
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Light metal pyrazolates excel in carbon dioxide uptake
Felix
Kracht
and
Reiner
Anwander
 *
*
Institut für Anorganische Chemie, Eberhard Karls Universität Tübingen, Auf der Morgenstelle 18, 72076 Tübingen, Germany. E-mail: reiner.anwander@uni-tuebingen.de
First published on 2nd February 2026
Abstract
The development of new carbon (CO2) capture materials has emerged as a top-priority transdisciplinary research field. Ideally, CO2 is not only captured and stored (CCS), but also transformed into more valuable organic compounds, because CO2 itself is a cheap, abundant, non-flammable gas and thus an attractive C1 building block. However, activation of this thermodynamically rather stable molecule requires high activation energies. To overcome this energy barrier, activation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O double bond is routinely achieved by exploiting a synergetic metal–ligand cooperativity. The most promising candidates from academia or industry revolve around amino-functionalized materials or components featuring metal–nitrogen bonds. Given their natural abundance, low prices and nontoxicity, environmentally friendly materials should ultimately involve light metals. Recently, we found that the cerium pyrazolate [Ce+IV(pzMe2)4]2 is able to insert CO2 exhaustively and reversibly. In general, such nitrogen-rich azolato ligands comprising pyrazolato, triazolato and tetrazolato derivatives exhibit five-membered aromatic ring systems with nucleophilic nitrogen coordination sites. Azolato ligands adopt a wide variety of coordination modes and especially light metal pyrazolates are a well-established class of compounds. Aiming at higher CO2 uptake capacities, the conceptual approach, developed for the heavy metal cerium, has been consequently adapted to the light metals magnesium, aluminium, scandium and titanium. This review gives an overview of light metal pyrazolates and their CO2 insertion behaviour as well as their catalytic activity in the cycloaddition reaction of CO2 and epoxides to cyclic carbonates. In addition, consideration is given to immobilized variants as well as exemplary complexes and metal–organic framework materials derived from nitrogen-richer azoles/azolates.
O double bond is routinely achieved by exploiting a synergetic metal–ligand cooperativity. The most promising candidates from academia or industry revolve around amino-functionalized materials or components featuring metal–nitrogen bonds. Given their natural abundance, low prices and nontoxicity, environmentally friendly materials should ultimately involve light metals. Recently, we found that the cerium pyrazolate [Ce+IV(pzMe2)4]2 is able to insert CO2 exhaustively and reversibly. In general, such nitrogen-rich azolato ligands comprising pyrazolato, triazolato and tetrazolato derivatives exhibit five-membered aromatic ring systems with nucleophilic nitrogen coordination sites. Azolato ligands adopt a wide variety of coordination modes and especially light metal pyrazolates are a well-established class of compounds. Aiming at higher CO2 uptake capacities, the conceptual approach, developed for the heavy metal cerium, has been consequently adapted to the light metals magnesium, aluminium, scandium and titanium. This review gives an overview of light metal pyrazolates and their CO2 insertion behaviour as well as their catalytic activity in the cycloaddition reaction of CO2 and epoxides to cyclic carbonates. In addition, consideration is given to immobilized variants as well as exemplary complexes and metal–organic framework materials derived from nitrogen-richer azoles/azolates.
 Felix Kracht | Felix Kracht, born in 1990, studied chemistry at the Eberhard Karls University of Tübingen, Germany, where he also finished his Master's thesis under the guidance of Reiner Anwander in 2019. During his PhD from 2019 to 2025 he investigated the carbon dioxide uptake by light metal pyrazolates, developing the concept of carboxophilicity, which provides the basis for this feature article. |
 Reiner Anwander | Reiner Anwander earned a Dr. rer. nat. degree in 1992 from the Technische Universität München (TUM), under the supervision of Wolfgang A. Herrmann. This was followed by postdoctoral research on organolanthanide chemistry with Bill Evans at the University of California, Irvine. Then, he spent three years at the Universität Stuttgart starting his habilitation on surface organometallic chemistry at nanoporous materials, which he completed in 2000 at TUM. From 2005 until 2008, he held a position (Heterogeneous Catalysis) at the University of Bergen, Norway. He joined the faculty of the Eberhard Karls Universität Tübingen, Germany, in 2009 and was a Fellow of the Japan Society for the Promotion of Science (JSPS) in 2012. His research interests include organometallic chemistry and nanostructured materials, with emphasis on sustainable chemistry and catalysis. |
Introduction
The inexorably rising carbon dioxide level in the Earth's atmosphere is mainly caused by the continuous burning of fossil fuels needed for increased energy production/consumption.1–13 Noteworthily, the associated anthropogenic climate change was originally theorized about as early as 1896 by Svante Arrhenius.14 The development of new technologies for CO2 management is one of the most pressing concerns of our times, particularly in the environmental, engineering, and chemical sectors. Carbon capture and storage (CCS) as well as direct air capture (DAC) display effective approaches in this regard.5,15–17 Aqueous amine scrubbers have been used as a sorbent for industrial CCS for almost a century, although low capacities (<15 wt% CO2) and high regeneration energies (“energy penalty”) reduce their effectiveness and sustainable use.18–20Another industrial approach exploits the precipitation of CaCO3 from aqueous Ca(OH)2 solutions, but the carbonate can be regenerated to CaO via calcination only at above 900 °C.16 The energy barrier for regeneration can be lowered when CO2 is captured with NaOH and the carbonate subsequently transferred to Ca(OH)2. This process which is called causticization still suffers from high regeneration energies and limited solubility of Ca(OH)2 in water (1 mol L−1).21
Newer technologies successfully draw on the nucleophilic characteristic of nitrogen-containing components as revealed by amine-containing ionic liquids22,23 or amine-functionalized porous high-surface materials such as silica, zeolites24,25 or metal–organic frameworks (MOFs).26–29 For example, the latter reach a record-high capacity of 35 wt% CO2 at ambient temperature for Mg2(dobdc) (dobdc = 2,5-dioxido-1,4-benzendicarboxylate) when adsorption and chemisorption are combined.26 Further to this, the related MOF Mg2(dobpdc) (dobpdc = 4,4′-dioxidobiphenyl-3,3′-dicarboxylate) currently seems to be the most effective absorbent for carbon capture from simulated flue gas.29
Generally, CCS largely depends on appropriate storage locations and the transportation of stored CO2, which makes its (on-spot) utilization/valorization highly desirable.30 As an inexpensive, “abundant” (byproduct in a lot of industrial processes), non-toxic, non-flammable gas, CO2 features an attractive C1 synthon for organic synthesis. However, because CO2 has the lowest energy level of organic compounds, its conversion necessitates a high activation energy. Therefore, coping with this energetic barrier and converting CO2 sustainably into higher-value “useful” chemicals is a top-priority research field.10,31–34
The cooperation of a nucleophilic ligand that attacks the electrophilic carbon atom of CO2 and a Lewis acidic metal centre that interacts with the nucleophilic oxygen atom (nc-MLC, non-classical metal–ligand cooperativity), can decrease the activation barrier of CO2 significantly (carboxophilicity, Fig. 1A).35 In nature calcium and magnesium carbonates (lithosphere) and bicarbonates (oceans) function as natural CO2 storage (Fig. 1B).2 Inspired by this role in nature, the natural abundancy of metal carbonates and the related industrial approach of causticization as well as the exceptional performance of amine-functionalized materials we developed the conceptional approach of pyrazolato-based carbamate complexes (Fig. 1C). By changing the functional group of a carbonate to a N-functionalized group the “energy penalty” gets reduced significantly.
 | ||
| Fig. 1 (A) Non-classical metal–ligand cooperativity (nc-MLC) involving an oxophilic metal and a nucleophilic ligand determines/impacts the affinity for CO2 and its activation, termed carboxophilicity. (B) Naturally occurring metal carbonates. (C) Conceptional approach and design strategy for reversible molecular CO2 adsorbers, resulting in carbamates for N-functional groups. | ||
Why pyrazolates?
The pyrazolato ligand (pz) as derived from deprotonation of the corresponding aromatic pyrazole (Hpz) stands out through its highly versatile coordination chemistry. This monoanionic ligand provides an equally stabilizing environment for both main group and early/late transition metals adopting a wide range of coordination modes. The free electron pairs of the adjacent nitrogen atoms can engage in terminal [κ1(N)]/[κ2(N,N′)], side-on [µ2: µ2(N,N′)] or end-on bridging [µ1: µ1(N,N′)] coordination, but also η1 to η5 π-interactions with the metal centre are feasible. In addition, the steric and electronic properties of the pyrazolato ligands can be fine-tuned by ring substitution in the 3/5 position. For example, molecular homoleptic [Ce+III(pzMe2)3]4 (1) features a cage structure with as many as six distinct pyrazolato coordination modes.36 Our interest in carbon dioxide valorization was triggered by the facile reversible insertion of benzophenone into homoleptic tetravalent [Ce+IV(pzMe2)4]2 (2).37 Along this line, we found that both tri- and tetravalent cerium pyrazolates, [Ce+III(pzMe2)3]4 (1) and [Ce+IV(pzMe2)4]2 (2), respectively, are capable of inserting CO2 reversibly into one Ce–Npz-bond of each pyrazolato ligand. Exhaustive CO2 insertion gave the carbamate complexes Ce+IV(CO2·pzMe2)4 (1-CO2) and [Ce+III(CO2·pzMe2)3]4 (2-CO2) featuring CO2 capacities of up to 25 wt% (Fig. 2).38 | ||
| Fig. 2 CO2 insertion products of homoleptic cerium pyrazolates [Ce+IV(pzMe2)4]2 (1) and [Ce+III(pzMe2)3]4 (2). | ||
These reactions were conducted with excess of CO2, but even equimolar reactions with the ceric pyrazolate, afforded isolable insertion products as revealed for [Ce+IV3(pzMe2)9(CO2·pzMe2)3(thf)] (3). Crucially, following the CO2 insertion into a classic metal-amide complex the resulting carbamato ligand coordinates routinely in a carboxylate-like κ2(O,O′) mode (Scheme 1). In contrast, metal pyrazolates insert CO2 only in one of the two κ2(N,N′) nitrogen-atoms resulting in CO2·pzR,R carbamato moieties in the κ2(N,O) coordination mode, with one nitrogen of the pyrazolato moiety still attached to the metal centre. Since the inserted CO2 coordinates with one oxygen atom only to the metal center, electron delocalization is hindered as revealed by distinct C–O single and double bonds. This allows for an easy CO2 releasing step which is why metal pyrazolates can insert CO2 reversibly. All cerium compounds in this study were also active catalysts in the cycloaddition of CO2 and epoxides to cyclic carbonates.38
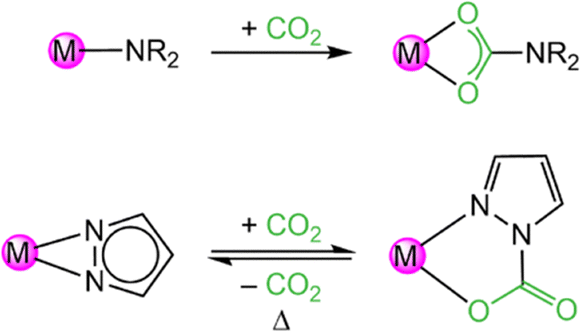 | ||
| Scheme 1 Comparison of irreversible CO2 insertion into metal amides (R = alkyl or aryl group) and reversible CO2 insertion into metal pyrazolates. | ||
Shortly thereafter, the group of Lu reported on the CO2 insertion into the iron pyrazolate [Fe(pzMe)(NO)2]2 (4). Upon initial reduction of 4 with KC8/18-crown-6 to [K(18-crown-6)]2[(NO)2Fe(µ-pzMe)2Fe(NO)2] (5) exposure to 1 bar CO2 gave the contact ion pair [K(18-crown-6)][Fe(CO2·pzMe)(NO)2] (5-CO2, see Scheme 2).39Via reductive coupling involving the ferrocenium/ferrocene couple and Ca(OTf)2, 5-CO2 could be converted into CaC2O4, reforming iron precursor 4.
