
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceProtonated bisphosphines: a new class of powerful hydride donors capable of CO2 reduction
Danijela
Barić
a,
Mario
Damjanović
a,
Zoran
Glasovac
b,
Ines
Despotović
 a and
Borislav
Kovačević
*a
a and
Borislav
Kovačević
*a
aRuđer Bošković Institute, Division of Physical Chemistry, 10000, Zagreb, Croatia. E-mail: Borislav.Kovacevic@irb.hr
bRuđer Bošković Institute, Division of Organic Chemistry and Biochemistry, 10000, Zagreb, Croatia
First published on 10th February 2026
Abstract
DFT calculations reveal that proton-sponge-like monoprotonated bisphosphines are exceptionally strong hydride donors, in some cases surpassing Super-Hydride. This work establishes a new design principle for metal-free hydride donors and highlights their potential for CO2 reduction.
Hydride transfer reactions are essential to a variety of chemical transformations, ranging from fundamental redox chemistry to large-scale industrial synthesis and enzyme catalysis.1–4 Conventional hydride donors, such as borohydrides and transition-metal hydride complexes, are highly effective but have several well-recognized limitations. In particular, reagents like NaBH4 and LiBH4 exhibit high reducing power but limited control over chemoselectivity. Their use typically requires anhydrous conditions and results in stoichiometric inorganic residues, posing challenges for sustainable synthesis and large-scale processing.3–5 A further limitation arises with metal-hydride catalysts, many of which rely on precious or rare transition metals (Ir, Rh, Ru, Pd) that are expensive and difficult to recover or recycle. Main-group alternatives such as ammonia-borane avoid the use of transition metals but still generate element-containing by-products, can be thermally unstable, and are difficult to regenerate.
Designing new organic, metal-free hydride donors can address some of these challenges by eliminating the use of critical metals, reducing environmental impact, and enabling milder, more chemoselective reductions. Well-known examples of metal-free organic donors include Hantzsch esters, NADH (and its synthetic dihydropyridine mimetics), and several recently investigated guanidine-based derivatives.6–8 These reagents mitigate some of the drawbacks mentioned above but possess their own limitations. Specifically, although several examples of benzimidazole-based strong organic hydrides with hydricities as low as ∼33 kcal mol−1 have been reported, they often display limited hydride-donating ability for challenging reductions and may be insufficient to reduce highly inert substrates. In addition, organic hydrides frequently suffer from kinetic limitations, as high activation barriers for hydride transfer result in sluggish reaction rates. Several of these NADH mimetics have recently been employed as homogeneous catalysts for the electro- and photochemical reduction of CO2 to formate and other reduced carbon species.9,10 Such studies not only demonstrate their catalytic versatility but also highlight the importance of hydride-donor strength as a key thermodynamic parameter governing reactivity.
The strength of a hydride donor determines which reactions it can promote, so hydricity has become an important parameter for comparing different hydride sources. Among the various transformations governed by hydride transfer, the reduction of CO2 has emerged as a particularly informative benchmark, as it requires a donor with well-defined and quantifiable hydricity. For CO2 reduction to formate, the hydride donor must be sufficiently strong to transfer H− to CO2; in thermodynamic terms, its hydricity should be more favorable than that of the formate anion, which is approximately 44 kcal mol−1 in acetonitrile.2
Hydricity, 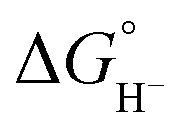 , (hydride donor strength), is defined thermodynamically as the Gibbs energy change for the reaction:
, (hydride donor strength), is defined thermodynamically as the Gibbs energy change for the reaction:
| A − H → A+ + H− |
Because hydride release is generally endergonic in the absence of a hydride acceptor, 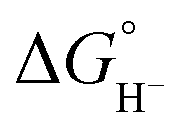 values are positive; thus, lower hydricity values correspond to stronger hydride donors with a greater tendency to transfer H− to an acceptor. According to the classification proposed by Glusac et al.,10 formate anion lies near the boundary between “strong” and “moderate” hydride donors, while the transition from “moderate” to “weak” donors occurs around 80 kcal mol−1 in acetonitrile.
values are positive; thus, lower hydricity values correspond to stronger hydride donors with a greater tendency to transfer H− to an acceptor. According to the classification proposed by Glusac et al.,10 formate anion lies near the boundary between “strong” and “moderate” hydride donors, while the transition from “moderate” to “weak” donors occurs around 80 kcal mol−1 in acetonitrile.
In recent years, neutral phosphines bearing strongly electron-donating substituents (whose protonated forms, 1b–1f, are shown in Fig. 1) have been studied as efficient Brønsted bases and as ligands for transition-metal complexes.11–14 Some synthesized examples (e.g., 1e and 1f) have set new records for basicity, even surpassing the classical Schwesinger bases.14 Their extraordinary basicity arises from stabilization of the conjugate phosphonium cation by the electron-donating substituents. Although the pKBH+ values of the cited protonated phosphines have been measured experimentally and characterized computationally, to the best of our knowledge, the potential for protonated phosphines to act as hydride donors has never been investigated. However, when a protonated phosphine releases a hydride, the resulting phosphorus-centered dication can be stabilized by electron donation from strong donor substituents into the vacant p orbital, making the protonated phosphine a potentially powerful hydride donor. Further, charge analysis of protonated phosphines 1a–1f indicates that the H atom bound to the central phosphorus carries a non-negligible fraction of negative charge (Table S1), opposite to the behavior of protonated nitrogen bases. Driven by these insights, we set out to probe phosphines bearing powerful electron-donating groups as hydride donors, and to test their capacity to transfer H− under mild, metal-free conditions. We began by computing the hydricities of phosphines 1a–1f (depicted in Fig. 1) in acetonitrile.
 | ||
| Fig. 1 Structures of protonated phosphines (1), bisphosphines (2) and monophosphines (3) examined in this study. | ||
Glusac9 and Lee8 showed that the ωB97X-D functional accurately reproduces experimental hydricities; thus, it is employed here (see SI for details). Results are presented in Table 1. The computed hydricities range from 71.8 to 95.3 kcal mol−1, placing these compounds among moderate-to-weak hydride donors. As with basicity, the strongest hydride donors are the phosphines bearing the most strongly electron-donating substituents (1e and 1f). As anticipated, the dication formed by hydride loss from the protonated phosphine is resonance-stabilized, which was confirmed by Natural Resonance Theory (NRT) analysis for compound 1f. The analysis shows that the three most significant resonance structures, each contributing equally (8.6%; 25.8% in total), feature a P–N bond with partial double-bond character between the central phosphorus atom and each of the three adjacent nitrogen atoms (Fig. S1).
| Phos |
|
Bis |
|
|
Mono |
|
|
|---|---|---|---|---|---|---|---|
| 1a | 95.3 | 2a | 41.5 | 53.8 | 3a | 103.8 | 62.3 |
| 1b | 84.4 | 2b | 37.5 | 46.9 | 3b | 96.1 | 58.6 |
| 1c | 77.2 | 2c | 26.5 | 50.7 | 3c | 93.2 | 66.7 |
| 1d | 84.3 | 2d | 38.9 | 45.4 | 3d | 98.4 | 59.5 |
| 1e | 78.3 | 2e | 24.6 | 53.7 | 3e | 94.2 | 69.6 |
| 1f | 71.8 | 2f | 17.4 | 54.4 | 3f | 93.2 | 75.8 |
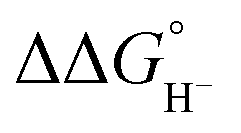
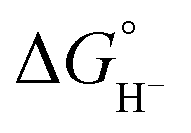
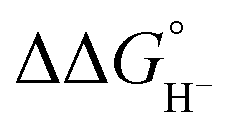
Although the present calculations show that protonated phosphines can behave as hydride donors, their hydricities remain far too high to be useful for CO2 reduction. This raises the question of whether the hydride-donating properties of phosphines can be further enhanced. To increase pKBH+, one strategy is to place two basic moieties in close proximity to each other. The first compound to demonstrate this effect was the so-called “proton sponge”, 1,8-bis(dimethylamino) naphthalene (DMAN), synthesized by Alder in 1965.15 The strategy of enhancing basicity by bringing two basic sites into proximity has since been widely applied in the design of strong organic bases, mostly nitrogen-based systems.16–22 Proton-sponge analogues based on alkyl- and arylphosphines have been synthesized and investigated in the context of Brønsted basicity and the strength of intramolecular P–H⋯P interactions.23–25 In this regard, both neutral and monoprotonated forms of bisphosphines have been experimentally prepared. However, it remains an open question whether incorporating two phosphine moieties into a proton-sponge-like framework influences the hydricity of their protonated forms. To the best of our knowledge, the hydricity of bisphosphine systems has never been systematically investigated. However, a one study by Stephan and co-workers demonstrated that a bisphosphine dication based on 1,8-bis(diphenylphosphino)naphthalene, in combination with tri-tert-butylphosphine (tBu3P), can heterolytically cleave H2, with the bisphosphine dication accepting a hydride to form the monoprotonated 1,8-bis(diphenylphosphino)naphthalene, while the proton is transferred to tBu3P.26 This experimental observation unequivocally demonstrates that the bisphosphine dication is capable of hydride acceptance, yielding the monoprotonated species–effectively the reverse of hydride donation reaction by monoprotonated bisphosphines studied here. To quantify the effect of incorporating of two phosphine moieties into a proton-sponge-like framework, we calculated the hydricities of bisphosphines 2a–2f (Fig. 1). The results are presented in Table 1. Compared with phosphines 1a–1f, the bisphosphines show a pronounced reduction in 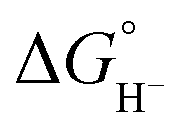 by 45.4–54.4 kcal mol−1, corresponding to a marked enhancement of hydride-donor ability. Surprisingly, compounds 2e and 2f appear to be even stronger hydride donors than Super-hydride [Et3BH]−, which has an experimentally determined
by 45.4–54.4 kcal mol−1, corresponding to a marked enhancement of hydride-donor ability. Surprisingly, compounds 2e and 2f appear to be even stronger hydride donors than Super-hydride [Et3BH]−, which has an experimentally determined 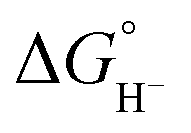 value of 26 kcal mol−1 in MeCN.2 This large decrease in
value of 26 kcal mol−1 in MeCN.2 This large decrease in  can be rationalized by thermodynamic stabilization of the resulting phosphorus-centered dication via formation of a donor–acceptor P–P interaction (dative bond) within the dication (Fig. 2a). The P–P distance in dications 2a–2f (Table S2) corresponds to the P–P single bond distance (2.21 Å) in diphosphines.27 To estimate the contribution of inter-phosphine interactions to the hydricity of bisphosphines 2a–2f, we computed the hydricities of the corresponding monophosphines 3a–3f (Fig. 1). As expected, their
can be rationalized by thermodynamic stabilization of the resulting phosphorus-centered dication via formation of a donor–acceptor P–P interaction (dative bond) within the dication (Fig. 2a). The P–P distance in dications 2a–2f (Table S2) corresponds to the P–P single bond distance (2.21 Å) in diphosphines.27 To estimate the contribution of inter-phosphine interactions to the hydricity of bisphosphines 2a–2f, we computed the hydricities of the corresponding monophosphines 3a–3f (Fig. 1). As expected, their  values are higher than those of 2a–2f (Table 1). The hydricity differences between series 3 and 2 range from 59.5 to 75.8 kcal mol−1, indicating strong interunit interactions. Table 1 shows that hydricity increases with the electron-donating strength of substituent R but exhibits a saturation trend, suggesting an upper limit to bisphosphine hydricity. Similar behavior and upper bounds have been reported for Brønsted basicity.28 The difference in hydricity between systems 2a–2f and 3a–3f is influenced by interactions between the phosphine moieties in both the protonated form and the dication. To further assess the impact of P–P bond formation on the stability of the dication, we also evaluated this interaction using a homodesmotic reaction (Fig. S3), with the results summarized in Table S3. The monophosphines 3a–3f also exhibit weaker hydride-donor ability than the phosphines 1a–1f, consistent with replacing one strongly electron-donating R group with a naphthalene unit, a weaker electron donor.
values are higher than those of 2a–2f (Table 1). The hydricity differences between series 3 and 2 range from 59.5 to 75.8 kcal mol−1, indicating strong interunit interactions. Table 1 shows that hydricity increases with the electron-donating strength of substituent R but exhibits a saturation trend, suggesting an upper limit to bisphosphine hydricity. Similar behavior and upper bounds have been reported for Brønsted basicity.28 The difference in hydricity between systems 2a–2f and 3a–3f is influenced by interactions between the phosphine moieties in both the protonated form and the dication. To further assess the impact of P–P bond formation on the stability of the dication, we also evaluated this interaction using a homodesmotic reaction (Fig. S3), with the results summarized in Table S3. The monophosphines 3a–3f also exhibit weaker hydride-donor ability than the phosphines 1a–1f, consistent with replacing one strongly electron-donating R group with a naphthalene unit, a weaker electron donor.
 | ||
| Fig. 2 (a) Formation of dative P–P bond in dication upon hydride donation from protonated bisphosphines. (b) Representation of in–in and in–out proton sponge. | ||
Inspection of the geometries of protonated bisphosphines 2a–2f revealed a noteworthy structural feature. Namely, nearly all so far designed and synthesized proton sponge analogues adopt so-called in–in substituent orientation (Fig. 2b), where the lone pairs on the basic nitrogen atoms are oriented inward while the substituents point outward. The only reported case in which a proton sponge adopts an in–out orientation in protonated form, despite the absence of 2,7-substitution on the naphthalene core that could sterically favor such a conformation,29,30 involves a combination of diisopropylamino cyclopropenimine and dimethylamino substituents at 1,8 positions of naphthalene in the so-called “Janus proton sponge”.21
Among the six studied bisphosphines (2a–2f), compounds 2a and 2d exhibited an in–out conformation. In such in–out proton sponges, the proton binds to the electron pair oriented outwards. A possible explanation for why 2a and 2d adopt the in–in conformation, whereas 2b, 2c, 2e, and 2f adopt the in–out conformation, is provided in the SI, together with a comparison of the relative stabilities of these conformers across all bisphosphine species (Table S4) and an NBO analysis of both conformations (Table S5).
From the obtained hydricity data, it appears that all studied bisphosphines have thermodynamic hydricities below 44 kcal mol−1, indicating they are capable of reducing CO2. The next important aspect is the kinetics of hydride transfer from protonated bisphosphines 2a–2f towards CO2. Applying the same computational model used for hydricity calculations, we explored the potential-energy surface for this process.
Dielmann et al. demonstrated that electron-rich phosphines (e.g., 1c) can bind CO2 to form air-stable adducts,31 (see Fig. S4 and related discussion in the SI) which represent a typical example of CO2 capture.32 Motivated by these findings, and by the possibility of achieving combined CO2 capture and conversion,33 we investigated the ability of the neutral phosphine moiety in 2a–2f to bind CO2. The Gibbs energy of binding is positive in all cases except 2e, which shows a marginally favorable value of −0.1 kcal mol−1 (Table S6), indicating that the studied protonated bisphosphines do not bind CO2. The fact that none of the bisphosphines 2[a–f] bind CO2 likely arises from the replacement of one strongly electron-donating group with a naphthalene unit, which renders the phosphine center less electron-rich. This interpretation is consistent with the findings of Dielmann and co-workers, who showed that only phosphines bearing three strongly electron-donating substituents bind CO2.31 Further, we found that, with the exception of 2a and 2d, the hydride-transfer reaction to CO2 proceeds in two steps, as depicted in Fig. 3. In the first step, a relatively high-energy intermediate (Int) in which the two phosphorus atoms are bonded is formed via transition state TS1. In the second step, a hydride is transferred to CO2via transition state TS2, yielding the bisphosphine dication and the formate anion (Products). The energies for these processes are reported in Table 2. Transition state TS1 corresponds to P–P bond formation, and the activation energies are relatively low, not exceeding ∼20 kcal mol−1. By contrast, the hydride-transfer barriers (except for 2a and 2d) are considerably high, mostly around or above 30 kcal mol−1, indicating slow kinetics. Interestingly, for bisphosphines 2a and 2d, no intermediate is formed; hydride transfer proceeds in a single step with relatively low barriers of 21.8 and 24.3 kcal mol−1, respectively. These values are comparable to the lowest hydride-transfer values reported for recently studied guanidine analogues8 and some benzimidazole-based hydrides.34 We note that 2a and 2d are the only protonated bisphosphines exhibiting an in–out orientation of the phosphine groups. It is evident that this phosphine-group orientation favorably affects the kinetics of hydride transfer. One likely explanation is steric in nature. In intermediate Int, the phosphine groups come into closer proximity to each other than in the starting structure (see above). Consequently, CO2 must insert between them to enable hydride transfer (with CO2 approaching from above relative to the bisphosphine), thereby increasing steric repulsion (see Fig. S5a). In 2a and 2d, CO2 approaches the bisphosphine laterally instead, avoiding this steric clash (Fig. S5b). Further, in both compounds, given the in–out orientation of the phosphine groups, a more favorable interaction is enabled between the lone pair on one phosphorus atom and the P–H antibonding orbital on the protonated phosphine, thereby enhancing the hydride character of the H atom on the protonated phosphine group. It should be noted that the reduction of CO2 by the protonated bisphosphines examined in this work is not regarded as a catalytic process but rather as a stoichiometric reaction. Whether the bisphosphine dications can be regenerated, either by reaction with H2 or electrochemically, and thus act as catalysts for CO2 reduction, remains to be explored in future studies. However, for the catalytic process to proceed, the phosphine dication formed upon hydride transfer should not be highly electrophilic and should not react favorably with the formate anion. Therefore, we calculated the Gibbs free energies of the dication–formate reactions, and the results are presented in Table S7. As can be seen, the reaction of the phosphine dication with formate is endergonic for all studied bisphosphine species.
 | ||
| Fig. 3 Reaction pathway for hydride transfer. | ||
| Bisphosphine | TS1 | Int | TS2 | Products |
|---|---|---|---|---|
| 2a | — | — | 21.8 | −3.0 |
| 2b | 14.9 | 13.2 | 36.7 | −6.5 |
| 2c | 14.9 | 11.4 | 37.7 | −17.5 |
| 2d | — | — | 24.3 | −5.1 |
| 2e | 19.9 | 13.6 | 29.4 | −13.3 |
| 2f | 17.4 | 10.3 | 32.8 | −26.4 |
In conclusion, our calculations show that proton-sponge-like protonated bisphosphines with strongly electron-donating substituents are exceptionally powerful hydride donors, in some cases surpassing Super-Hydride. Their kinetic properties depend on phosphine orientation, with the in-out form exhibiting relatively low barriers (21.8–24.3 kcal mol−1). These findings open new avenues for designing strong metal-free hydride donors.
Conflicts of interest
There are no conflicts to declare.Data availability
All data supporting this article are included in the main text and the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc06952a.Acknowledgements
The authors acknowledge funding by the Croatian Science Foundation grant No. IP-2024-05-7730 (Bis-phosphines as Metal-Free Catalysts for Small Molecules' Activation – Design and Synthesis). This research was conducted using the Advanced Computing Service provided by the University of Zagreb Computing Centre – SRCE (the Supek supercomputer).References
- R. A. Marcus and N. Sutin, Biochim. Biophys. Acta, 1985, 811, 265 CrossRef CAS.
- E. S. Wiedner, M. B. Chambers, C. L. Pitman, R. M. Bullock, A. J. M. Miller and A. M. Appel, Chem. Rev., 2016, 116, 8655 CrossRef CAS PubMed.
- U. Wietelmann, M. Felderhoff and P. Rittmeyer, Hydrides, Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH, Weinheim, 2016 Search PubMed.
- S. Hammes-Schiffer and J. Watney, Philos. Trans. R. Soc., B, 2006, 361, 1365 CrossRef CAS PubMed.
- D. Wang and D. Astruc, Chem. Rev., 2015, 115, 6621 CrossRef CAS PubMed.
- C. Zheng and S.-L. You, Chem. Soc. Rev., 2012, 41, 2498 RSC.
- R. J. Mayer and J. Moran, Org. Biomol. Chem., 2023, 21, 85 RSC.
- J. Chen, H. Yu, D. Tan and R. Lee, Chem. Commun., 2023, 59, 5201 RSC.
- S. Ilic, J. L. Gesiorski, R. B. Weerasooriya and K. D. Glusac, Acc. Chem. Res., 2022, 55, 844 CrossRef CAS PubMed.
- S. Ilic, A. Alherz, C. B. Musgrave and K. D. Glusac, Chem. Soc. Rev., 2018, 47, 2809 RSC.
- P. Mehlmann, C. Mück-Lichtenfeld, T. T. Y. Tan and F. Dielmann, Chem. – Eur. J., 2017, 23, 5929 CrossRef CAS PubMed.
- F. Buß, M. B. Röthel, J. A. Werra, P. Rotering, L. F. B. Wilm, C. G. Daniliuc, P. Löwe and F. Dielmann, Chem. – Eur. J., 2021, 28, e202104021 CrossRef.
- P. Rotering, L. F. B. Wilm, J. A. Werra and F. Dielmann, Chem. – Eur. J., 2019, 26, 406 CrossRef PubMed.
- S. Ullrich, B. Kovačević, X. Xie and J. Sundermeyer, Angew. Chem., Int. Ed., 2019, 58, 10335 CrossRef CAS PubMed.
- R. W. Alder, P. S. Bowman, W. R. S. Steele and D. R. Winterman, Chem. Commun., 1968, 723 RSC.
- V. Raab, J. Kipke, R. M. Gschwind and J. Sundermeyer, Chem. – Eur. J., 2002, 8, 1682 CrossRef CAS PubMed.
- J. F. Kögel, B. Oelkers, B. Kovačević and J. Sundermeyer, J. Am. Chem. Soc., 2013, 135, 17768 CrossRef PubMed.
- V. Raab, E. Gauchenova, A. Merkoulov, K. Harms, J. Sundermeyer, B. Kovačević and Z. B. Maksić, J. Am. Chem. Soc., 2005, 127, 15738 Search PubMed.
- J. F. Kögel, X. Xie, E. Baal, D. Gesevicius, B. Oelkers, B. Kovačević and J. Sundermeyer, Chem. – Eur. J., 2014, 20, 7670 CrossRef PubMed.
- L. Belding and T. Dudding, Chem. – Eur. J., 2014, 20, 1032 Search PubMed.
- L. Belding, P. Stoyanov and T. Dudding, J. Org. Chem., 2016, 81, 6 Search PubMed.
- M. Guest, R. Le Sueur, M. Pilkington and T. Dudding, Chem. – Eur. J., 2020, 26, 8608 CrossRef CAS PubMed.
- S. A. Reiter, S. D. Nogai, K. Karaghiosoff and H. Schmidbaur, J. Am. Chem. Soc., 2004, 126, 15833 CrossRef CAS PubMed.
- A. Karacar, H. Thonnessen, P. G. Jones, R. Bartsch and R. Schmutzler, Heteroat. Chem., 1997, 8, 539 CrossRef CAS.
- P. Kilian, F. R. Knight and J. D. Woollins, Chem. – Eur. J., 2011, 17, 2302 CrossRef CAS PubMed.
- M. H. Holthausen, J. M. Bayne, I. Mallov, R. Dobrovetsky and D. W. Stephan, J. Am. Chem. Soc., 2015, 137, 7298 Search PubMed.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brammer, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- I. Leito, I. A. Koppel, I. Koppel, K. Kaupmees, S. Tshepelevitsh and J. Saame, Angew. Chem., Int. Ed., 2015, 54, 9262 Search PubMed.
- A. F. Pozharskii, O. V. Ryabtsova, V. A. Ozeryanskii, A. V. Degtyarev, O. N. Kazheva, G. G. Alexandrov and O. A. Dyachenko, J. Org. Chem., 2003, 68, 10109 CrossRef CAS PubMed.
- A. F. Pozharskii, A. V. Degtyarev, O. V. Ryabtsova, V. A. Ozeryanskii, M. W. Kletskii, Z. A. Starikova, L. Sobczyk and A. Filarowski, J. Org. Chem., 2007, 72, 3006 Search PubMed.
- F. Buß, P. Mehlmann, C. Mück-Lichtenfeld, K. Bergander and F. Dielmann, J. Am. Chem. Soc., 2016, 138, 1840 CrossRef PubMed.
- B. Vujic and A. P. Lyubartsev, Chem. Eng. Sci., 2017, 174, 174 Search PubMed.
- S. Kar, A. Goeppert and G. K. Surya Prakash, Acc. Chem. Res., 2019, 52, 2892 CrossRef CAS PubMed.
- R. B. Weerasooriya, J. L. Gesiorski, A. Alherz, S. Ilic, G. N. Hargenrader, C. B. Musgrave and K. D. Glusac, J. Phys. Chem. Lett., 2021, 12, 2306 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |