction yield progressively increased as the temperature decreased, reaching a maximum at −78 °C, at which the highest yield and selectivity were obtained (Fig. 1). These findings underscore the critical role of low-temperature operation in optimizing reaction efficiency under flow conditions. Importantly, to evaluate the practical applicability of the protocol, a gram-scale experiment was performed under the optimized flow conditions, affording product 3aa in 87% isolated yield. This result demonstrates the scalability and synthetic utility of the methodology (see SI, p. S4, for details). A series of experiments were conducted wherein the total flow rate was maintained constant at 10.5 mL min−1, while the internal diameter of micromixer M2 was systematically varied. As the diameter increased, both the yield of product 3aa and the overall selectivity decreased (Fig. 2), underscoring the critical importance of efficient mixing. Notably, the highest yield was obtained using a micromixer with an inner diameter of 250 µm at a flow rate of 10.5 mL min−1.
 | ||
| Scheme 3 A flow microreactor system for reaction of benzoyl cyanide 1a and an aryllithium reagent generated from aryl halide 2a. T1 = 0 °C. When total flow rate is 10.5 mL min−1, t1 = 1.7 s, t2 = 2.2 s. | ||
 | ||
| Fig. 1 A flow microreactor system for reaction of benzoyl cyanide (1a) and PhLi generated from bromobenzene (2a): the effect of the temperature. | ||
 | ||
Fig. 2 A flow microreactor system for reaction of benzoyl cyanide (1a) and PhLi generated from bromobenzene (2a): the effect of the mixing. Flow rate at M2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10.5 mL min−1. Reaction's temperature: T2 = −78 °C. :10.5 mL min−1. Reaction's temperature: T2 = −78 °C. | ||
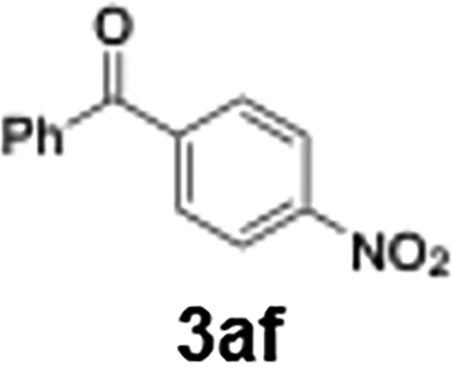
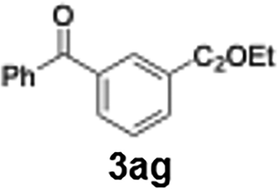
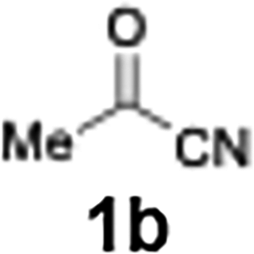
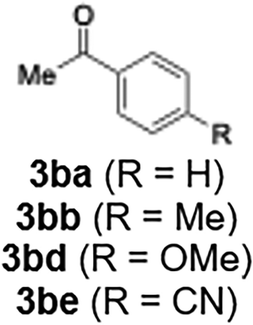
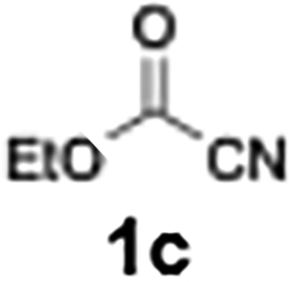
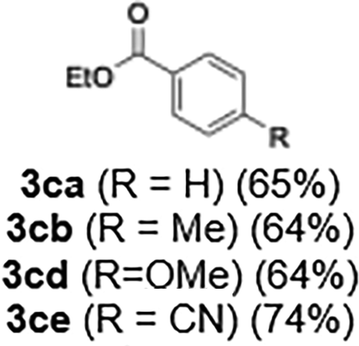
With the optimized conditions established, we subsequently explored the scope of the reaction by employing a range of aryllithium reagents bearing various electrophilic functional groups, with particular emphasis on evaluating the influence of steric hindrance (Table 1). Reactions involving benzoyl cyanide and aryllithium reagents substituted with either electron-donating or electron-withdrawing groups furnished the desired products in yields ranging from 71% to 90%. Aryllithium species bearing electron-donating substituents such as methyl, methoxy and isobutyl groups were well tolerated under the optimized flow conditions and consistently delivered the corresponding diaryl ketones in high yields, indicating that increased electron density on the aromatic ring does not adversely affect the nucleophilic acylation process. For example, the reaction of (4-cyanophenyl)lithium with benzoyl cyanide afforded product 3ae in 71% yield. These high yields are attributed to the advantages of the flash microflow system, which facilitates the efficient generation and immediate consumption of short-lived aryllithium intermediates through rapid mixing and precise control over temperature and residence time, thereby minimizing byproduct formation. Furthermore, the applicability of this methodology was confirmed by extending the protocol to other α-keto nitriles, such as acetyl cyanide and ethyl cyanoformate, which similarly afforded high yields under flow conditions.
| α-Keto nitriles | Aryl Halide | Product | Yield (%) |
|---|---|---|---|
| Reaction Temp.: T1 = 0 °C. When total flow rate is 10.5 mL min−1, t1 = 1.7 s, t2 = 2.2 s. | |||
|
|

|
90 |
| 74 | |||
| 80 | |||
| 75 | |||
| 71 | |||
|
|
77 | |
|
|
53 | |
|
|
54 | |
|
|
|
76 |
| 72 | |||
| 70 | |||
| 62 | |||
|
|
61 | |
|
|
|
65 |
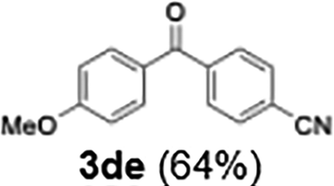
| 64 | |||
| 64 | |||
| 74 | |||
|
|
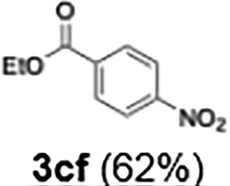
62 | |
|

|
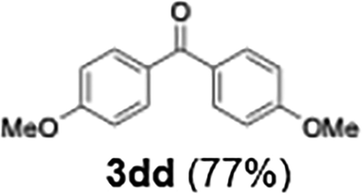
|
77 |
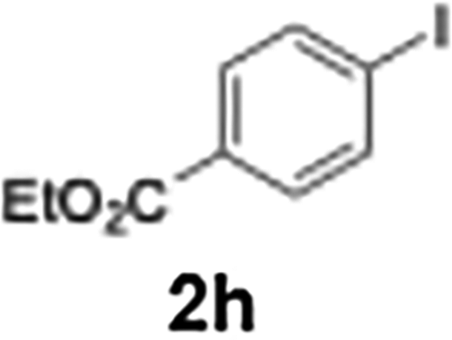
|
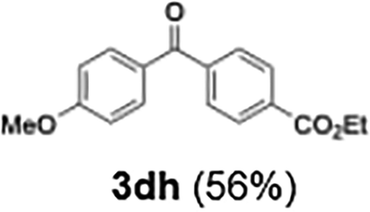
|
56 | |
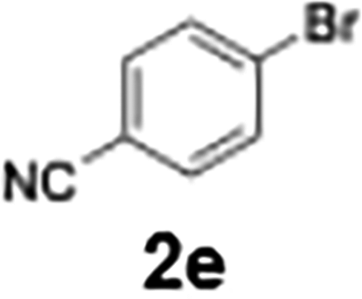
|
|
64 | |
|
|
|
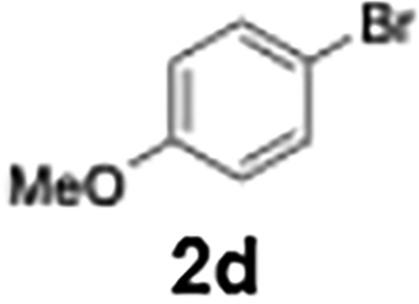
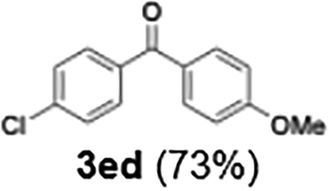
73 |
|
|
51 | |
|
|
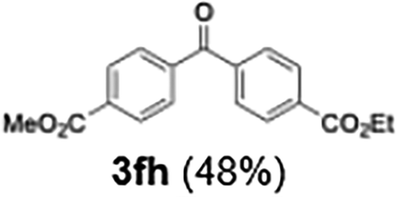
|
48 |
|
|
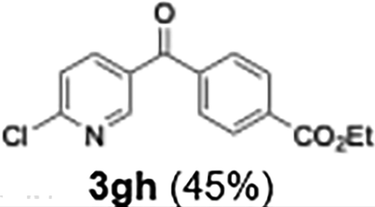
|
45 |
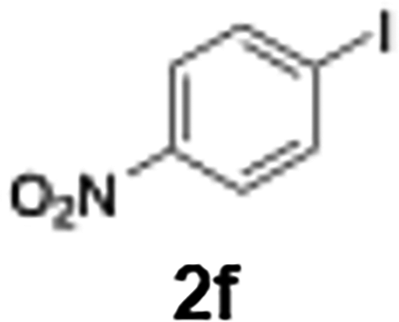
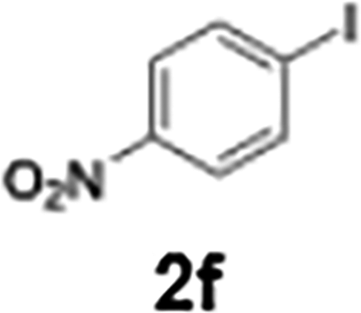
The generation of highly short-lived aryllithium species involving nitro and alkoxycarbonyl moieties presented considerable challenges due to the well-documented instability of the corresponding nucleophilic intermediates, which are prone to rapid decomposition under conventional batch conditions.65 For the generation of nitrophenyllithium species, freshly generated PhLi with microflow mixing was immediately transferred to micromixer M2, maintained at −40 °C, where it underwent halogen–lithium exchange with p-nitrophenyl iodide (2f), affording the desired aryllithium intermediate (Scheme 4a).
 | ||
| Scheme 4 Flash-flow generation of short-lived aryllithium intermediates. (a) p-nitrophenyllithium; (b) (ethoxycarbonyl)phenyllithium. | ||
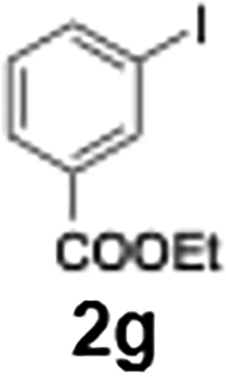
The acylation of meta- and para-substituted ethyl iodobenzoates presented significant synthetic challenges, largely due to their reduced reactivity under conventional halogen–lithium exchange conditions.
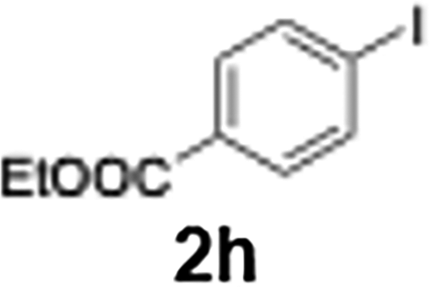
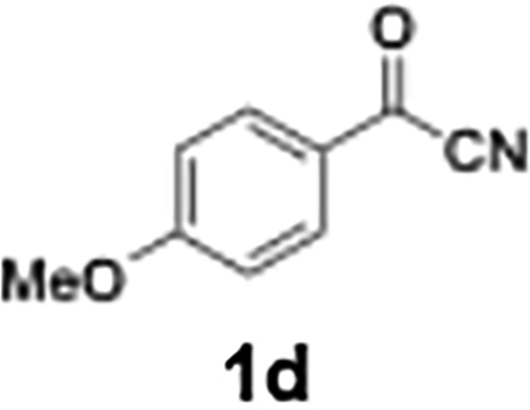
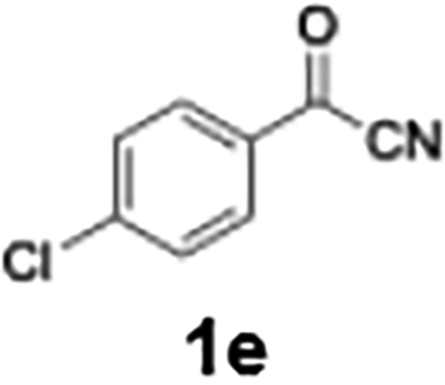
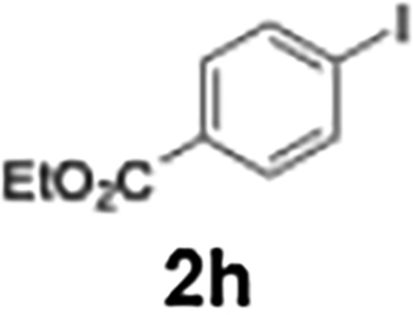
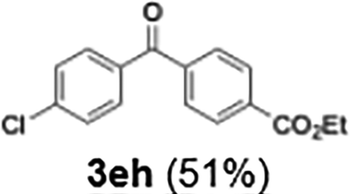
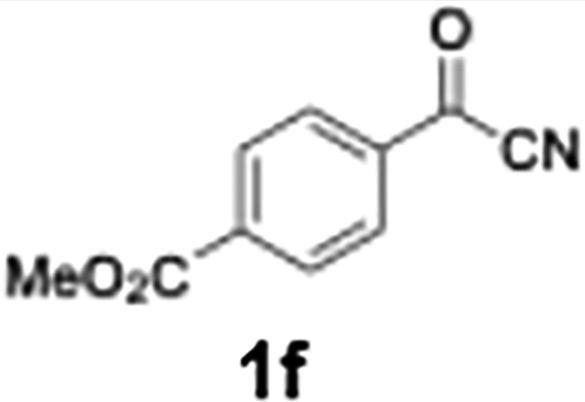
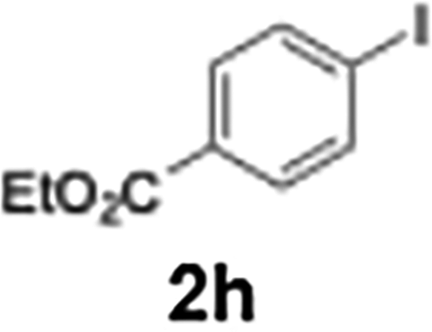
Efficient activation of these substrates could not be achieved using standard protocols. Subsequent nucleophilic addition to α-ketonitriles (1) was performed at micromixer M3 under a range of temperatures, with optimal results obtained at −78 °C, affording the desired product in excellent yields of 61–77% with high chemoselectivity. Specifically, ethyl iodobenzoates required activation with sBuLi instead of BuLi, and the exchange needed to be performed at −60 °C rather than 0 °C to ensure efficient generation of the corresponding aryllithium intermediates (Scheme 4b). Furthermore, we extended the methodology to reactions between ethyl 4-iodobenzoate (2h) and α-keto nitriles bearing electronically diverse substituents, including 4- methoxybenzoyl cyanide (1d) as an electron-donating group and 4-chlorobenzoyl cyanide (1e) as an electron-withdrawing group. In both cases, the corresponding products were obtained in good yields without the use of any catalyst, demonstrating the generality and robustness of the catalyst-free flow protocol across electronically distinct substrates.
In summary, we have developed a highly efficient and chemoselective method for the synthesis of polyfunctional ketones through the nucleophilic addition of aryllithium reagents to acyl cyanides under continuous flow conditions. By utilizing a flow microreactor system without the need for any external catalysts, we achieved significant improvements in reaction yield and selectivity compared to conventional batch processes. This study demonstrates the utility of flow microreactor technology as a powerful tool for controlling highly reactive organolithium intermediates, enabling catalyst-free access to polyfunctional ketones with high efficiency and selectivity, and offers a valuable approach for future applications in complex molecule synthesis.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc07035j.Acknowledgements
This work was supported by MEXT KAKENHI number JP24H01051 (Digi-TOS), and JSPS KAKENHI numbers, JP23K04744, JP20KK0121, and JP21H01936. This work was also partially supported by AMED PJ24126027 (GAP-FREE), PJ22230021 (BINDS), NEDO PJ22240036, JKA Foundation, and Takahashi Industrial and Economic Foundation.References
- Modern Organocopper Chemistry, ed. N. Krause, Wiley, 2002 Search PubMed.
- N. J. Lawrence, J. Chem. Soc., Perkin 1, 1998, 1739–1750 RSC.
- R. K. Dieter, Tetrahedron, 1999, 55, 4177–4236 CrossRef CAS.
- G. Cahiez, Tetrahedron Lett., 1981, 22, 1239–1242 CrossRef CAS.
- G. Cahiez and B. Laboue, Tetrahedron Lett., 1989, 30, 3545–3546 CrossRef CAS.
- G. Cahiez and B. Laboue, Tetrahedron Lett., 1989, 30, 7369–7372 CrossRef CAS.
- G. Cahiez and B. Laboue, Tetrahedron Lett., 1992, 33, 4439–4442 CrossRef CAS.
- G. Cahiez, P.-Y. Chavant and E. Metais, Tetrahedron Lett., 1992, 33, 5245–5248 CrossRef CAS.
- P. Knochel, N. Millot, A. L. Rodriguez and C. E. Tucker, Organic Reactions, Wiley, 2001, pp. 417–759 Search PubMed.
- M. J. Rozema, A. Sidduri and P. Knochel, J. Org. Chem., 1992, 57, 1956–1958 CrossRef CAS.
- P. Knochel, M. C. P. Yeh, S. C. Berk and J. Talbert, J. Org. Chem., 1988, 53, 2390–2392 CrossRef CAS.
- D. Wang and Z. Zhang, Org. Lett., 2003, 5, 4645–4648 CrossRef CAS PubMed.
- E. Negishi, V. Bagheri, S. Chatterjee, F.-T. Luo, J. A. Miller and A. T. Stoll, Tetrahedron Lett., 1983, 24, 5181–5184 CrossRef CAS.
- T. N. Majid and P. Knochel, Tetrahedron Lett., 1990, 31, 4413–4416 CrossRef CAS.
- H. Fillon, C. Gosmini and J. Périchon, Tetrahedron, 2003, 59, 8199–8202 CrossRef CAS.
- I. Kazmierski, C. Gosmini, J.-M. Paris and J. Périchon, Tetrahedron Lett., 2003, 44, 6417–6420 CrossRef CAS.
- H. Fillon, C. Gosmini and J. Périchon, J. Am. Chem. Soc., 2003, 125, 3867–3870 CrossRef CAS PubMed.
- R. M. Wehmeyer and R. D. Rieke, Tetrahedron Lett., 1988, 29, 4513–4516 CrossRef CAS.
- R. D. Rieke, Science, 1989, 246, 1260–1264 CrossRef CAS PubMed.
- A. Guijarro, D. M. Rosenberg and R. D. Rieke, J. Am. Chem. Soc., 1999, 121, 4155–4167 CrossRef CAS.
- A. E. Jensen, W. Dohle, I. Sapountzis, D. M. Lindsay, V. A. Vu and P. Knochel, Synthesis, 2002, 565–569 CrossRef CAS.
- P. Knochel, W. Dohle, N. Gommermann, F. F. Kneisel, F. Kopp, T. Korn, I. Sapountzis and V. A. Vu, Angew. Chem., Int. Ed., 2003, 42, 4302–4320 CrossRef CAS PubMed.
- H. Liu, Y.-X. Chai, J.-J. Yang, D. Miao, M.-Y. Wang, T. Jiang, Y.-Y. Sun, X. Lu, Q. Sun, J.-H. Li and Y.-P. Zhu, Org. Lett., 2024, 26(2), 477–482 CrossRef CAS PubMed.
- W. Dohle, F. Kopp, G. Cahiez and P. Knochel, Synlett, 2001, 1901–1904 CrossRef CAS.
- C. K. Reddy and P. Knochel, Angew. Chem., Int. Ed. Engl., 1996, 35, 1700–1701 CrossRef CAS.
- A. Fürstner, D. De Souza, L. Parra-Rapado and J. T. Jensen, Angew. Chem., Int. Ed., 2003, 42, 5358–5360 CrossRef PubMed.
- A. Fürstner and M. Méndez, Angew. Chem., Int. Ed., 2003, 42, 5355–5357 CrossRef PubMed.
- A. Fürstner and A. Leitner, Angew. Chem., Int. Ed., 2002, 41, 609–612 CrossRef.
- A. Fürstner, A. Leitner, M. Méndez and H. Krause, J. Am. Chem. Soc., 2002, 124, 13856–13863 CrossRef PubMed.
- G. Cahiez and H. Avedissian, Synthesis, 1998, 1199–1205 CrossRef CAS.
- G. Cahiez and S. Marquais, Tetrahedron Lett., 1996, 37, 1773–1776 CrossRef CAS.
- A. J. Birch, Pure Appl. Chem., 1996, 68, 553–556 CrossRef CAS.
- R. S. Smith and J. K. Kochi, J. Org. Chem., 1976, 41, 502–509 CrossRef CAS.
- M. Tamura and J. Kochi, J. Organomet. Chem., 1971, 31, 289–309 CrossRef CAS.
- M. Tamura and J. Kochi, Synthesis, 1971, 303–305 CrossRef CAS.
- J. K. Kochi and M. Tamura, J. Am. Chem. Soc., 1971, 93, 1485–1487 CrossRef CAS.
- V. Fiandanese, G. Marchese and L. Ronzini, Tetrahedron Lett., 1983, 24, 3677–3680 CrossRef CAS.
- V. Fiandanese, G. Marchese, V. Martina and L. Ronzini, Tetrahedron Lett., 1984, 25, 4805–4808 CrossRef CAS.
- C. Cardellicchio, V. Fiandanese, G. Marchese and L. Ronzini, Tetrahedron Lett., 1987, 28, 2053–2056 CrossRef CAS.
- S. Hünig and R. Schaller, Angew. Chem., Int. Ed. Engl., 1982, 21, 36–49 CrossRef.
- A. J. Bellamy and L. Maclean, J. Chem. Soc., Perkin 1, 1979, 204 RSC.
- J. Thesing, D. Witzel and A. Brehm, Angew. Chem., 1956, 68, 425–435 CrossRef CAS.
- C. Duplais, F. Bures, I. Sapountzis, T. J. Korn, G. Cahiez and P. Knochel, Angew. Chem., Int. Ed., 2004, 43, 2968–2970 CrossRef CAS PubMed.
- J. Yoshida, H. Kim and A. Nagaki, ChemSusChem, 2011, 4, 331–340 CrossRef CAS PubMed.
- D. Webb and T. F. Jamison, Chem. Sci., 2010, 1, 675 RSC.
- S. Marre and K. F. Jensen, Chem. Soc. Rev., 2010, 39, 1183 RSC.
- W.-Y. Lin, Y. Wang, S. Wang and H.-R. Tseng, Nano Today, 2009, 4, 470–481 CrossRef CAS PubMed.
- P. Watts and C. Wiles, Chem. Commun., 2007, 443–467 RSC.
- B. Ahmed-Omer, J. C. Brandt and T. Wirth, Org. Biomol. Chem., 2007, 5, 733–740 RSC.
- B. P. Mason, K. E. Price, J. L. Steinbacher, A. R. Bogdan and D. T. McQuade, Chem. Rev., 2007, 107, 2300–2318 CrossRef CAS PubMed.
- M. Brivio, W. Verboom and D. N. Reinhoudt, Lab Chip, 2006, 6, 329 RSC.
- J. Kobayashi, Y. Mori and S. Kobayashi, Chem. – Asian J., 2006, 1, 22–35 CrossRef CAS PubMed.
- A. J. deMello, Nature, 2006, 442, 394–402 CrossRef CAS PubMed.
- K. Geyer, J. D. C. Codée and P. H. Seeberger, Chem. – Eur. J., 2006, 12, 8434–8442 CrossRef CAS PubMed.
- P. Watts and S. J. Haswell, Chem. Soc. Rev., 2005, 34, 235 RSC.
- P. Natho and R. Luisi, Tetrahedron, Green Chem., 2023, 2, 100015 Search PubMed.
- T. Zhao, L. Micouin and R. Piccardi, Helv. Chim. Acta, 2019, 102, DOI:10.1002/hlca.201900172.
- F. Lima, M. Meisenbach, B. Schenkel and J. Sedelmeier, Org. Biomol. Chem., 2021, 19, 2420–2424 RSC.
- L. Brassart, M. Manneveau, T. Poisson, J. Legros, P. Huez, M. Forcato, P. Jubault and L. Chausset-Boissarie, Adv. Synth. Catal., 2025, 367(10), e20241438 CrossRef.
- P. Natho, M. Colella, M. Andresini, L. Degennaro and R. Luisi, Org. Lett., 2024, 26, 3032–3036 CrossRef CAS PubMed.
- P. He, P. Watts, F. Marken and S. J. Haswell, Angew. Chem., Int. Ed., 2006, 45, 4146–4149 CrossRef CAS PubMed.
- K. Tanaka, S. Motomatsu, K. Koyama, S. Tanaka and K. Fukase, Org. Lett., 2007, 9, 299–302 CrossRef CAS PubMed.
- H. R. Sahoo, J. G. Kralj and K. F. Jensen, Angew. Chem., Int. Ed., 2007, 46, 5704–5708 CrossRef CAS PubMed.
- C. H. Hornung, M. R. Mackley, I. R. Baxendale and S. V. Ley, Org. Process Res. Dev., 2007, 11, 399–405 CrossRef CAS.
- M. S. Qenawy, K. Okamoto and A. Nagaki, Chem. – Eur. J., 2025, 31, e202500299 CrossRef CAS PubMed.
- C. Wiles, P. Watts, S. J. Haswell and E. Pombo-Villar, Chem. Commun., 2002, 1034–1035 RSC.
| This journal is © The Royal Society of Chemistry 2026 |