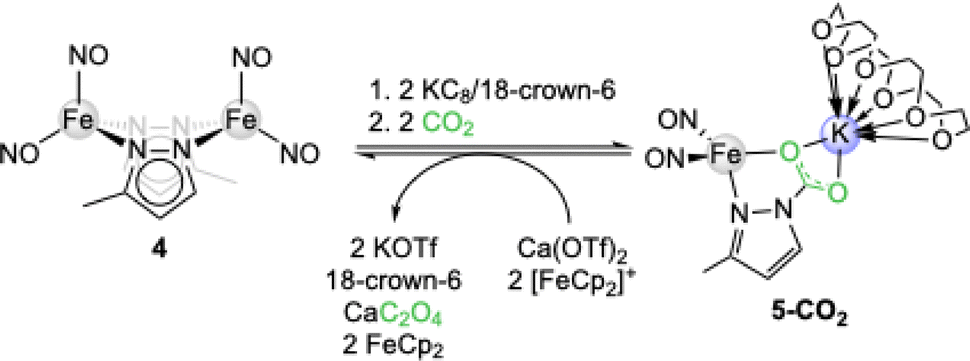 | ||
| Scheme 2 CO2 insertion and reduction to oxalate with iron pyrazolate [Fe(pzMe)(NO)2]2 (4) via the potassium iron carbamate [K(18-crown-6)][Fe(CO2·pzMe)(NO)2] (5-CO2). | ||
Later, this CO2-insertion protocol could be adapted to ceric pyrazolates with bulkier alkyl moieties at the pyrazolato ligand, comprising Ce+IV(pztBu2)4 (6), Ce+IV(pzPh2)4 (7) and Ce+IV(pztBu,Me)4 (8) respectively, although the efficiency decreased with increasing steric bulk.40 However, when grafted onto periodic mesoporous silica SBA-15500, the cerium dimethylpyrazolates kept their CO2 insertion properties, affording capacities of up to 20 wt% CO2, which, to the best of our knowledge, features the highest uptake for silica materials.41 This immobilization study produced hybrid materials denoted as [Ce+IV(pzMe2)4]2@SBA-15500 (1-SBA-15), Ce+IV(pzMe2)4(thf)@SBA-15500 (9-SBA-15), Ce+III4(pzMe2)12@SBA-15500 (2-SBA-15), and [Ce+III(pzMe2)3(thf)]2@SBA-15500 (10-SBA-15) including the lanthanum-based [La(pzMe2)3(thf)]2@SBA-15500 (11-SBA-15).
Light metal pyrazolates by nature should provide high weight percentages of inserted CO2 and hence a low “mass penalty” (light metal density: ρ < 5 g cm−3). Herein, CO2 capacity refers to the CO2 mass percentage of the inserted pyrazolates measurable by weight gain. Moreover, most light metals are abundant, inexpensive, nontoxic and thus favourably environmentally benign. In this review, we give an overview of light metal pyrazolates as carbon capture systems and point to their use in the catalytic conversion of CO2 (CCC).
Choice of light metal pyrazolates
Light metal pyrazolates are already a well-established class of compounds. Given their favourable thermal stability and volatility, early studies on light metal pyrazolates focused on their potential use as precursors for metal nitrides via chemical vapour deposition (CVD). Initial examples include Winter's monomeric titanium complex Ti+IV(pzMe2)4 (12, Fig. 3)42 and the homoleptic magnesium dimer [Mg(pztBu2)2(thf)x]2 (13, x = 0, 1; Fig. 4)43 as well as the mixed pyrazolato/pyrazole magnesium complex [Mg2(pziPr2)4(HpziPr2)2] (14) reported by Ruhlandt-Senge.44 Other examples were synthesized as potential precursors for polynuclear species like the mixed pyrazolato/pyrazole magnesium complex [Mg(pztBu2)2(HpztBu2)2] (15) described by Mösch-Zanetti.45 | ||
| Fig. 3 Examples of monomeric light metal pyrazolates. | ||
 | ||
| Fig. 4 Examples of dimeric light metal pyrazolates. | ||
Most of these studies focused on the structural chemistry re-emphasizing the feasibility of ample pyrazolato coordination modes. The structurally authenticated monomeric homoleptic complexes comprise Ti+IV(pzMe2)4 (12, Fig. 3),42 Al(pztBu2)3 (16, Fig. 3),46 Ti+III(pztBu2)3 (17, Fig. 3),47 and Sc(pztBu2)3 (18).48 Dimeric derivatives feature [Mg(pztBu2)2]2 (13)43 and adduct [Y(pzMe2)3(thf)]2 (19, Fig. 4).49 Strikingly, except for titanium complex Ti+IV(pzMe2)4 (12)42 all structurally elucidated homoleptic light metal pyrazolates bear tBu moieties on the pyrazolato ring. Further examples include alkali-metal complexes, tetrameric [Li(pztBu2)]4 (20) and the ladder-like [K(pztBu2)]n (21).50
In the case of smaller pyrazolato substituents solubility issues occur and donor stabilization is necessary to yield crystalline material. For this reason, we targeted the synthesis of several new light metal pyrazolates bearing smaller alkyl substituents on the pyrazolato ring such as iPr, Me and H (Fig. 5). The “unsubstituted” parent pyrazolates seemed particularly desirable, because of their easy accessibility and expected highest CO2 capacity (minimum mass penalty).
 | ||
| Fig. 5 Examples of recently reported light metal pyrazolates with tailor-made modified pyrazolato ligand substitution. | ||
For magnesium, a wide variety of pyrazolato ligands was probed, affording the mixed pyrazolato/pyrazole complex [Mg3(pziPr2)6(HpziPr2)2] (22), [Mg2(pziPr2)4(thf)2] (23), and [Mg(pztBu,Me)2(thf)]2 (24) containing a pyrazolato ligand with distinct alkyl substituents, the fluorinated complex [Mg2(pzCF3,CF3)4(thf)3] (25, Fig. 5), and the two amorphous compounds [Mg(pzMe2)2]n (26, Fig. 5) and [Mg(pz)2]n (27, Fig. 5).51 The highest CO2 capacity was to be expected for the latter two compounds, assuming (exhaustive) CO2 insertion in all Mg–pzR (RH, Me) bonds of these low-mass pyrazolates (0.5 M(13) = 454.99 g mol−1, M(13-CO2) = 615.11 g mol−1 ≙ 14.3wt% CO2; M(26) = 214.56 g mol−1, M(26-CO2) = 302.57 g mol−1 ≙ 29.1wt% CO2; M(27) = 158.04 g mol−1; M(27-CO2) = 246.47 g mol−1 ≙ 35.7wt% CO2).
Access to crystalline 26 and 27 was hampered because of their insolubility in all common solvents, however, their formation was confirmed by 13C CP/MAS (magic-angle spinning) NMR experiments. The fluorinated complex 25 was envisaged because of the strongly electron-withdrawing substituents which might contribute to a better understanding of the nc-MLC.
Furthermore, a series of new trivalent light metal pyrazolates was obtained, comprising dimeric [Al(pziPr2)3]2 (28, Fig. 5), monomeric Sc(pztBu2)3(thf) (29), which was synthesized over a mercury-free salt-metathesis protocol, monomeric Y(pztBu2)3(thf)2 (30), and the two amorphous [Al(pz)3]n (31, Fig. 5) and [Sc(pz)3]n (32, Fig. 5) which both contain the parent pyrazolato ligand.52 Compounds 31 and 32 were accessed via a transamination protocol from the corresponding di-tBu-pyrazolate derivatives 16 and 29, respectively. All listed light metal pyrazolates, except for the alkali metal derivatives, were tested for their capability to take up CO2 as well as their catalytic behaviour in the cycloaddition of CO2 and epoxides to cyclic carbonates.
Reversible carbon dioxide insertion into light metal pyrazolates
Magnesium pyrazolates
Reacting complex [Mg(pztBu2)2(thf)]2 (13-thf) with 1 bar CO2 in THF led to insertion of CO2 into both Mg–(pyrazolato) moieties and the formation of carbamate complex [Mg(CO2·pztBu2)2(thf)2] (13-CO2,thf) (Scheme 3).51 This equals an uptake of 14.3 wt% or 3.3 mmol CO2 per g. An overview of CO2 capacities and selected interatomic distances and angles of metal carbamates can be found in Table 1. This process is completely reversible in solution (VT NMR: 70–105 °C) and in the solid state (TGA: 134–233 °C).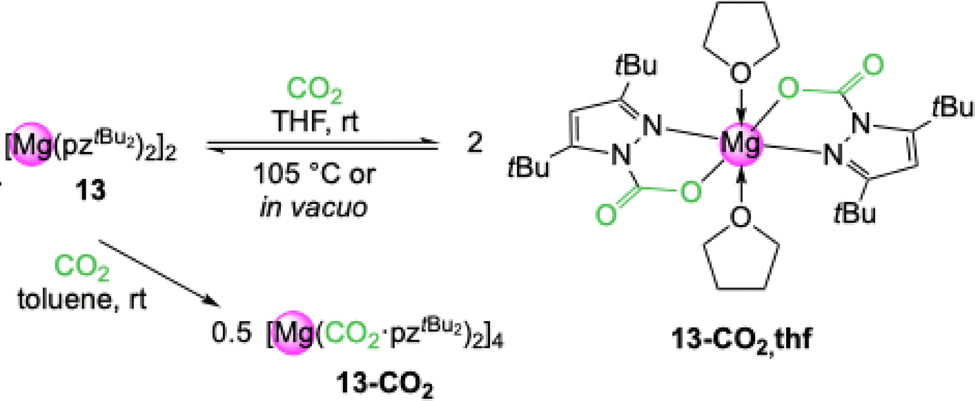 | ||
| Scheme 3 Reversible CO2 insertion into [Mg(pztBu2)2(thf)x]2 (13) to yield monomeric [Mg(CO2·pztBu2)2(thf)2] (13-CO2,thf) in THF and donor-free tetrameric [Mg(CO2·pztBu2)2]4 (13-CO2) in toluene. | ||
| Compound | Metal | R2 | C–O [Å] | CO [Å] |
O–CO [°] |
N–M–O [°] | wt% CO2a | mmol CO2 per g | Ref. |
|---|---|---|---|---|---|---|---|---|---|
| a Calculated from molecular formula. b A crystal structure could not be obtained. c Donor-free. d Connectivity only. | |||||||||
| 13-CO2,thf | Mg | tBu | 1.2621 (11) | 1.2165 (12) | 129.23 (9) | 75.33 (3) | 14.3 | 3.3 | 51 |
| 13-CO2 | Mg | tBuc | — | — | — | — | 18.7 | 4.3 | 51 |
| 23-CO2,thf | Mg | iPr | — | — | — | — | 15.7 | 3.6 | 51 |
| 33 | Mg, Li | iPr | 1.244 (9)–1.278 (9) | 1.203 (9)–1.248 (9) | 128.4 (8)–130.2 (8) | 73.3 (2)–77.0 (2) | 21.2 | 4.8 | 51 |
| 26-CO2 | Mg | Me | — | — | — | — | 29.1 | 6.6 | 51 |
| 27-CO2 | Mg | H | — | — | — | — | 35.7 | 8.1 | 51 |
| 16-CO2 | Al | tBu | 1.288 (2), 1.285 (2) | 1.199 (2), 1.203 (3) | 127.47 (19), 127.4 (2) | 81.22 (7), 80.57 (7) | 13.5 | 3.1 | 52 |
| 28-CO2 | Al | iPr | — | — | — | — | 21.5 | 4.9 | 52 |
| 12-CO2 | Ti+IV | Me | 1.275 (3) | 1.207 (3) | 129.0 (3) | 72.82 (8) | 17.0 | 3.8 | 55 |
| 17-CO2 | Ti+III | tBu | 1.276 (6)–1.290 (6) | 1.201 (6)–1.217 (6) | 126.6 (5)–128.6 (4) | 74.05 (14)–75.45 (14) | 13.1 | 3.0 | 55 |
| 29-CO2 | Sc | tBu | 1.2991 (18) | 1.2035 (19) | 126.24 (14) | 70.30 (4) | 7.0 | 1.6 | 52 |
| 1-CO2 | Ce+IV | Me | 1.276 (6)–1.289 (2) | 1.201 (3)–1.212 (3) | 126.6 (5)–128.6 (4) | 64.06 (5)–64.68 (5) | 25.3 | 5.7 | 38 |
| 2-CO2 | Ce+III | Me | 1.26 (1)–1.301 (9) | 1.21 (1)–1.235 (9) | 123.8 (8)–131.0 (8) | 59.43 (19)–63.1 (2) | 23.6 | 5.4 | 38 |
| 5-CO2 | Fe | Me, H | 1.2608 (4) | 1.2257 (4) | 130.1 (3) | 76.81 (9) | 8.1 | 1.8 | 39 |
An overview of CO2 release temperatures of the pyrazolate-based carbamate complexes can be found in Table 2. Solid 13-CO2,thf slowly releases CO2 over time, which can be expedited under reduced pressure or elevated temperature. Solution NMR studies of 13-CO2,thf revealed a stepwise de-insertion, in accordance with two distinct CO2-releasing steps in the thermogravimetric analysis (TGA). However, putative [Mg(CO2·pztBu2)(pztBu2)(thf)2] could not be isolated. In toluene, donor-free 13 showed also an immediate and complete reaction with CO2, however, no crystalline material could be isolated. A 1H DOSY NMR study revealed the exclusive formation of one distinct product with a molar mass M = 1875 g mol−1 which is close to tetrameric [Mg(CO2·pztBu2)2]4 (13-CO2, M = 1883.59 g mol−1). This would equal an uptake of 18.7 wt% CO2.
| Compound | CO2 release temperature [°C] | Ref. | |
|---|---|---|---|
| 1H NMR (start→end)a | TGA (start → end)b | ||
| a VT NMR experiment: solution of compound in THF-d8 (toluene-d8 for Ti and Ce) placed in a J. Young-valved NMR tube and stepwise heated with 20 °C per step and steps of 5–10 °C close to the CO2 release temperature (first cooled to 10 °C for 1-CO2). After each step a 1H NMR spectrum was recorded upon temperature equilibrium. Afterwards the sample was cooled applying similar temperature steps and 1H NMR spectra were recorded at each temperature stage. b TGA samples were heated in the range of ambient temperature to 1000 °C with a heating ratio of 1 K min−1 under constant argon flow. c VT IR experiments were used due to the insolubility of 27. d No complete CO2 release because of the temperature limit of the device. | |||
| Mg:13-CO2 | 70–105 | 134–233 | 51 |
| Mg:26-CO2 | — | rt–210 | 51 |
| Mg:27-CO2 | 70–240c | 50–220 | 51 |
| Al:16-CO2 | 100–120d | 114–196 | 52 |
| TiIV:12-CO2 | — | 90–150 | 55 |
| TiIII:17-CO2 | 40–70 | 77–110 | 55 |
| Y:19-CO2 | rt–70 | 60–154 | 52 |
| CeIV:1-CO2 | 10–60 | 55–95 | 38 |
| CeIII:2-CO2 | — | 52–90 | 38 |
| Sc:29-CO2 | rt | rt | 52 |
Complex 23 is also capable of inserting CO2 in THF solution to afford [Mg(CO2·pziPr2)2(thf)2] (23-CO2). Although the structure was not verified by SCXRD analysis, both the 1H and 13C NMR spectra confirmed its formation. In toluene, the reaction of 23 with CO2 gave a rather complicated 1H NMR spectrum, however, an SCXRD analysis of the few obtained crystals revealed the ate-complex [LiMg4(CO2·pziPr2)9] (33). The lithium atom most likely originated from residual lithium contained in Mg(nBu)2 as verified by a 7Li NMR experiment. The formation of 33 most likely was induced by the lithium contaminated trimer [Mg3(pziPr2)6(thf)2] (23). The exhaustive CO2 insertion revealed by complex 33 (21.3 wt% CO2 or 4.5 mmol CO2 per g) might be exploited for the targeted synthesis of light metal ate complexes and their use for CO2 uptake.
Treatment of the mixed pyrazolato/pyrazole magnesium complexes 14, 15 and 22 with 1 bar CO2 led to the formation of 13-CO2 and 23-CO2, respectively (Scheme 4). In addition, NMR signal sets were observed that can be assigned to the respective carbamic acids HO2CpzR2 (R = tBu, iPr). The pyrazole donor dissociates after CO2 insertion. For complexes 14 and 15 the produced amounts of carbamate and carbamic acid reflect the different ratios of pyrazolato and pyrazole donor in both complexes. In contrast to the metal carbamates, CO2 release was not observed for the carbamic acids. Noteworthy, the carbamic acids also occur as decomposition products, when light metal carbamates are exposed to ambient atmosphere and react with moisture.
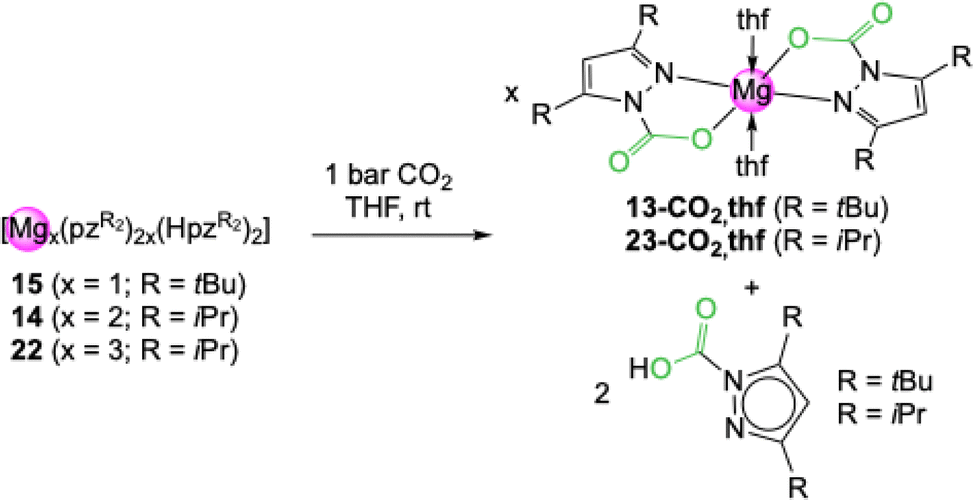 | ||
| Scheme 4 Reactivity of mixed pyrazolato/pyrazole magnesium complexes towards an excess of CO2 under the formation of the corresponding carbamates and dissociation of the corresponding carbamic acid. | ||
Due to their insolubility in common organic solvents, solid powders of [Mg(pzMe2)2]n (26) and [Mg(pz)2]n (27) were exposed to an atmosphere of 1 bar CO2 in the absence of solvent. Surprisingly, an immediate heat evolution and a mass gain in accordance with full CO2 insertion was observed. Successful gas/solid-state reactions were proven by FTIR and 13C CP/MAS NMR signatures showing the quantitative formation of [Mg(CO2·pzMe2)2]n (26-CO2) and [Mg(CO2·pz)2]n (27-CO2), respectively (Fig. 6). Both compounds exhibit exceptionally high CO2 uptakes of 29.1 wt% (measured mass gain: 29.4 wt%; TGA: 29. 5 wt%) and 35.7 wt% (measured mass gain: 36.3 wt%; TGA: 34.3 wt%), respectively. Complex 26-CO2 even became soluble in THF and solution NMR experiments confirmed its formation as well. Unfortunately, no crystalline material was obtained. Both compounds display a reversible CO2 insertion at elevated temperatures, which was confirmed by TGA and IR (see Table 2). A complete reversibility was also observed under reduced pressure as monitored by 13C CP/MAS experiments.
 | ||
| Fig. 6 Top: Solvent-free CO2 insertion into solid magnesium pyrazolates with small substituents (RH, M) at the pyrazolato ligand. Middle: 13C CP/MAS NMR spectra (75.47 MHz) of [Mg(pz)2]n (27) and [Mg(CO2·pz)2]n (27-CO2). Bottom: TGA of 27-CO2. The sample was heated from 28 °C to 1000 °C with a heating ratio of 1 K min−1 under constant argon flow. | ||
The mixed substituted complex [Mg(pztBu,Me)2(thf)]2 (24) also inserted CO2, but no crystalline material was obtained and the NMR spectra were rather complicated, ruling out the assignment of a precise structure. In contrast, fluorinated [Mg2(pzCF3,CF3)4(thf)3] (25) did not indicate any perceptible CO2 insertion in NMR experiments. It can be hypothesized that the reduced nucleophilicity of the pyrazolato ligand disfavours interaction with CO2.
Group III and XIII light metal pyrazolates
Exhaustive CO2 insertion was not detected for the aluminium complex 16 but the doubly inserted [Al(CO2·pztBu2)2(pztBu2)] (16-CO2) formed in THF (Scheme 5).52 This equals a CO2 uptake of 13.5 wt% or 3.1 mmol CO2 per g (TGA: 15.8 wt%).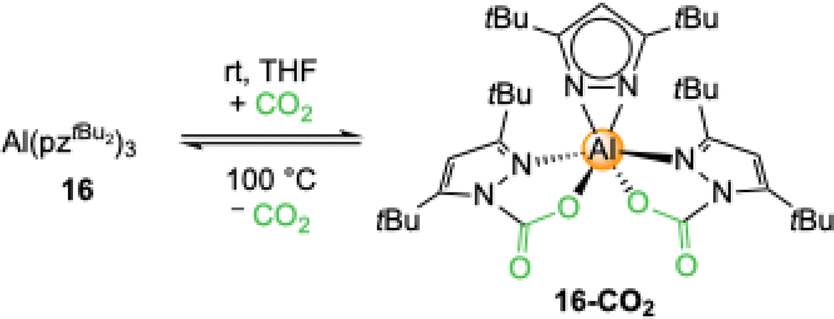 | ||
| Scheme 5 Twofold CO2 insertion into complex Al(pztBu2)3 (16) to [Al(CO2·pztBu2)2(pztBu2)] (16-CO2). | ||
A close examination of the crystal structure revealed that the third CO2 insertion might be hindered by the steric bulk of the tBu groups. The necessary tilt for the insertion of the third pyrazolato ligand is hindered by the two carbamato ligands. The insertion process of complex 16-CO2 is reversible which was proven by a 1H VT NMR study and TGA (Table 2). Striking is that the required energy for the release of CO2 is significantly higher than for the magnesium congener 13-CO2, but also that solid 16-CO2 did not show any de-insertion over time, even under reduced pressure. The less sterically demanding 28 showed complete CO2 insertion to [Al(CO2·pziPr2)3]n (28-CO2) in THF, equaling a capacity of 21.5 wt% CO2. Although no crystalline material was obtained, NMR studies unambiguously revealed full CO2 insertion by showing three distinct signal sets for the carbamato ligands.
In THF, the scandium complex 29 inserted CO2 only into one Sc–pyrazolato moiety to yield donor-stabilized [Sc(CO2·pztBu2)(pztBu2)2(thf)]n (29-CO2,thf), as detectable in the NMR spectra (Scheme 6).52 However, no crystalline material was obtained, due to immediate CO2 de-insertion at ambient temperature upon solvent removal. This is in stark contrast to the aluminium congener 16-CO2, which did not release CO2 even under reduced pressure. A 1H VT NMR study uncovered a fully reversible equilibrium between 29 and 29-CO2,thf. In toluene, two different insertion species were initially observed in the 1H NMR spectrum. Also, a crystalline material was formed and an SCXRD measurement applying permanent cooling revealed the dimer [Sc(CO2·pztBu2)(pztBu2)2]2 (29-CO2). This equals a CO2 uptake of 7.0 wt%. Complex 29-CO2 could be assigned to one of the two species found in the 1H NMR spectrum. The second species is most likely the twofold inserted [Sc(CO2·pztBu2)2(pztBu2)] (29a-CO2), which immediately releases one CO2 to convert into 29-CO2 upon opening of the NMR tube. An IR spectrum of 29-CO2 could only be obtained with an in situ experiment in an CO2 atmosphere. The overall CO2 affinity (carboxophilicity) of the scandium pyrazolate proved to be rather weak.
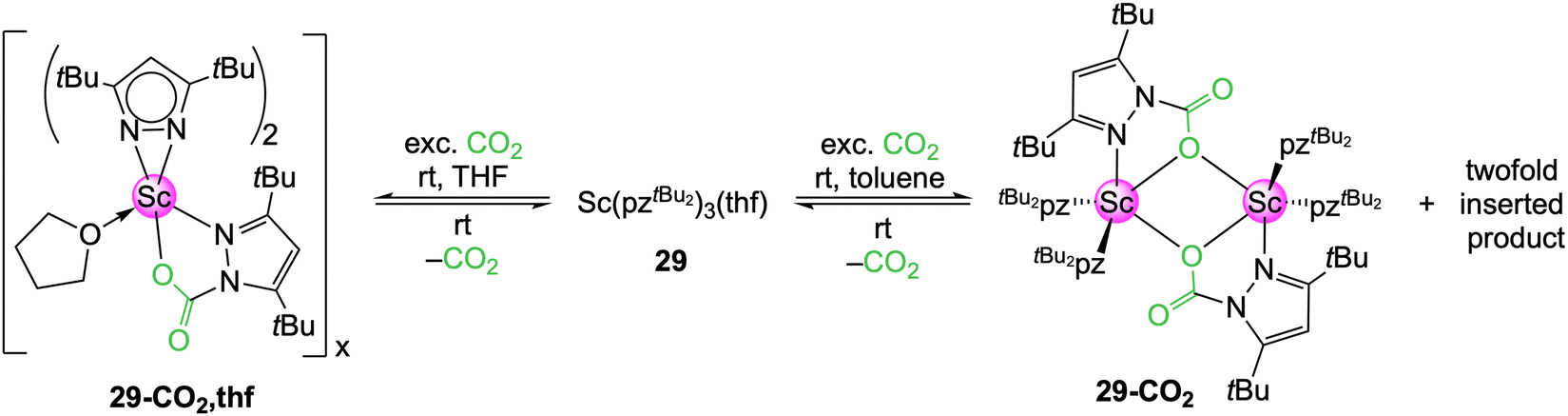 | ||
| Scheme 6 Solvent-dependent reactivity of scandium complex Sc(pztBu2)3(thf) (29) towards CO2. | ||
Even less carboxophilicity was observed for yttrium complex Y(pztBu2)3(thf)2 (30) showing no detectable CO2 insertion reminiscent of fluorinated [Mg2(pzCF3,CF3)4(thf)3] (25). In contrast, yttrium complex [Y(pzMe2)3(thf)]2 (19) bearing less bulky pyrazolato ligands engaged in CO2 insertion as detected by NMR spectroscopy. One signal set could be assigned to fully inserted [Y(CO2·pzMe2)3(thf)]n (19-CO2) while the other signals appeared too complicated to be assigned to one distinct product. A VT 1H NMR study showed the reversibility of 19-CO2 whereas the other species remained unchanged.
In contrast to compound [Mg(pz)2]n (27), the parent pyrazolate derivatives of aluminium (31) and scandium (32) displayed no extensive CO2 insertion. When exposed to a CO2 atmosphere mass gains of only 8.2 wt% (Al) and 6.9 wt% (Sc) were observed. This is far off from an exhaustive CO2 uptake which would correspond to 36.7 and 34.9 wt%, respectively. Further analytics of putative 31-CO2 and 32-CO2 like FTIR and 13C CP/MAS NMR experiments indicated only traces of CO2-inserted products. This is in accordance with either a fast CO2 release in non-CO2 atmospheres or CO2 adsorption only at the solid surface. Apparently, combining the trivalent metal centres Al3+ and Sc3+ with the less nucleophilic parent pyrazolato ligand results in a low carboxophilicity. Hence, the enhanced oxophilicity of these two metals compared to magnesium (Mg: 0.6, Al: 0.8, Sc: 0.8) seems to be overcompensated by the lower electronegativity of magnesium (Mg: 1.31, Al: 1.61, Sc: 1.36).53,54 The combination Mg2+ centre/parent pyrazolato ligand seems to achieve an optimal nc-MLC for high CO2 uptake.
Titanium pyrazolates
The tetravalent titanium complex Ti+IV(pzMe2)4 (12) displayed a twofold CO2 insertion in toluene to yield Ti+IV(CO2·pzMe2)2(pzMe2)2 (12-CO2), which was verified by NMR studies and experiments with labeled 13CO2 (Scheme 7).55 However, after one day a second signal set appeared and over time 12-CO2 was fully converted into this new compound in addition to several other new species. This “decomposition” process is reproducible at ambient temperature but was not observed when a solution of 12-CO2 was stored in the cold (0 °C). After one week at ambient temperature the original solution of 12-CO2 produced yellow crystals of the oxy-bridged dimer O[Ti+IV(CO2·pzMe2)2(pzMe2)]2 (34). Two carbamato and one pyrazolato ligands are coordinated to each titanium centre. Oxy formation in 34 can be rationalized by terms of hydrolysis and a subsequent dissociation of pyrazole/carbamic acid or deoxygenation of inserted CO2. For related deoxygenation reactions some examples exist in the literature.56,57 A light-induced reaction could be excluded in the case of 34 by experiments with amber glass. The urea derivative OC(pzMe2)2 could be detected by mass spectrometry as a possible deoxygenation side product. After two months orange crystals were obtained from the original 12-CO2 (ambient temperature) solution. SCXRD analysis revealed the formation of trinuclear [Ti+IV(µ-O)2(µ-pzMe2)4(Ti+IV{CO2·pzMe2}{pzMe2})2] (35, Scheme 7) featuring a prolonged oxy chain. This suggests that over time the reaction might result in the formation of titania, TiO2.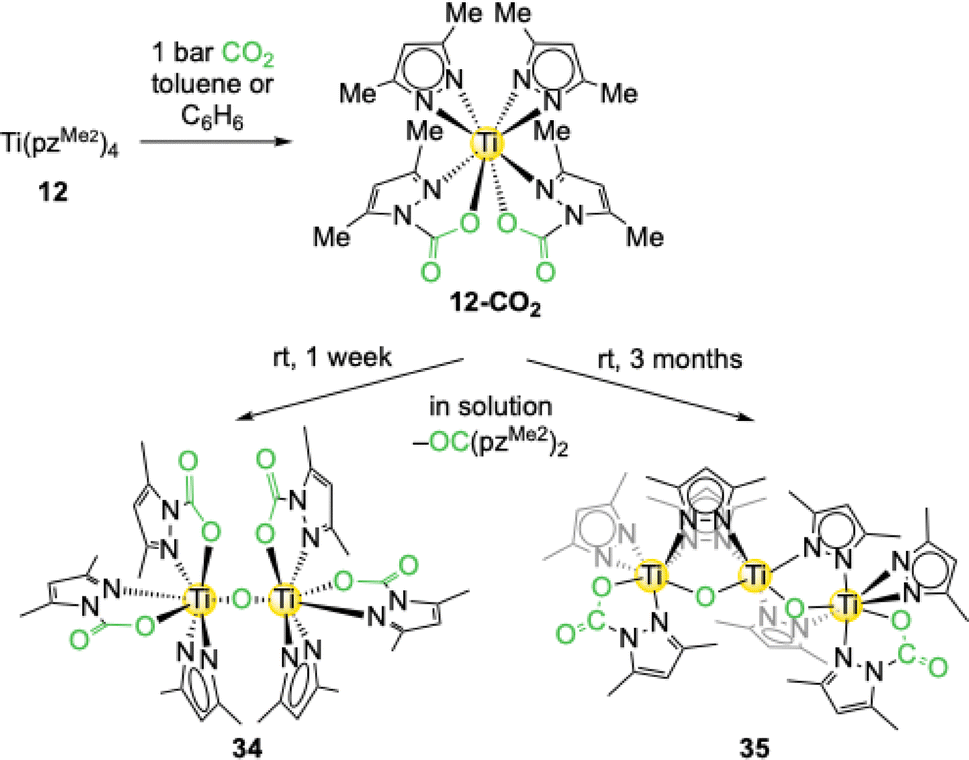 | ||
| Scheme 7 CO2 insertion into the tetravalent titanium complex Ti+IV(pzMe2)4 (12) to yield Ti+IV(CO2·pzMe2)2(pzMe2)2 (12-CO2) and subsequent deoxygenative decomposition. | ||
Conducting and instant storage of the 12/CO2 reaction at −40 °C produced a yellow amorphous precipitate. After an extended storage time at −40 °C red crystalline needles grew from the original solution. An SCXRD measurement confirmed the proposed structure of [Ti+IV(CO2·pzMe2)2(pzMe2)2] (12-CO2) with an uptake of 17.0 wt% CO2. While complete CO2 release could be observed by TGA (17.5 wt%), a VT 1H NMR experiment was inexpedient, due to a fast decomposition of 12-CO2 at elevated temperatures. In contrast, solid 12-CO2 displayed an exceptional high stability under an ambient atmosphere, showing only traces of hydrolysis after one month. Therefore, it can be hypothesized that solvated CO2 initiates the deoxygenation decomposition pathway of 12-CO2. The high stability of 12-CO2 at ambient atmosphere is in stark contrast to the CO2-inserted magnesium(II) and aluminium(III)/lanthanide(III) congeners which were only stable for a few days or decomposed almost instantly, respectively.
Complex Ti+IV(pzMe2)4 (12) is also capable of inserting the related heteroallene CS2.55 The insertion reaction is rather slow under ambient conditions but reached completion after three days at 60 °C. In contrast to CO2, the heavier homologue CS2 only inserts into one of the four possible pyrazolato ligands to afford complex [Ti+IV(CS2·pzMe2)(pzMe2)3] (12-CS2) (Scheme 8).
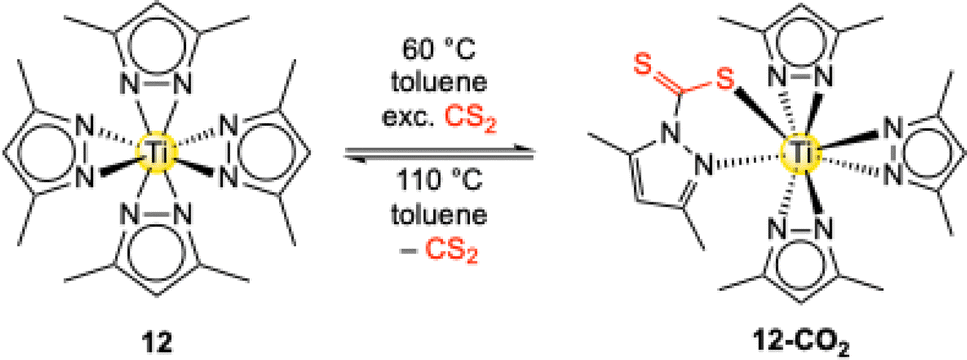 | ||
| Scheme 8 Reactivity of complex Ti+IV(pzMe2)4 (12) towards an excess of the heteroallene CS2 under formation of [Ti+IV(CS2·pzMe2)(pzMe2)3] (12-CS2). | ||
This reaction behaviour is consistent with the carboxophilicity criteria stated in Fig. 1. First, CS2 is a weaker electrophile than CO2.58 Together with the low thiophilicity of titanium (S = 0.0),53 the combination of the hard Lewis-acid Ti4+ and soft Lewis-base CS2 and the increased steric bulk of the sulfur atoms results in a mismatch of absorber and sorbent.59 Moreover, at 110 °C no decomposition to sulfur-bridging compounds was observed, but CS2 release with the recovery of complex 12 was noted, again reflecting the almost “non-existent” thiophilicity versus high oxophilicity (O = 1.0) of titanium.53
The reaction of the trivalent titanium complex Ti+III(η2-pztBu2)3 (17) with an excess of CO2 led to an immediate colour change from blue over green and yellow to finally red. Surprisingly, the crystal structure revealed that the titanium stayed in the oxidation state of +III and that the complex Ti+III(CO2·pztBu2)2(pztBu2) (17-CO2) formed (Scheme 9).55 Monomeric 17-CO2 is isostructural to the aluminium complex 16-CO2 and thus only two of the possible three pyrazolato ligands inserted a CO2 molecule (13.1 wt% CO2, TGA: 13.9 wt%). Compared to 16-CO2, complex 17-CO2 shows a full release of CO2 already at 70 °C in 1H VT NMR experiments.
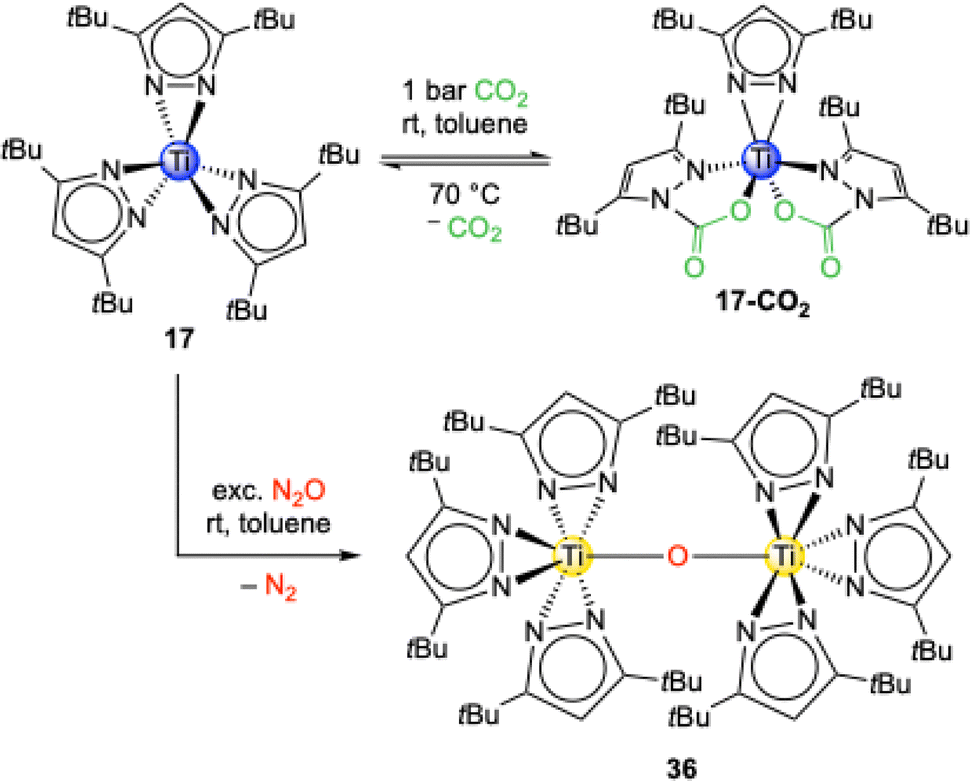 | ||
| Scheme 9 Reactivity of the trivalent titanium complex Ti+III(pztBu2)3 (17) toward CO2 and N2O and the formation of trivalent Ti+III(CO2·pztBu2)2(pztBu2) (17-CO2) and tetravalent oxo-bridged O[Ti+IV(η2-pztBu2)3]2 (36), respectively. | ||
Complex 17 reacts with N2O under N2 elimination and oxidation of the titanium centre to the oxo-bridged tetravalent dimer O[Ti+IV(η2-pztBu2)3]2 (36). This is also indicated by the color change from dark blue to yellow and the change of a paramagnetic to a diamagnetic signal set in the NMR experiments. The core structure of 36 is similar to O[Ti+IV(CO2·pzMe2)2(pzMe2)]2 (34) without inserted CO2. However, exposure of 36 to an excess of CO2 did not lead to a putative tBu analogue of 34. This is caused by the sterically bulky tBu moieties which shield the titanium centres towards any insertion reactions. Tetravalent Ti+IV(pzMe2)4 (12) did not show any reactivity towards N2O.
Catalytic conversion of epoxides and carbon dioxide to cyclic carbonates
The cycloaddition of epoxides and CO2 to cyclic carbonates is a carbon dioxide transformation well established in industry. The cyclic carbonates are highly sought-after and versatile products, since ethylene carbonate (EC) and propylene carbonate (PrC) are used as electrolytes in lithium-ion batteries.60–64 PrC was used for the first commercially available lithium-ion battery, but was later outperformed by the smaller derivative EC. Not only has EC a higher dielectric constant (ε = 78), which is necessary to dissolve the corresponding salts, but also a better conductivity.65,66 Further applications of cyclic carbonates are the replacement of phosgene in the synthesis of polyurethanes67 and as fuel additives.68–71 When cyclic carbonates are added to diesel fuel the amount of unburned hydrocarbons, carbon monoxide and particle pollution can be reduced remarkably.Given our previous work on cerium pyrazolate promoted conversion of epoxides and CO2 to cyclic carbonates, we were interested in the catalytic performance (if existing) of the light metal pyrazolates. The scope of the catalytic study was limited to the four epoxides propylene oxide (PO), styrene oxide (SO), 2-tert-butyloxirane (tBO), and 1,2-epoxyhexane (EH). The protocol for the catalytic conversion and proposed mechanistic scenario are stated in Table 3 and Scheme 10. In general, the pyrazolate complexes show a similar conversion as their corresponding carbamate complexes.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | PO (%) | SO (%) | tBO (%) | EH (%) |
| a Reaction conditions not stated in the table: 0.5 mol% [Cat.] (referred to metal centre), 24 h, neat in epoxide. b Conversion determined by comparison of the proton integrals in the α-position of the epoxide and the corresponding cyclic carbonate (except for tBO where the integral of the tBu moieties was used). c Only TBAB is used as the catalyst. | |||||
| 1 | TBABc | 3 | — | — | — |
| 2 | 13-Mg | 56 | 4 | 4 | 7 |
| 3 | 22-Mg | 45 | 4 | 3 | 11 |
| 4 | 23-Mg | 42 | — | — | — |
| 5 | 24-Mg | 43 | — | — | — |
| 6 | 25-Mg | 59 | 8 | 4 | 18 |
| 7 | 12-TiIV | 59 | 8 | 2 | 7 |
| 8 | 17-TiIII | 27 | — | — | — |
| 9 | 16-Al | 65 | 5 | 2 | 7 |
| 10 | 28-Al | 43 | 8 | 3 | 11 |
| 11 | 29-Sc | >99 | 27 | 24 | 29 |
| 12 | 19-Y | 84 | 20 | 5 | 22 |
| 13 | 30-Y | 97 | 22 | 8 | 18 |
| 14 | 1-CeIV | 88 | 23 | 13 | 25 |
| 15 | 2-CeIII | 96 | 13 | 5 | 9 |
For PO, the magnesium complexes showed only a moderate catalytic activity with conversions between 45% and 59% (entries 1–6). The highest conversion was achieved with the fluorinated derivative [Mg2(pzCF3,CF3)4(thf)3] (25) (entry 6), however, impaired by a polymeric side product. An almost equally high conversion was achieved with the tBu congener [Mg(pztBu2)2]2 (13) (56%, entry 2). The presence of donor molecules such as THF or HpzR2 slightly reduced the conversion (entries 3–5). Apparently, the donor molecules compete with the epoxide for coordination sites at the metal centre. In addition, the formation of carbamic acid from the respective pyrazole donors affects the transformation. The group 13 and titanium(IV) pyrazolates 16 (65%) and 12 (59%) displayed slightly higher conversions (entries 7 and 9). In general, the rare-earth metal-based catalysts revealed the highest catalytic activity, which is in line with their fast ligand exchange characteristics (entries 11–15).72 The scandium pyrazolate 29 performed best of all molecular/soluble pyrazolates with an almost quantitative conversion (entry 11), equaling a TON of 199. TOF studies revealed a starting TOF of 120 h−1 and an almost complete conversion after 6 h. The TON could be further increased to 4600 when 0.01 mol% of 29 is used at 90 °C and 10 bar CO2.
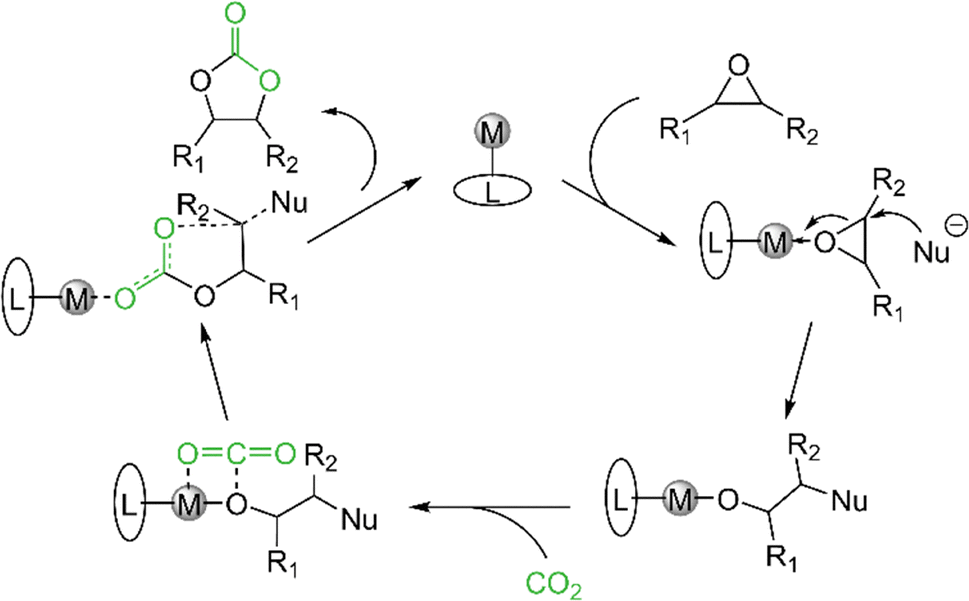 | ||
| Scheme 10 General proposed mechanism for the cycloaddition of CO2 and epoxides to cyclic carbonates. | ||
Dimeric compounds of the same metal perform slightly worse than their monomeric compounds despite having a less bulky alkyl moiety at the pyrazolato ligand (entries 9 vs. 10 and 12 vs. 13). As expected for the sterically more demanding epoxides SO, tBO and EH, an overall lower catalytic conversion was observed compared to PO with tBO being converted the least. Here, only the rare-earth-metal catalysts achieved significant conversions between 5–29%, with scandium complex 29 again as the most active catalyst. Not surprisingly, in the case of magnesium and aluminium, the complexes with less sterically demanding ligands performed slightly better than those with tBu substituents for all three bulky epoxides (entries 2 vs. 3 and 9 vs. 10). For yttrium this was only the case for EH (entries 12 vs. 13).
Interestingly, an inverse correlation between the catalytic conversion and the carboxophilicity can be stated (Table 2). The carboxophilicity is expressed by the CO2 release temperature of the corresponding CO2 insertion complexes which can be determined by VT NMR and TGA experiments. The magnesium and aluminum compounds 13 and 16 require the highest temperature to release CO2 but exhibit the lowest catalytic activity in cyclic carbonate formation. In contrast, the highly active rare-earth-metal complexes 29 and 30 do not insert CO2 at all at ambient temperature but release CO2 immediately. The trend in/origin of carboxophilicity is multifaceted and cannot be assigned only to the effects of oxophilicity, electronegativity, ionic radii or the Lewis acidity. Most likely it is a combination of these four metal properties and the nucleophilicity, the steric bulk and the coordination mode of the corresponding pyrazolato ligand. For example, the best-performing magnesium catalyst in this study, [Mg2(pzCF3,CF3)4(thf)3] (25), shows no visible CO2 insertion under ambient conditions. This originates from the fluorinated pyrazolato ligand, which has the lowest nucleophilicity of all used pyrazoles. Another example is the dual effect of the tBu moiety at the pyrazolato ligand which on the one hand increases the nucleophilicity of the ligand and thus enhances activation of CO2, but on the other hand can also prevent further CO2 insertion due to steric bulk.
In context with other reported catalyst systems, light metal pyrazolates show only a moderate catalytic activity. Two outstanding examples are the triple magnesium porphyrin complex Ph[(Ar3porphyrin)Mg]3 (Ar = Ph[m-O(CH2)6N(nBu)3]+[Br]−) which features the tetraalkyl ammonium salt cocatalyst unit incorporated intramolecularly into the ligand scaffold and the tetraarylporphyrin aluminium complex [({2,4-Cl}Ph)4porphyrin]AlCl (with bis(triphenylphosphine)-iminium chloride as the cocatalyst) exhibiting the highest catalytic activity not only for light metals but also for catalysts in general.73,74 These porphyrin complexes accomplish TOFs of 46![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 and 185200 h−1, respectively, although under much harsher conditions (120 °C, 17 and 30 bar CO2).
000 h−1 and 185200 h−1, respectively, although under much harsher conditions (120 °C, 17 and 30 bar CO2).
Heterogenized light metal pyrazolates for the catalytic formation of cyclic carbonates
Metal-amide grafting has emerged as a prolific branch of surface organometallic chemistry, especially when periodic mesoporous silicas (PMSs) are used as support materials.75,76 Considering the pKa criterion (vide infra) the related pyrazolates ideally qualify for monofunctional protonolytic surface reactions. This was successfully shown for the immobilization of La/Ce pyrazolates onto the mesoporous silica SBA-15500.41 In general, such surface grafting of metal complexes on chemically robust and surface-rich supports like PMS ensure a high population of active surface species. Moreover, surface confinement and site isolation minimize (intermolecular) deactivation processes, while the grafted metal centres can exhibit increased reactivity/Lewis acidity originating from distorted coordination geometry and the electron-withdrawing effect of the activated silica surface. Large-pore PMSs favour an efficient grafting of comparatively large pyrazolate complexes, minimizing any diffusion limitations during the grafting reaction (pore blockage as the worst case scenario) and promote any envisaged surface-promoted follow-up chemistry. Consequently, the surface chemistry of light metal pyrazolates 13, 16, 12 and 17 was investigated on large-pore PMS SBA-15. As for the rare-earth-metal pyrazolates, the grafting reaction was conducted with a slight excess of the light metal pyrazolates on dehydrated silica SBA-15500 in n-hexane/toluene, yielding [Mg(pztBu2)2]2@SBA-15500 (13@SBA-15), Al(pztBu2)3@SBA-15500 (16@SBA-15) and Ti+IV(pzMe2)4@SBA-15500 (12@SBA-15) and Ti+III(pztBu2)3@SBA-15500 (17@SBA-15).77Complete consumption of the surface silanol sites was confirmed for the grafted materials by the disappearance of the Si–O–H stretching vibration signal and the appearance of C–H vibration bands in the DRIFT spectra. Similar to the La/Ce protocols any released pyrazole was not detected in the supernatant solution, which indicates the formation of mixed pyrazolate/pyrazole species at the surface. This is further emphasized by signals for NH protons in the 1H MAS NMR spectra. Elemental analyses led to the conclusion that the surface species of magnesium hybrid species 13@SBA-15 mainly consists of bipodal species of the type [(![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) SiO)2Mg(HpztBu2)2] coordinated with two pyrazole donor molecules (Fig. 7). For 16@SBA-15 the monopodal species [(SiO)2Al(pztBu2)(HpztBu2)2] and for 12@SBA-15 the bipodal [(SiO)2Ti(pzMe2)2(HpzMe2)] seem plausible. Trivalent 17@SBA-15 most likely consists of a mixture of monopodal [(SiO)Ti+III(pztBu2)2(HpztBu2)] and bipodal [(SiO)2Ti+III(pztBu2)(HpztBu2)]. For all four materials the surface area and the pore volume decreased by ca. 50%. The pore diameter was reduced from 8.2 nm to 6.2–6.5 nm.
SiO)2Mg(HpztBu2)2] coordinated with two pyrazole donor molecules (Fig. 7). For 16@SBA-15 the monopodal species [(SiO)2Al(pztBu2)(HpztBu2)2] and for 12@SBA-15 the bipodal [(SiO)2Ti(pzMe2)2(HpzMe2)] seem plausible. Trivalent 17@SBA-15 most likely consists of a mixture of monopodal [(SiO)Ti+III(pztBu2)2(HpztBu2)] and bipodal [(SiO)2Ti+III(pztBu2)(HpztBu2)]. For all four materials the surface area and the pore volume decreased by ca. 50%. The pore diameter was reduced from 8.2 nm to 6.2–6.5 nm.
 | ||
| Fig. 7 Proposed surface species of silica-grafted light metal pyrazolates. | ||
Exposing the magnesium(II)/aluminium(III)/titanium(IV) hybrid materials to an atmosphere of 1 bar CO2 afforded mass gains accounting for 7% (13@SBA-15), 6% (16@SBA-15) and 11% (12@SBA-15), respectively. The obtained CO2-inserted materials are denoted CO2@[Mg(pztBu2)2]2@SBA-15500 (CO2@13@SBA-15), CO2@Al(pztBu2)3@SBA-15500 (CO2@16@SBA-15) and CO2@Ti+IV(pzMe2)4@SBA-15500 (CO2@12@SBA-15). In contrast, the titanium(III) hybrid material 17@SBA-15 was oxidized when exposed to CO2 while CO2 insertion was not observed. The oxidized surface species of 17@SBA-15 is reminiscent of oxo-bridged tetravalent dimer O[Ti+IV(pztBu2)3]2 (36) which also did not engage in CO2 insertion.
The mesoporous hybrid materials 13@SBA-15, 16@SBA-15, 12@SBA-15 and 17@SBA-15 were tested as heterogeneous catalysts. For PO, the magnesium material 13@SBA-15 yielded a mixture of the desired cyclic carbonate and a polymeric side product at ambient temperature and 1 bar CO2 (see Table 4, entry 1). Strikingly, when TBAI instead of TBAB was used as a cocatalyst the selectivity of the transformation shifted to the cyclic carbonate exclusively. Moreover, grafted 13@SBA-15 outperformed the molecular precursor 13 under standard conditions converting 88% of PO (entry 2). At 90 °C and 10 bar CO2 a quantitative conversion (>99%) of PO was achieved (entry 4). Under these conditions, even for the bulky epoxides SO and EH a conversion of 99% was obtained. This performance is similar to the cerium material 1@SBA-15 (entry 8). With a catalyst load of 13@SBA-15 of 0.01 mol% and conditions of 90 °C/10 bar CO2 a TON of 2100 was achieved. This remarkably increased catalytic activity can be ascribed to the surface species [(SiO)2Mg(HpztBu2)2], which is only coordinated by donor molecules, thus affording highly active magnesium centres. In the absence of cocatalyst, 13@SBA-15 yielded almost exclusively a polymeric product which was not further characterized (entry 3). Overall, this indicates that the selectivity of 13@SBA-15 is tunable via the use of (distinct) cocatalysts. In addition, 13@SBA-15 displayed a high reusability and the catalytic conversion of PO remained stable after eight consecutive runs. The titanium material 12@SBA-15 gave a conversion of PO of 15%, in the absence of any side product (entry 5), which could be increased to 97% at 90 °C and 10 bar CO2 (entry 6). Under these conditions a moderate to good conversion even for the bulky epoxides could be achieved. The trivalent titanium material 17@SBA-15 converted 46% of PO. However, when contacted with PO instant oxidation of the material was observed by a color change from purple to yellow.
| Entry | Catalyst | PO | SO | tBO | EH | Cocat. |
|---|---|---|---|---|---|---|
| a Reaction conditions if not stated otherwise: 0.5 mol% [catalyst] (referred to metal centre), 0.5 mol% cocatalyst, 24 h, neat in epoxide, ambient temperature and 1 bar CO2. b Conversion determined by comparison of the proton integrals in the α-position of the epoxide and the corresponding cyclic carbonate (except for tBO where the integral of the tBu moieties was used). c Not representative due to polymer formation as a side reaction. d Polymer as the main product. e 90 °C, 10 bar CO2. f Oxidation to a Ti+IV species occurred. Values adapted from ref. 41 and 77. | ||||||
| 1 | 13@SBA-15 | 63%c | — | — | — | TBAB |
| 2 | 13@SBA-15 | 88% | — | — | — | TBAI |
| 3 | 13@SBA-15 | <1d | — | — | — | — |
| 4e | 13@SBA-15 | >99% | 99% | 60% | 99% | TBAI |
| 5 | 12@SBA-15 | 15% | — | — | — | TBAB |
| 6e | 12@SBA-15 | 97% | 91% | 41% | 74% | TBAB |
| 7 | 17@SBA-15 | 46%f | — | — | — | TBAB |
| 8 | 1@SBA-15 | >99% | 91% | 66% | 94% | TBAB |
Pyrazole-based metal–organic frameworks for CO2 activation
Metal–organic framework (MOF) formation features an alternative approach to access periodic porous metal-containing materials with high intrapore surface.78 Bridged bis- and tris(pyrazolyl) ligands emerged as robust linkers in MOF compounds qualifying them as potential candidates for carbon capture materials.79–84 However, mostly transition metal-based MOFs have been targeted and examples involving light metals are rather scarce. For example, the zinc-based BUT-31 (37) with 2,5-bis(4-pyrazole)benzaldehyde as a linker hit a CO2 uptake of 23.2 wt% at 0 °C (15.8 wt% at 26 °C, Fig. 8).85 Another example features MOF [Co8.5(µ4-O)(bpdc)3(bpz)3(Hbpz)3](dmf)6(CH3OH)9(H2O)15 (38) with a mix of 3,3′,5,5′-tetramethyl-4,4-bipyrazolate (bpz) and 4,4′-biphenyldicarboxylate (bpdc) linkers.86 Capacities of up to 5.9 wt% CO2 at 40 °C were achieved with a high selectivity over N2 (58–188) in a CO2/N2 mixture of 50:50.
 | ||
| Fig. 8 Representative examples of pyrazolato/pyrazole derived linkers employed in MOF design. | ||
MOF-303 [Al(OH)(pdc)]n (39) features a light metal MOF where aluminium(III) centres are linked by 3,5-(dicarboxylate)-pyrazole.87 MOF 39 was incorporated into a polymer matrix which afforded a microporous membrane (40). Material 40 accomplishes a CO2 uptake of over 11 wt% with a high selectivity over N2 (25.6) at 35 °C. Additionally, bispyrazolato-based materials, like [Zn(azbp)]n (41) with a dianionic 4,4′-azobis(3,5-dimethylpyrazolato) linker, were used for gas separations showing no preference for CO2 in mixtures with acetylene (selectivity of 2.6 for C2H2 over CO2).88
The aluminium-based MOF-303 (39) displays a highly versatile material, which not only can be used as a water absorber, but it also achieves CO2 uptakes as high as 22.4 wt% (5.1 mmol g−1; 25 °C) with a high selectivity over N2 (18) and CH4 (5).89,90 In addition, MOF 39 is also capable of reducing CO2 photocatalytically to CO and CH4.91 DFT calculations revealed that the pyrazole ligand plays an active role in the reduction step by hydrogen bonding to the oxygen (CO2) and nucleophilic bonding of the pyrazole-nitrogen to the carbon (CO2). This was further confirmed by in situ DRIFT experiments showing characteristic stretching vibrations for the carbamate unit NCOO.
The iron-based MOF Fe2[1,4-bis(4-pyrazolate)phenyl]3 (42) achieved an adsorption capacity of 6.4 wt% at 0 °C using an 85:15 mixture of N2 and CO2. Computational studies revealed that CO2 molecules mainly interact with the aromatic linker.92 MOF Fe2[1,4-bis(pyrazolate-4-ethynyl)benzene]3 (43) features a linker which is extended by two alkyne moieties to allow for a higher porosity.93 The increased porosity resulted in a significantly increased adsorption capacity of 20.6 wt% under similar conditions. At 10 bar and 25 °C the capacity could be increased to 40.5 wt%.
Pyrazole-based MOFs like the bispyrazolato linked Ni[1,4-bis(4-pyrazolate)phenyl] (44) and the trispyrazolato linked Ni3[1,3,5-tris(4-pyrazolate)phenyl]2 (45) were tested as catalysts in the cycloaddition of epoxides and CO2.94 Material 44 with TBAB as a cocatalyst, showed a quantitative conversion at 80 °C/5 bar. Material 45 contains less active metal sites than 44 and thus accomplishes a lower conversion of 70%. An overview of pyrazole-based MOFs as well as tri- and tetrazole compounds as catalysts in the cycloaddition of epoxides and CO2 to cyclic carbonates can be found in Table 5.
| Compound (metal) | T/p/t (°C/bar/h) | M/TBAX (mol%) | Conversion (%) | Cocatalyst | Ref. |
|---|---|---|---|---|---|
| Values adapted from the corresponding references. Pressure is converted to bar for an easier comparison. | |||||
| 44 (Ni) | 80/5/24 | 0.5/8 | >99 | TBAB | 94 |
| 45 (Ni) | 80/5/24 | 0.5/8 | 70 | TBAB | 94 |
| 47 (Cu) | rt/1/48 | 0.2/10 | 96 | TBAB | 108 |
| 48 (Zn) | 40/1/24 | 0.5/1 | >99 | TBAB | 109 |
| 49 (Ti) | 75/22/4.5 | 0.1/0.1 | 39 | TBAI | 110 |
| 50 (Ti) | 75/22/4.5 | 0.1/0.1 | 29 | TBAI | 110 |
| 51 (Ti) | 75/22/4.5 | 0.1/0.1 | 86 | TBAI | 110 |
| 54 (Cd) | 80/1/4 | 0.2/2 | 98 | TBAB | 113 |
| 55 (Zn) | 60/40/24 | 0.2/0.4 | 99 | TBAB | 114 |
Triazolates and tetrazolates for CO2 activation
The nucleophilicity of the N-heterocyclic pyrazolato ligand can be effectively modified by substituents in the 2,5-ring positions but to an even greater extent by introducing additional nitrogen atoms into the aromatic ring. Consequently, the nitrogen-richer azolates comprising 1,2,4-triazolate, 1,2,3-triazolate and tetrazolate display potential CO2 activation compounds as well. The changed basicity of the azolato ligands is nicely quantified by comparison of the pKa values of the respective proligands, encompassing a wide range of at least 10 orders of magnitude (Scheme 11, pyrazole/19.8 → tetrazole/8.2).95 Such distinct nucleophilicity makes triazolate and tetrazolate complexes interesting candidates for examining nc-MLC in CO2 activation. A lower nucleophilicity of the ligands should lead to an increased stability of the complexes towards hydrolysis. Similar to bridged pyrazoles, both (deprotonated) triazoles and tetrazoles display robust linkers in framework materials acting as the coordination site as well as bridging backbone.96 However, the interaction of the framework with CO2 was noticed almost exclusively of adsorptive nature and only rarely a chemisorption behaviour was reported. | ||
| Scheme 11 pKa values for azoles and N-heterocycles (values are listed for R = H). Consideration is also given to common amines and alcohols, indicating trends in equilibrium acidities. Depending on the solvent, pKa values differ dramatically. Unfortunately, pKa values of the depicted compounds (“proligands”) determined in the same solvent are not available. | ||
Metal-free carbon capture materials based on triazoles including functionalized porous organic polymers or 1,2,3-triazole-based deep eutectic solvents have been reported.97,98 Triazole itself is also capable of activating CO2 as reported by Zare for its reduction to formic acid by using 1,2,3-triazole-containing water microdroplets.99 Accordingly, gas-phase CO2 is captured by triazole at the gas–liquid interface which is significantly increased for water microdroplets.
A number of MOF compounds with triazole-containing linkers were reported as carbon capture materials, however exclusively for transition metals.100–107 The highest CO2 uptake was achieved with the copper–triazolyl MOF [Cu(L)]n (46, L = 5-(3-methyl-5-(pyridine-4-yl)-4H-1,2,4-triazol-4-yl)isophthalate) which achieves 26.8 wt% at ambient temperature.103 Several of these MOFs were tested as heterogenous catalysts in the cycloaddition of CO2 and epoxides. The copper–triazole-based MOF [Cu4L]n (47, L = 5,5′,5″,5‴-((methanetetrayltetrakis(benzene-4,1-diyl)) tetrakis(1H-1,2,3-triazole-4,1-diyl))tetra-iso-phthalic acid) displayed a 96% conversion of PO to propylene carbonate (PC) (Table 5).108 The triazole-functionalized tetracarboxylate zinc MOF [(Zn2{L}{H2O})·4(H2O)]n (48, L = Me2Si[1,4-Ph-(1,2,3-triazol-4,1-diyl)-1,4-Ph(COO)2]2) gave a quantitative yield of PC under mild conditions.109 The carboxylato groups function as metal-binding sites while the nucleophilic triazole sites activate CO2.
Regarding the application of tetrazoles, the titanium complexes CpTiCl2(L) (49, L = N,O-5-(2-hydroxyphenyl)-1H-tetrazole), CpTiCl3(L)(thf) (50) and CpTiCl2(L)2 (51) were tested as homogenous catalysts in the conversion of PO (Table 5).110 Complex 51 showed the highest conversion (86%) and a maximum TOF of 422 h−1 (0.01 mol% catalyst), however under harsher conditions (80 °C and 22 bar). A number of tetrazole-containing frameworks were reported either as CO2 storage/separation materials or for catalytic conversion of CO2, but again mostly based on transition metals, especially zinc. For example, the MOF [(Zn{L})·dmf·0.5H2O]n (52, L = 1,5-bis(5-tetrazolo)-3-oxopentane) can achieve CO2 uptakes of up to 22 wt% at ambient temperature.111 The zeolitic zinc tetrazolate framework [Zn(H2Ntet)2]n (53) exhibited a CO2 uptake as high as 24.6 wt%.112 The additional amino group at the tetrazolato linker enhances the adsorptive properties. More recently, tetrazole-based MOFs were also tested as catalysts in the cycloaddition of CO2 and epoxides (Table 5). The cadmium MOF [M2NH2]+[Cd(dtztp)0.5(HCOO)]−·1.5(dmf)·H2O (2,5-di(5-tetrazole)-terephthalate) (54) achieved a conversion of 98% for PO.113 The zinc MOF [Zn2(L)2(dmf)2]n·nH2O (55, L = 4-[(4-{1H-tetrazol-5-yl}phenyl)carbamoyl]benzoate) converted PO quantitatively, albeit under much harsher conditions (60 °C, 40 bar).114
Conclusions and outlook
An in-depth understanding of the interaction of carbon dioxide with carbon capture materials is crucial for designing more efficient adsorber components and catalysts. A synergetic non-classical metal–ligand cooperativity (nc-MLC) can be exploited to tailor metal complexes for CO2 capture as well as transformation. The carboxophilicity of the metal complexes generally depends on the interplay of various factors including the nucleophilicity and steric bulk of the ligand as well as the Lewis acidity, oxophilicity and electronegativity of the metal centre. Monoanionic N-functional ligands stand out and in particular pyrazolato ligands provide a perfect fit for controlling reversible insertion/de-insertion scenarios.Metalorganic amide complexes M(NR2)x (R = aliphatic or aromatic group) are able to insert CO2 quantitatively, however, this process is irreversible in most cases, since the formed carbamato ligands engage in a stable carboxylato coordination. In contrast, (light) metal pyrazolates insert CO2 not only exhaustively and most weight efficiently but also reversibly under the formation of pyrazole-based carbamato ligands. The parent magnesium pyrazolate excels in CO2 uptake, accomplishing a record high CO2 chemisorption of 35.7 wt% CO2. This is comparable to the most effective metal–organic frameworks which combine CO2 adsorption and chemisorption. Fine-tuning of the carboxophilicity is nicely revealed by the trivalent complexes Al(pztBu2)3 and Sc(pztBu2)3(thf). The former aluminium derivative combines a hard Lewis acid and strongly nucleophilic pyrazolato ligands (tBu substituents), and hence, imparts a strong carboxophilicity, but it exhibits only moderate catalytic conversion in the cycloaddition of CO2 and epoxides. On the other hand, the same ligand on trivalent scandium (Sc(pztBu2)3(thf)) does not lead to an isolable carbamato derivative but excels as a catalyst. This is facilitated by a comparatively softer Lewis acidity and more rapid ligand exchange rates. The apparent inverse correlation of the catalytic activity and carboxophilicity makes light metal pyrazolates highly attractive and versatile CO2 capture and transformation components. Consequently, supported light metal pyrazolates have been exploited as heterogeneous catalysts. In particular, favourable basicity of the pyrazolate complexes makes silica-grafted variants of the type M(pzR2)x@silica easily accessible materials. Such hybrid materials take up CO2 to generate CO2@M(pzR2)x@silica or act as reusable catalysts for cyclocarbonate formation.
Although pyrazolate complexes featuring high stability towards hydrolysis have been discovered, like the mixed carbamato/pyrazolato titanium complex Ti+IV(CO2·pzMe2)2(pzMe2)2, most light metal pyrazolates are sensitive towards moisture. Since framework materials based on the nitrogen-richer azoles – 1,2,4-triazole, 1,2,3-triazole and tetrazole – are stable towards hydrolysis and adsorb CO2, it should be enlightening to study the respective reaction behaviour of molecular metal-azolate complexes. Our initial studies on cerium triazolate and tetrazolate chemistry revealed the feasibility of CO2 insertion but it was less pronounced than for pyrazolate derivatives.115
Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We are grateful to the VECTOR foundation for the generous support (grant P2021-0099). R. A. thanks his former and present students Daniel Werner, Uwe Bayer, Jitpisut Poolwong, Felix Kracht, Jonas Riedmaier and Paul Preisenberger for their enthusiasm as well as great commitment and accomplishments as part of the “CO2 project”. Along this line, many thanks are given to Yucang Liang and Valerio D’Elia for exciting collaborations.Notes and references
- H. Lee and J. Romero, IPCC, 2023: Climate Change 2023: Synthesis Report, Contribution of Working Groups I, II and III to the Sixth Assessment Report of the Intergovernmental Panel on Climate Change, Geneva, Switzerland, 2023, pp. 35–115.
- P. Falkowski, R. J. Scholes, E. Boyle, J. Canadell, D. Canfield, J. Elser, N. Gruber, K. Hibbard, P. Högberg, S. Linder, F. T. Mackenzie, B. Moore III, T. Pedersen, Y. Rosenthal, S. Seitzinger, V. Smetacek and W. Steffen, Science, 2000, 290, 291–296 CrossRef PubMed.
- S. Solomon, G.-K. Plattner, R. Knutti and P. Friedlingstein, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 1704–1709 CrossRef PubMed.
- R. S. Haszeldine, Science, 2009, 325, 1647–1652 CrossRef PubMed.
- D. W. Keith, Science, 2009, 325, 1654–1655 CrossRef PubMed.
- G. Centi and S. Perathoner, Catal. Today, 2009, 148, 191–205 CrossRef.
- M. Cokoja, C. Bruckmeier, B. Rieger, W. A. Herrmann and F. E. Kühn, Angew. Chem., Int. Ed., 2011, 50, 8510–8537 Search PubMed.
- N. von der Assen, P. Voll, M. Peters and A. Bardow, Chem. Soc. Rev., 2014, 43, 7982–7994 RSC.
- M. Aresta, A. Dibenedetto and A. Angelini, Chem. Rev., 2014, 114, 1709–1742 CrossRef PubMed.
- Q. Liu, L. Wu, R. Jackstell and M. Beller, Nat. Commun., 2015, 6, 5933 Search PubMed.
- R. E. Siegel, S. Pattanayak and L. A. Berben, ACS Catal., 2023, 13, 766–784 CrossRef.
- M. J. Prather, Science, 1998, 279, 1339–1341 CrossRef PubMed.
- T. S. Ledley, E. T. Sundquist, S. E. Schwartz, D. K. Hall, J. D. Fellows and T. L. Killeen, EOS, Trans., Am. Geophys. Union, 1999, 80, 453–458 CrossRef.
- S. Arrhenius, London, Edinburgh Dublin Philos. Mag. J. Sci., 1896, 41, 237–276 Search PubMed.
- D. M. D’Alessandro, B. Smit and J. R. Long, Angew. Chem., Int. Ed., 2010, 49, 6058–6082 CrossRef.
- E. S. Sanz-Pérez, C. R. Murdock, S. A. Didas and C. W. Jones, Chem. Rev., 2016, 116, 11840–11876 Search PubMed.
- A. C. Forse and P. J. Milner, Chem. Sci., 2021, 12, 508–516 RSC.
- G. T. Rochelle, Science, 2009, 325, 1652–1654 CrossRef PubMed.
- P. Brœder and H. F. Svendsen, Energy Procedia, 2012, 23, 45–54 CrossRef.
- D. J. Heldebrant, P. K. Koech, V.-A. Glezakou, R. Rousseau, D. Malhotra and D. C. Cantu, Chem. Rev., 2017, 117, 9594–9624 CrossRef PubMed.
- L. B. Miller and J. C. Witt, J. Phys. Chem., 1929, 33, 285–289 CrossRef.
- S. Zeng, X. Zhang, L. Bai, X. Zhang, H. Wang, J. Wang, D. Bao, M. Li, X. Liu and S. Zhang, Chem. Rev., 2017, 117, 9625–9673 CrossRef PubMed.
- B. Yoon, S. Chen and G. A. Voth, J. Am. Chem. Soc., 2024, 146, 1612–1618 Search PubMed.
- P. D. Jadhav, R. V. Chatti, R. B. Biniwale, N. K. Labhsetwar, S. Devotta and S. S. Rayalu, Energy Fuels, 2007, 21, 3555–3559 CrossRef.
- H. Lee, D. Xie, S. I. Zones and A. Katz, J. Am. Chem. Soc., 2024, 146, 68–72 CrossRef.
- S. R. Caskey, A. G. Wong-Foy and A. J. Matzger, J. Am. Chem. Soc., 2008, 130, 10870–10871 CrossRef PubMed.
- J. A. Mason, K. Sumida, Z. R. Herm, R. Krishna and J. R. Long, Energy Environ. Sci., 2011, 4, 3030–3040 RSC.
- T. M. McDonald, J. A. Mason, X. Kong, E. D. Bloch, D. Gygi, A. Dani, V. Crocellà, F. Giordanino, S. O. Odoh, W. S. Drisdell, B. Vlaisavljevich, A. L. Dzubak, R. Poloni, S. K. Schnell, N. Planas, K. Lee, T. Pascal, L. F. Wan, D. Prendergast, J. B. Neaton, B. Smit, J. B. Kortright, L. Gagliardi, S. Bordiga, J. A. Reimer and J. R. Long, Nature, 2015, 519, 303–308 CrossRef PubMed.
- E. J. Kim, R. L. Siegelman, H. Z. H. Jiang, A. C. Forse, J.-H. Lee, J. D. Martell, P. J. Milner, J. M. Falkowski, J. B. Neaton, J. A. Reimer, S. C. Weston and J. R. Long, Science, 2020, 369, 392–396 CrossRef.
- S. Chu, Science, 2009, 325, 1599 CrossRef PubMed.
- E. Alper and O. Yuksel Orhan, Petroleum, 2017, 3, 109–126 CrossRef.
- A. Behr, Chem. Eng. Technol., 1987, 10, 16–27 CrossRef.
- T. Sakakura, J.-C. Choi and H. Yasuda, Chem. Rev., 2007, 107, 2365–2387 CrossRef.
- I. Omae, Coord. Chem. Rev., 2012, 256, 1384–1405 CrossRef.
- R. Ayyappan, I. Abdalghani, R. C. D. Costa and G. R. Owen, Dalton Trans., 2022, 51, 11582–11611 RSC.
- D. Werner, U. Bayer, N. E. Rad, P. C. Junk, G. B. Deacon and R. Anwander, Dalton Trans., 2018, 47, 5952–5955 RSC.
- D. Werner, G. B. Deacon, P. C. Junk and R. Anwander, Eur. J. Inorg. Chem., 2017, 3419–3428 CrossRef.
- U. Bayer, D. Werner, C. Maichle-Mössmer and R. Anwander, Angew. Chem., Int. Ed., 2020, 59, 5830–5836 CrossRef PubMed.
- Y.-T. Tseng, W.-M. Ching, W.-F. Liaw and T.-T. Lu, Angew. Chem., Int. Ed., 2020, 59, 11819–11823 CrossRef CAS PubMed.
- U. Bayer, A. Jenner, J. Riedmaier, C. Maichle-Mössmer and R. Anwander, Molecules, 2021, 26, 1957 CrossRef CAS PubMed.
- U. Bayer, Y. Liang and R. Anwander, Inorg. Chem., 2020, 59, 14605–14614 CrossRef CAS.
- I. A. Guzei, A. G. Baboul, G. P. A. Yap, A. L. Rheingold, H. B. Schlegel and C. H. Winter, J. Am. Chem. Soc., 1997, 119, 3387–3388 CrossRef CAS.
- D. Pfeiffer, M. J. Heeg and C. H. Winter, Angew. Chem., Int. Ed., 1998, 37, 2517–2519 CrossRef CAS.
- J. Hitzbleck, G. B. Deacon and K. Ruhlandt-Senge, Eur. J. Inorg. Chem., 2007, 592–601 CrossRef CAS.
- N. C. Mösch-Zanetti, M. Ferbinteanu and J. Magull, Eur. J. Inorg. Chem., 2002, 950–956 CrossRef.
- G. B. Deacon, E. E. Delbridge, C. M. Forsyth, P. C. Junk, B. W. Skelton and A. H. White, Aust. J. Chem., 1999, 52, 733–740 CrossRef CAS.
- N. C. Mösch-Zanetti, R. Krätzner, C. Lehmann, T. R. Schneider and I. Usón, Eur. J. Inorg. Chem., 2000, 13–16 CrossRef.
- G. B. Deacon, A. Gitlits, P. W. Roesky, M. R. Bürgstein, K. C. Lim, B. W. Skelton and A. H. White, Chem. – Eur. J., 2001, 7, 127–138 CrossRef CAS PubMed.
- X. Zhou, L. Zhang, R. Ruan, L. Zhang, R. Cai and L. Weng, Chin. Sci. Bull., 2001, 46, 723–726 CrossRef CAS.
- S.-Á. Cortés-Llamas, R. Hernández-Lamoneda, M.-Á. Velázquez-Carmona, M.-A. Muñoz-Hernández and R. A. Toscano, Inorg. Chem., 2006, 45, 286–294 CrossRef.
- F. Kracht, P. Rolser, P. Preisenberger, C. Maichle-Mössmer and R. Anwander, Adv. Sci., 2024, 11, 2403295 CrossRef CAS.
- F. Kracht, P. Rolser, K. Eichele, C. Maichle-Mössmer and R. Anwander, Inorg. Chem. Front., 2024, 11, 6948–6959 RSC.
- K. P. Kepp, Inorg. Chem., 2016, 55, 9461–9470 CrossRef CAS PubMed.
- L. Pauling, The chemical bond: a brief introduction to modern structural chemistry, Cornell University Press, Ithaca, N.Y., 1967 Search PubMed.
- F. Kracht, S. Mayer, C. Maichle-Mössmer and R. Anwander, Dalton Trans., 2025, 54, 10890–10897 RSC.
- M. Anpo and K. Chiba, J. Mol. Catal., 1992, 74, 207–212 CrossRef CAS.
- M. Anpo, H. Yamashita, Y. Ichihashi and S. Ehara, J. Electroanal. Chem., 1995, 396, 21–26 CrossRef.
- Z. Li, R. J. Mayer, A. R. Ofial and H. Mayr, J. Am. Chem. Soc., 2020, 142, 8383–8402 CrossRef CAS.
- R. D. Shannon, Acta Crystallogr., Sect. A, 1976, 32, 751–767 CrossRef.
- W. S. Harris, Electrochemical Studies in Cyclic Esters, University of California Radiation Laboratory, 1958 Search PubMed.
- G. Pistoia, M. D. Rossi and B. Scrosati, J. Electrochem. Soc., 1970, 117, 500 CrossRef CAS.
- S. Tobishima, M. Arakawa, T. Hirai and J. Yamaki, J. Power Sources, 1989, 26, 449–454 CrossRef CAS.
- United States, US4959281A, 1990.
- R. S. McMillan and M. W. Juzkow, J. Electrochem. Soc., 1991, 138, 1566 CrossRef CAS.
- G. Pistoia, J. Electrochem. Soc., 1971, 118, 153 CrossRef CAS.
- X. You, M. Chaudhari, S. Rempe and L. R. Pratt, ECS Trans., 2015, 69, 107 CrossRef CAS.
- H. Blattmann, M. Fleischer, M. Bähr and R. Mülhaupt, Macromol. Rapid Commun., 2014, 35, 1238–1254 CrossRef CAS.
- United States, US2331386A, 1943.
- P. Rounce, A. Tsolakis, P. Leung and A. P. E. York, Energy Fuels, 2010, 24, 4812–4819 CrossRef CAS.
- M. O. Sonnati, S. Amigoni, E. P. T. de Givenchy, T. Darmanin, O. Choulet and F. Guittard, Green Chem., 2013, 15, 283–306 RSC.
- M. Szőri, B. Raj Giri, Z. Wang, A. E. Dawood, B. Viskolcz and A. Farooq, Sustainable Energy Fuels, 2018, 2, 2171–2178 RSC.
- S. Kobayashi, S. Nagayama and T. Busujima, J. Am. Chem. Soc., 1998, 120, 8287–8288 CrossRef CAS.
- C. Maeda, T. Taniguchi, K. Ogawa and T. Ema, Angew. Chem., Int. Ed., 2015, 54, 134–138 CrossRef CAS PubMed.
- Y. Qin, H. Guo, X. Sheng, X. Wang and F. Wang, Green Chem., 2015, 17, 2853–2858 RSC.
- R. Anwander, Chem. Mater., 2001, 13, 4419–4438 CrossRef CAS.
- Y. Liang and R. Anwander, Dalton Trans., 2013, 42, 12521–12545 RSC.
- F. Kracht, J. Poolwong, N. Roth, Y. Liang, C. Maichle-Mössmer and R. Anwander, Inorg. Chem., 2026 DOI:10.1021/acs.inorgchem.5c05578.
- R. Anwander, Immobilization of Molecular Catalysts, in Handbook of Heterogeneous Catalysis, ed. G. Ertl, H. Knözinger, F. Schüth and J. Weitkamp, Wiley-VCH Verlag GmbH, Weinheim, 2nd edn, 2008, vol. 8, pp. 583–614 Search PubMed.
- H. J. Choi, M. Dincă and J. R. Long, J. Am. Chem. Soc., 2008, 130, 7848–7850 Search PubMed.
- M. Tonigold, Y. Lu, B. Bredenkötter, B. Rieger, S. Bahnmüller, J. Hitzbleck, G. Langstein and D. Volkmer, Angew. Chem., Int. Ed., 2009, 48, 7546–7550 CrossRef CAS.
- V. Colombo, S. Galli, H. J. Choi, G. D. Han, A. Maspero, G. Palmisano, N. Masciocchi and J. R. Long, Chem. Sci., 2011, 2, 1311–1319 RSC.
- N. M. Padial, E. Quartapelle Procopio, C. Montoro, E. López, J. E. Oltra, V. Colombo, A. Maspero, N. Masciocchi, S. Galli, I. Senkovska, S. Kaskel, E. Barea and J. A. R. Navarro, Angew. Chem., Int. Ed., 2013, 52, 8290–8294 CrossRef CAS.
- C. Heering, I. Boldog, V. Vasylyeva, J. Sanchiz and C. Janiak, CrystEngComm, 2013, 15, 9757–9768 RSC.
- M. Parshad, D. Kumar and V. Verma, Inorg. Chim. Acta, 2024, 560, 121789 CrossRef CAS.
- T. He, Y.-Z. Zhang, B. Wang, X.-L. Lv, L.-H. Xie and J.-R. Li, ChemPlusChem, 2016, 81, 864–871 CrossRef CAS PubMed.
- H.-H. Wang, L.-N. Jia, L. Hou, W. Shi, Z. Zhu and Y.-Y. Wang, Inorg. Chem., 2015, 54, 1841–1846 CrossRef CAS.
- Q. Shen, S. Cong, J. Zhu, Y. Zhang, R. He, S. Yi and Y. Zhang, J. Membr. Sci., 2022, 664, 121107 CrossRef CAS.
- G. Berkbigler, Q. Liu, N. Hoefer, Y. Xie, J. S. Hilliard, D. W. McComb and C. R. Wade, Eur. J. Inorg. Chem., 2024, e202300548 CrossRef CAS.
- Z. Li, K. Shi, L. Zhai, Z. Wang, H. Wang, Y. Zhao and J. Wang, Sep. Purif. Technol., 2023, 307, 122725 CrossRef CAS.
- N. Hanikel, X. Pei, S. Chheda, H. Lyu, W. Jeong, J. Sauer, L. Gagliardi and O. M. Yaghi, Science, 2021, 374, 454–459 CrossRef CAS PubMed.
- K. Li, S. Ge, X. Wei, W. Zou, X. Wang, Q. Qian, B. Gao and L. Dong, Inorg. Chem., 2023, 62, 15824–15828 CrossRef CAS PubMed.
- R. Vismara, S. Terruzzi, A. Maspero, T. Grell, F. Bossola, A. Sironi, S. Galli, J. A. R. Navarro and V. Colombo, Adv. Mater., 2024, 36, 2209907 CrossRef CAS.
- C. Giacobbe, E. Lavigna, A. Maspero and S. Galli, J. Mater. Chem. A, 2017, 5, 16964–16975 RSC.
- Z. Wang, Q. Xie, Y. Wang, Y. Shu, C. Li and Y. Shen, New J. Chem., 2020, 44, 18319–18325 RSC.
- (a) R. K. Iller, The Chemistry of Silica, Wiley-Interscience, New York, 1979 Search PubMed; (b) K. Unger, Porous Silica, Elsevier, Amsterdam, 1979 Search PubMed; (c) R. R. Fraser, T. S. Mansour and S. Sevard, J. Org. Chem., 1985, 50, 3232–3234 CrossRef CAS; (d) F. G. Bordwell, Acc. Chem. Res., 1988, 21, 456–463 CrossRef CAS; (e) R. Duchateau, U. Cremer, R. J. Harmsen, S. I. Mohamud, H. C. L. Abbenhuis, R. A. van Santen, A. Meetsma, S. K.-H. Thiele, M. F. H. van Tol and M. Kranenburg, Organometallics, 1999, 18, 5447–5459 CrossRef CAS.
- P.-Z. Li, X.-J. Wang and Y. Zhao, Coord. Chem. Rev., 2019, 380, 484–518 CrossRef CAS.
- L.-H. Xie and M. P. Suh, Chem. – Eur. J., 2013, 19, 11590–11597 CrossRef CAS.
- Z. Wang, C. Wu, Z. Wang, S. Zhang and D. Yang, Chem. Commun., 2022, 58, 7376–7379 RSC.
- X. Song, Y. Meng and R. N. Zare, J. Am. Chem. Soc., 2022, 144, 16744–16748 CrossRef CAS PubMed.
- J.-B. Lin, J.-P. Zhang and X.-M. Chen, J. Am. Chem. Soc., 2010, 132, 6654–6656 CrossRef CAS PubMed.
- S.-M. Zhang, Z. Chang, T.-L. Hu and X.-H. Bu, Inorg. Chem., 2010, 49, 11581–11586 CrossRef CAS PubMed.
- J.-P. Zhang, A.-X. Zhu, R.-B. Lin, X.-L. Qi and X.-M. Chen, Adv. Mater., 2011, 23, 1268–1271 CrossRef CAS PubMed.
- D. Lässig, J. Lincke, J. Moellmer, C. Reichenbach, A. Moeller, R. Gläser, G. Kalies, K. A. Cychosz, M. Thommes, R. Staudt and H. Krautscheid, Angew. Chem., Int. Ed., 2011, 50, 10344–10348 CrossRef.
- R.-B. Lin, D. Chen, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Inorg. Chem., 2012, 51, 9950–9955 CrossRef CAS PubMed.
- S. Seth, G. Savitha and J. N. Moorthy, Inorg. Chem., 2015, 54, 6829–6835 CrossRef CAS.
- B. Liu, S. Yao, C. Shi, G. Li, Q. Huo and Y. Liu, Chem. Commun., 2016, 52, 3223–3226 RSC.
- H.-P. Li, S.-N. Li, H.-M. Sun, M.-C. Hu, Y.-C. Jiang and Q.-G. Zhai, Cryst. Growth Des., 2018, 18, 3229–3235 CrossRef CAS.
- P.-Z. Li, X.-J. Wang, J. Liu, J. S. Lim, R. Zou and Y. Zhao, J. Am. Chem. Soc., 2016, 138, 2142–2145 CrossRef CAS PubMed.
- V. Gupta and S. K. Mandal, Chem. – Eur. J., 2020, 26, 2658–2665 CrossRef CAS.
- M. J. Go, K. M. Lee, C. H. Oh, Y. Y. Kang, S. H. Kim, H. R. Park, Y. Kim and J. Lee, Organometallics, 2013, 32, 4452–4455 CrossRef CAS.
- P. Cui, Y.-G. Ma, H.-H. Li, B. Zhao, J.-R. Li, P. Cheng, P. B. Balbuena and H.-C. Zhou, J. Am. Chem. Soc., 2012, 134, 18892–18895 CrossRef CAS PubMed.
- T. Panda, P. Pachfule, Y. Chen, J. Jiang and R. Banerjee, Chem. Commun., 2011, 47, 2011–2013 RSC.
- G.-D. Wang, Y.-Z. Li, W.-J. Shi, L. Hou, Z. Zhu and Y.-Y. Wang, Inorg. Chem. Front., 2020, 7, 1957–1964 RSC.
- A. Paul, I. M. Garazade, A. Karmakar, R. A. Khan, M. F. C. Guedes da Silva, A. V. M. Nunes and A. J. L. Pombeiro, Catalysts, 2024, 14, 6 CrossRef CAS.
- J. Riedmaier, C. Maichle-Mössmer and R. Anwander, Inorg. Chem., 2025, 64, 22238–22250 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |