
An amorphous MOF conversion strategy for membrane fabrication with a unique surface architecture
Zilun
Guo
a,
Hiroto
Maruta
a and
Shunsuke
Tanaka
 *bcd
*bcd
aGraduate School of Science and Engineering, Kansai University, 3-3-35 Yamate-cho, Suita-shi, Osaka 564-8680, Japan
bDepartment of Chemical, Energy, and Environmental Engineering, Faculty of Environmental and Urban Engineering, Kansai University, 3-3-35 Yamate-cho, Suita-shi, Osaka 564-8680, Japan. E-mail: shun_tnk@kansai-u.ac.jp
cOrganization for Research and Development of Innovative Science and Technology (ORDIST), Kansai University, 3-3-35 Yamate-cho, Suita-shi, Osaka 564-8680, Japan
dCarbon Neutrality Research Center (CNRC), Kansai University, 3-3-35 Yamate-cho, Suita-shi, Osaka 564-8680, Japan
First published on 24th February 2026
Abstract
We demonstrate an innovative amorphous-to-crystalline MOF transformation strategy that enables the fabrication of separation membranes with a distinctive surface architecture and domain structure not observed in conventional MOF membranes. This work highlights the utility of amorphous MOFs as versatile precursors and provides a powerful platform for designing advanced MOF-based separation membranes.
In recent years, pressure-driven gas separation membranes have attracted considerable attention as an energy-efficient technology for mitigating global warming. Metal–organic frameworks (MOFs),1–4 composed of metal ions coordinated with organic ligands, have emerged as promising membrane materials owing to their exceptionally high specific surface areas compared with conventional porous materials such as zeolites.5–7 Among various MOFs, zeolitic imidazolate frameworks (ZIF)—featuring zeolite-like structures formed by the coordination of metal ions (typically Zn or Co) with imidazole-based ligands—have shown particular promise for gas separation. ZIF-8 (Zn(2-methylimidazole)2) has received significant attention due to its high chemical and thermal stability, facile aqueous synthesis, and tuneable effective pore size (4.0–4.2 Å), which results from ligand rotation.9–11
In MOF membrane synthesis, it is essential to balance two conflicting requirements: reducing membrane thickness to achieve high gas permeability and minimizing grain boundaries and intercrystalline defects to ensure gas selectivity with molecular sieving capability. This challenge has motivated extensive research, leading to methodologies for fabricating defect-free MOF membranes with well-intergrown crystals.12–18 Several synthesis approaches, including direct synthesis and seeded secondary growth, have been reported.19–24 However, these conventional strategies face inherent limitations: (1) difficulty in achieving uniform, defect-free membranes due to uncontrolled nucleation, (2) formation of grain boundaries that compromise sieving performance, and (3) limited scalability. Overcoming these issues requires a fundamentally different synthesis pathway.
Here, we propose a method to fabricate MOF membranes through amorphous-to-crystalline conversion. Recent studies have increasingly explored amorphization as a membrane synthesis strategy.25–28 For example, Zhou et al. reported ball-milling-induced amorphization of ZIF-8 into a melt-quenched glass with preserved local coordination,25 and Frentzel-Beyme et al. showed that pressure-induced amorphization generates dense glass phases with modified mechanical properties.26 Wang et al. used controlled partial amorphization of ZIF-8 membranes to heal grain-boundary defects, improving H2/CO2 selectivity,27 whereas Su et al. demonstrated improved processability of ZIF-67 through amorphization followed by recrystallization.28 Although these studies revealed advantages of amorphous MOF phases—such as defect healing and enhanced formability—they require energy-intensive crystallization followed by controlled amorphization (ball milling, thermal treatment, or high pressure), increasing process complexity and energy consumption. Inspired by these findings, but adopting a fundamentally different strategy, we exploit the intrinsically amorphous Zn–2-methylimidazole (Hmim) coordination network (amp-Zn–Hmim) that spontaneously forms during precursor mixing, prior to crystallization in conventional ZIF-8 synthesis. Rather than using the traditional crystallization–amorphization–recrystallization sequence, we intercept the synthesis at the amorphous stage and apply alcohol vapor treatment to directly convert the uniform amorphous membrane into continuous crystalline ZIF-8. This vapor-induced crystallization offers three key advantages: (1) elimination of the energy-intensive crystallization–amorphization cycle, (2) simplification to a single coating step, and (3) suppression of grain boundaries through homogeneous crystallization from a uniform amorphous phase rather than heterogeneous nucleation. Specifically, we demonstrate that the amp-Zn–Hmim network, prepared via simple mixing in alcohol and deposited as a thin membrane, can be converted into well-intergrown ZIF-8 through controlled alcohol vapor exposure (Scheme 1). This results in continuous membrane formation without the intercrystalline defects commonly encountered in solvothermal synthesis.
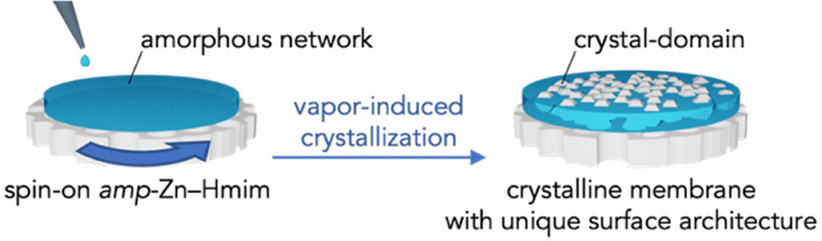 | ||
| Scheme 1 Schematic illustration of the amorphous-to-crystalline transformation strategy and a model representation of the structure after crystallization. | ||
To prepare the amp-Zn–Hmim precursor, we evaluated how the choice of metal salt affects the ability to obtain a uniform and homogeneous transparent solution. Using the stoichiometric ratio for ZIF-8 (Hmim/Zn = 2), zinc nitrate hexahydrate induced immediate precipitation, whereas zinc acetate dihydrate generated a transparent solution in 1-butanol (Fig. S1). This contrasts with conventional ZIF-8 synthesis, which uses a large amount of solvent (solvent/Zn ≥ 500),28,29 often leading to particle formation and turbidity. We therefore selected the clear Zn–Hmim solution prepared with zinc acetate for membrane fabrication.
Fig. 1 shows the XRD patterns of membranes prepared by coating amp-Zn–Hmim solution onto AAO supports followed by 1-butanol vapor treatment. The as-deposited amp-Zn–Hmim membrane showed a broad diffraction pattern, suggesting an amorphous structure. Pair distribution function (PDF) analysis is a powerful tool for investigating the structural characteristics of amorphous materials. Regarding the evaluation of structural order, PDF analysis revealed that both amp-Zn–Hmim and ZIF-8 exhibit distinct Zn–N (2.0 Å) correlations, indicating that they share a common short-range order. In contrast, no significant correlations are observed beyond ∼6 Å in amp-Zn–Hmim, suggesting the absence of medium- or long-range order and confirming that the precursor is genuinely amorphous.
 | ||
| Fig. 1 (a) XRD patterns of amp-Zn-Hmim membranes before and after exposure to 1-butanol vapor for 2, 4 and 12 h. (b) X-ray pair diffraction function spectra. Comparison of G(r) profiles for ZIF-8 and amp-Zn–Hmim. (c) Schematic illustration of preferentially oriented (222) planes with normals aligned along the membrane thickness direction. | ||
Fig. S2 shows the TGA curve for the amp-Zn–Hmim solution. Pure Hmim exhibits complete evaporation near its boiling point (approximately 270 °C). Zinc acetate dihydrate undergoes dehydration and decomposes around 300 °C, ultimately converting to ZnO. In contrast, the amp-Zn–Hmim shows a weight loss exceeding 50% by 400 °C due to the removal of residual 1-butanol and acetic acid. However, owing to the strength of the Zn–mim coordination bond (where mim denotes deprotonated Hmim), it exhibits higher thermal stability (approximately 400 °C) than either individual component. To further elucidate the chemical state of the amorphous precursor, FTIR analysis was performed (Fig. S3). An absorption band observed near 420 cm−1 can be assigned to Zn–N stretching vibrations, indicating coordination between Zn2+ and deprotonated mim species. In addition, the distinct ring vibration band around 1580 cm−1 can be clearly identified, suggesting the formation of a coordination environment structurally related to ZIF-8. However, the presence of Zn–O related bands and COO− stretching vibrations derived from zinc acetate indicates that residual acetate species and Zn–O coordination sites remain in the amorphous network. These results suggest that the amorphous precursor is not a simple Zn–Hmim assembly but rather a partially coordinated Zn–(mim/Hmim) network containing residual acetate species.
With increasing 1-butanol vapor exposure time, diffraction peaks characteristic of the ZIF-8 structure appeared and increased in intensity, indicating progressing crystallization. Comparative treatments with methanol and ethanol showed negligible or limited crystallization, whereas 1-butanol effectively induced the amorphous-to-crystalline transformation (Fig. S4 and S5). Notably, the (222) peak was more pronounced than the (110) peak observed in simulated ZIF-8.30–33 This qualitative observation suggests preferential orientation of the (222) planes (Fig. 1). The (222) plane exists parallel to the hexagonal rings defining the ZIF-8 pore openings, suggesting that the pore-forming planes are oriented parallel to the membrane surface. The dried amp-Zn–Hmim sample showed negligible nitrogen adsorption, indicating its non-porous nature. In contrast, the crystallized product obtained from amp-Zn–Hmim exhibited a Type I adsorption isotherm (Fig. S6), confirming the formation of a microporous structure. The BET area and micropore volume reached 1700 m2 g−1 and 0.74 cm3 g−1, respectively, demonstrating high crystallinity and well-developed porosity.
Fig. 2 and Fig. S7 show the surface and cross-sectional FESEM images of membranes before and after vapor-induced crystallization. The AAO support exhibited uniform cylindrical pores, and after coating with amp-Zn–Hmim solution, the surface became smooth and featureless. The cross-sectional image confirms the formation of a continuous amorphous layer tightly adhered to the substrate without delamination and cracking.8 After 2 h of 1-butanol vapor treatment, the surface transformed significantly, forming radially patterned domains. Cross-sectional analysis shows that the membrane thickness is maintained, indicating in situ structural rearrangement without material loss. Crystal-domain mapping revealed tight intergrowth without large voids. This domain architecture with a radial pattern is markedly different from the typical polyhedral crystalline membranes obtained in conventional membrane preparation.8–11 The distinctive morphology arises from vapor-induced amorphous-to-crystalline transformation, where nucleation initiates at discrete sites within the homogeneous amorphous phase and, due to limited local precursor availability, propagates predominantly in the plane of the substrate. Because crystallization proceeds under spatial confinement within the amp-Zn–Hmim network, the supply of coordinatively active species becomes locally depleted as crystal growth advances (Fig. S8). As each domain expands radially, neighbouring crystals interlock to generate a continuous, highly entangled membrane structure. The membrane thickness can in principle be tuned by adjusting the spin-coating conditions and precursor concentration. A systematic investigation of the gas separation performance as a function of membrane thickness is currently under investigation and will be reported in future work. Overall, vapor-induced crystallization within the amp-Zn–Hmim network enables controlled morphological evolution of the MOF layer while effectively suppressing intracrystalline void formation.
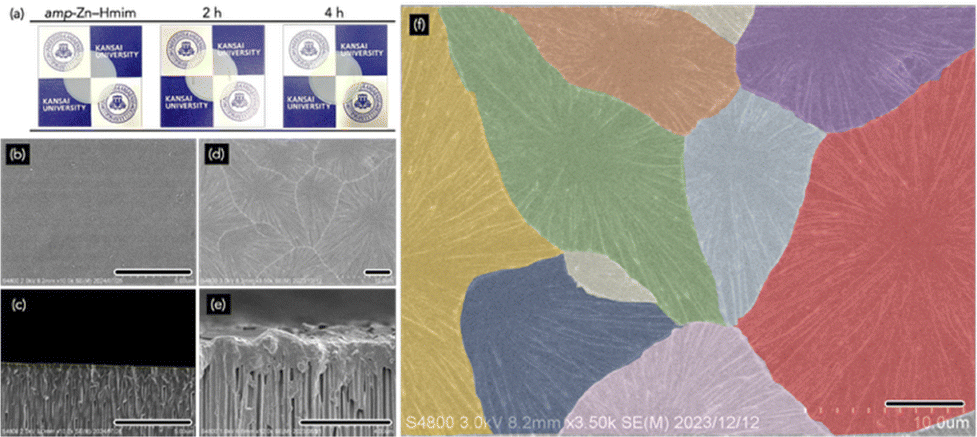 | ||
| Fig. 2 (a) Photographs of the amp-Zn–Hmim-coated AAO substrate and the membranes after 1-butanol vapor treatment for 2 and 4 h. (b)–(f) FESM images of the amp-Zn–Hmim-coated AAO substrate (b) and (c) and the membrane after 2 h of 1-butanol vapor treatment (d)–(f): surface views (b), (d) and (f) and cross-sectional views (c) and (e). (g) Enlarged view of (e) with color-coded domains. Scale bar: 5 µm. | ||
Fig. 3 shows the correlation between membrane structure and gas transport properties. The as-coated amp-Zn–Hmim (0 h) exhibited non-selective gas permeability, consistent with the N2 adsorption results (Fig. S6), confirming its negligible porosity (N2 uptake < 20 cm3 g−1 STP; BET area < 50 m2 g−1). The BET area difference of less than one-twentieth indicates that amorphization collapses the pore network despite retention of the local coordination structure. Thus, the amp-Zn–Hmim membrane behaves as a sealed barrier rather than a molecular sieve. Upon vapor-induced crystallization (2 and 4 h), porosity and permselectivity were dramatically changed. The permeabilities for small gases (H2, CO2, N2, CH4) increased, whereas C3H6 permeability remained nearly unchanged and C3H8 permeability decreased. This behavior reflects the formation of micropores, demonstrating molecular sieving,34–38 and the influence of gas solubility of the amorphous polymer, amp-Zn–Hmim. The elevated C3H8 permeance of the amp-Zn–Hmim membrane (0 h) relative to the crystallized microporous membranes (2 and 4 h) is presumably due to the contribution of gas solubility in the amp-Zn–Hmim. The newly formed ZIF-8 pores via vapor-induced crystallization (3.4–4.2 Å) enable the kinetic exclusion of C3H8, whose dynamic diameter is approximately 4.3 Å.37,38
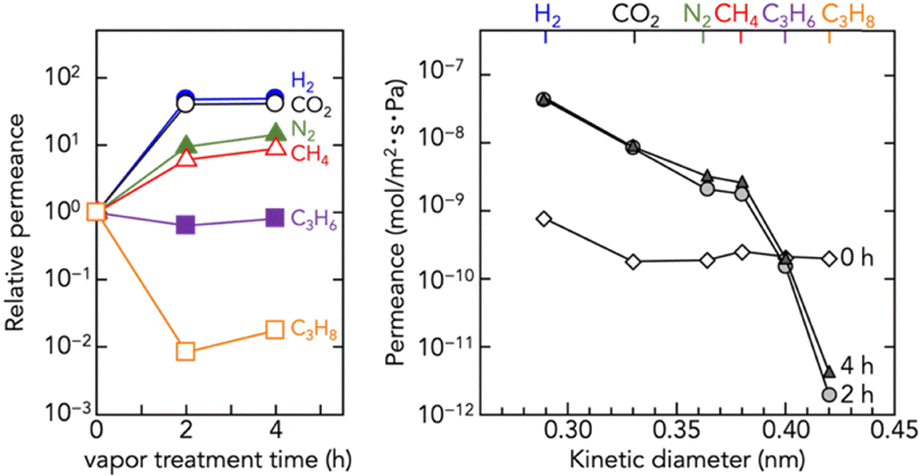 | ||
| Fig. 3 Single gas permeance of the amp-Zn–Hmim membrane (0 h) and the membranes after 1-butanol vapor treatment for 2 and 4 h. | ||
The amorphous-to-crystalline transformation proceeds through structural rearrangement facilitated by 1-butanol vapor. Although the amorphous network retains local Zn–Hmim coordination, it lacks long-range order (Fig. 1). Exposure to 1-butanol vapor introduces molecular mobility and hydrogen-bonding interactions, enabling reorganization into the sodalite framework of ZIF-8. Crystalline ZIF-8 exhibits intrinsic framework flexibility regardless of the crystallization route. Although its pore aperture is 3.4 Å, –Zn–mim–N– torsional motion enables reversible linker rotation, leading to gate-opening and transient expansion of the aperture to approximately 4.0–4.2 Å. This dynamic behaviour allows C3H6 (4.0 Å) to permeate, while excluding C3H8 (4.3 Å) is rate-limited. This provides an intrinsic molecular recognition mechanism explaining the observed C3H6/C3H8 selectivity. The amp-Zn–Hmim membrane exhibited negligible gas selectivity, with H2/CH4, C3H6/C3H8 and H2/C3H8 selectivities of 2.83, 1.02 and 4.69, respectively. On the other hand, the membrane treated with 1-butanol vapor for 2 h exhibited H2/CH4, C3H6/C3H8 and H2/C3H8 selectivities of 24.6, 78.1 and 22![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 814, respectively—substantially exceeding the Knudsen diffusion values of 2.83, 1.02 and 4.69. Literature benchmarking (Fig. S9) indicates that the obtained C3H6/C3H8 selectivity falls within the upper range of reported ZIF-8 membranes (30–100, with the highest value of 300) although the permeance is markedly lower than that of conventional membranes. Notably, the H2/C3H8 selectivity of 22814 exceeds most reported ZIF-8 membranes (100–5000), ZIF-67 membranes (30–60), and even state-of-the-art zeolite, carbon, and polymer membranes. Such exceptional performance is attributable to the uniform and morphology-controlled ZIF-8 layers produced by the amorphous-to-crystalline transformation. In contrast, conventional ZIF-8 membranes frequently contain non-selective defects that deteriorate separation performance, whereas the current approach effectively suppresses defect formation, thereby enhancing molecular sieving selectivity.32,39,40 The exceptionally high selectivity combined with low permeance can be attributed to the highly dense nature of the membrane, as shown in Fig. 4, where the presence of stacked defects and amorphous barrier layers within the structure likely restricts gas transport. The relatively low propylene permeation flux may also be related to structural imperfections formed during the vapor-induced crystallization process. As discussed above, crystallization proceeds under limited local precursor availability, and radially growing domains intergrow and compete at their boundaries. Such intergrowth interfaces and reconstructed regions may introduce junction defects or non-ideal stacking arrangements, which could increase mass transport resistance for propylene molecules.
814, respectively—substantially exceeding the Knudsen diffusion values of 2.83, 1.02 and 4.69. Literature benchmarking (Fig. S9) indicates that the obtained C3H6/C3H8 selectivity falls within the upper range of reported ZIF-8 membranes (30–100, with the highest value of 300) although the permeance is markedly lower than that of conventional membranes. Notably, the H2/C3H8 selectivity of 22814 exceeds most reported ZIF-8 membranes (100–5000), ZIF-67 membranes (30–60), and even state-of-the-art zeolite, carbon, and polymer membranes. Such exceptional performance is attributable to the uniform and morphology-controlled ZIF-8 layers produced by the amorphous-to-crystalline transformation. In contrast, conventional ZIF-8 membranes frequently contain non-selective defects that deteriorate separation performance, whereas the current approach effectively suppresses defect formation, thereby enhancing molecular sieving selectivity.32,39,40 The exceptionally high selectivity combined with low permeance can be attributed to the highly dense nature of the membrane, as shown in Fig. 4, where the presence of stacked defects and amorphous barrier layers within the structure likely restricts gas transport. The relatively low propylene permeation flux may also be related to structural imperfections formed during the vapor-induced crystallization process. As discussed above, crystallization proceeds under limited local precursor availability, and radially growing domains intergrow and compete at their boundaries. Such intergrowth interfaces and reconstructed regions may introduce junction defects or non-ideal stacking arrangements, which could increase mass transport resistance for propylene molecules.
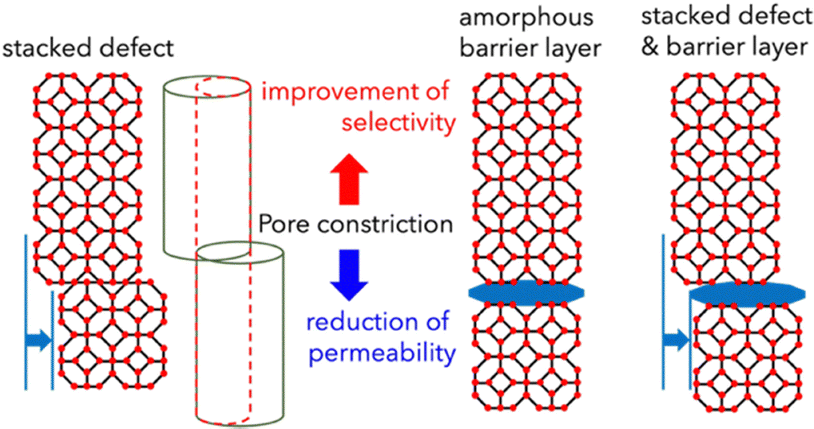 | ||
| Fig. 4 Schematic illustration of pore-connectivity defects within the crystals, which enhance selectivity while reducing permeance. | ||
In conclusion, we first fabricated an amorphous Zn–Hmim membrane and subsequently converted it into crystalline ZIF-8 membranes through 1-butanol vapor-induced crystallization. This process generated a distinct domain-architecture-based morphology, in contrast to hydrothermal or solution-based routes that typically produce polycrystalline ZIF-8 membranes. This strategy enables facile fabrication of MOF membranes with uniform molecular sieving properties, while allowing membrane thickness and crystallinity to be readily tuned. Moreover, extending this approach to other amorphous metal–ligand networks may provide precise control over MOF morphology and functionality across applications such as gas separation, adsorption, and catalysis.
Z. G.: investigation, validation, data curation, formal analysis, and writing – original draft; H. M.: investigation, validation, data curation, and formal analysis; S. T.: conceptualization, methodology, formal analysis, data curation, funding acquisition, project administration, supervision, writing – original draft, and writing – review and editing.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included in the supplementary information (SI). The supplementary information includes additional experimental details, characterization data, and supplementary figures supporting the results discussed in the main text. See DOI: https://doi.org/10.1039/d5cc07010d.Acknowledgements
This study was financially supported by JSPS KAKENHI, Grant Number JP24K21706, the Japan Science and Technology Agency (JST) SPRING, Grant number JPMJSP2150, the Reiwa Environment Foundation, 2024, the Kansai University Fund for Supporting Outlay Research Centers, 2021 and 2024, JKA and its Promotion Funds from KEIRIN RACE, Grant No. 2023M-412, and the FY2023 Research Grant Program of the Carbon Recycling Fund Institute, Japan. Z. G., H. M. and S. T. are grateful for access to the BL02B2 of the Spring-8 synchrotron radiation facility with the approval of the Japan Synchrotron Radiation Research Institute (JASRI; proposal numbers 2023B1570 and 2023B2019) and for the assistance provided by Dr Yuki Mori and Dr Shogo Kawaguchi for the X-ray pair distribution function measurements.References
- M. Kondo, T. Yoshitomi, H. Matsuzaka, S. Kitagawa and K. Seki, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725–1757 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O’Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- O. M. Yaghi, M. O’Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- K. S. Park, Z. Ni, A. P. Côté, J. Y. Choi, R. Huang, F. J. Uribe-Romo, H. K. Chae, M. O'Keeffe and O. M. Yaghi, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 10186–10191 CrossRef CAS PubMed.
- R. Banerjee, A. Phan, B. Wang, C. Knobler, H. Furukawa, M. O’Keeffe and O. M. Yaghi, Science, 2008, 319, 939–943 CrossRef CAS PubMed.
- A. Phan, C. J. Doonan, F. J. Uribe-Romo, C. B. Knobler, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2010, 43, 58–67 CrossRef CAS PubMed.
- K. Kida, M. Okita, K. Fujita, S. Tanaka and Y. Miyake, CrystEngComm, 2013, 15, 1794–1801 RSC.
- D. Fairen-Jimenez, S. A. Moggach, M. T. Wharmby, P. A. Wright, S. Parsons and T. Düren, J. Am. Chem. Soc., 2011, 133, 8900–8902 CrossRef CAS PubMed.
- H. Tanaka, S. Ohsaki, S. Hiraide, D. Yamamoto, S. Watanabe and M. T. Miyahara, J. Phys. Chem. C, 2014, 118, 8445–8454 CrossRef CAS.
- C. Zhang and W. J. Koros, J. Phys. Chem. Lett., 2015, 6, 3841–3849 CrossRef CAS PubMed.
- V. V. Guerrero, Y. Yoo, M. C. McCarthya and H.-K. Jeong, J. Mater. Chem., 2010, 20, 3938–3943 RSC.
- Y. X. Hu, X. L. Dong, J. P. Nan, W. Q. Jin, X. M. Ren, N. P. Xu and Y. M. Lee, Chem. Commun., 2011, 47, 737–739 RSC.
- N. Y. Wang, A. Mundstock, Y. Liu, A. S. Huang and J. Caro, Chem. Eng. Sci., 2015, 124, 27–36 CrossRef CAS.
- Z. Li, P. P. Yang, S. C. Yan, Q. R. Fang, M. Xue and S. L. Qiu, ACS Appl. Mater. Interfaces, 2019, 11, 15748–15755 CrossRef CAS PubMed.
- K. Kida, K. Fujita, T. Shimada, S. Tanaka and Y. Miyake, Dolton Trans., 2013, 42, 11128–11135 RSC.
- S. Tanaka, T. Shimada, K. Fujita, Y. Miyake, K. Kida, K. Yogo, J. F. M. Denayer, M. Sugita and T. Takewaki, J. Membr. Sci., 2014, 472, 29–38 CrossRef CAS.
- S. Tanaka, K. Okubo, K. Kida, M. Sugita and T. Takewaki, J. Membr. Sci., 2017, 544, 306–311 CrossRef CAS.
- H. Guo, G. Zhu, I. J. Hewitt and S. Qiu, J. Am. Chem. Soc., 2009, 131, 1646–1647 Search PubMed.
- J. Gascon, S. Aguado and F. Kapteijn, Microporous Mesoporous Mater., 2008, 113, 132–138 CrossRef CAS.
- A. Huang, H. Bux, F. Steinbach and J. Caro, Angew. Chem., Int. Ed., 2010, 49, 4958–4961 Search PubMed.
- H. Bux, F. Liang, Y. Li, J. Cravillon, M. Wiebcke and J. Caro, J. Am. Chem. Soc., 2009, 131, 16000–16001 Search PubMed.
- Y. Liu, G. Zeng, Y. Pan and Z. Lai, J. Membr. Sci., 2011, 379, 46–51 Search PubMed.
- M. C. McCarthy, V. Varela-Guerrero, G. V. Barnett and H.-K. Jeong, Langmuir, 2010, 26, 14636–14641 Search PubMed.
- C. Zhou, L. Longley and A. Krajnc, et al. , Nat. Commun., 2018, 9, 5042 CrossRef PubMed.
- L. Frentzel-Beyme, M. Kloß, P. Kolodzeiski, R. Pallach and S. Henke, J. Am. Chem. Soc., 2019, 141, 12362–12371 Search PubMed.
- Y. Wang, H. Jin, Q. Ma, K. Mo, H. Mao, A. Feldhoff, X. Cao, Y. Li, F. Pan and Z. Jiang, Angew. Chem., Int. Ed., 2020, 59, 4365–4369 CrossRef CAS PubMed.
- P. Su, H. Tang, M. Jia, Y. Lin and W. Li, AIChE J., 2022, 68, 17576 CrossRef.
- D. N. Ta, H. K. D. Nguyen, B. X. Trinh, Q. T. N. Le, H. N. Ta and H. T. Nguyen, Canadian J. Chem. Eng., 2018, 96, 1441–1644 CrossRef.
- K. Y. Cho, H. An, X. H. Do, K. Choi, H. G. Yoon, H.-K. Jeong, J. S. Lee and K.-Y. Baek, J. Mater. Chem. A, 2018, 6, 18912–18919 Search PubMed.
- S. Pang, Z. Si, G. Li, H. Wu, Y. Cui, C. Zhang, C. Ren, S. Yang, S. Pang and P. Qin, J. Membr. Sci., 2022, 611, 120920 CrossRef.
- J. B. James and Y. S. Lin, J. Membr. Sci., 2017, 532, 9–19 CrossRef CAS.
- W. Hu, L. Liu, J. Yan, Y. Gao, T. Ji, K. Yu, S. Meng, M. Wu, X. Fan, W. Dong and Y. Liu, J. Membr. Sci., 2024, 707, 123025 Search PubMed.
- S. Qiu, M. Xue and G. Zhu, Chem. Soc. Rev., 2014, 43, 6116–6140 Search PubMed.
- X. Ma, B. Lin, J. Kniep and Y. Y. Lin, Ind. Eng. Chem. Res., 2013, 52(11), 4297–4305 CrossRef CAS.
- K. Li, D. H. Olson, J. Seidel, T. J. Emge, H. Gong, H. Zeng and J. Li, J. Am. Chem. Soc., 2009, 131(30), 10368–10369 CrossRef CAS PubMed.
- H. Jin and Y. Li, Chem. Eng., 2018, 20, 107–113 Search PubMed.
- L. Lang, F. Banihashemi, J. B. James, J. Miao and Jerry Y. S. Lin, J. Membr. Sci., 2021, 619, 118743 Search PubMed.
- J. Wang, Y. Wang, Y. Liu, H. Wu, M. Zhao, Y. Ren, Y. Pu, W. Li, S. Wang, S. Song, X. Liang, G. He, Y. Han and Z. Jiang, Adv. Funct. Mater., 2022, 32, 2208064 Search PubMed.
- L. Liu, T. Ji, W. Hu, Y. Sun, Y. He, J. Yan, G. He and Y. Liu, J. Membr. Sci., 2023, 669, 121300 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |