
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDevelopment of tin oxide catalysts promoting the ring-closing depolymerization of poly(ε-caprolactone) under mild conditions
Tetsu Nakataniab,
Kohei Mishimaa,
Sho Yamaguchi c,
Akira Miurad,
Takato Mitsudome*ae and
Tomoo Mizugaki*aef
c,
Akira Miurad,
Takato Mitsudome*ae and
Tomoo Mizugaki*aef
aDepartment of Materials Engineering Science, Graduate School of Engineering Science, The University of Osaka, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan. E-mail: mitsudom@cheng.es.osaka-u.ac.jp; mizugaki.tomoo.es@osaka-u.ac.jp
bFunctional Products BU, Smart SBU, Daicel Corporation, 1239 Shinzaike, Aboshi-ku, Himeji, Hyogo 671-1283, Japan
cDepartment of Chemical Science and Engineering, Graduate School of Engineering, Kobe University, 1-1 Rokkodai, Nada-ku, Kobe 657-8501, Japan
dFaculty of Engineering, Hokkaido University, Kita 13-jo, Nishi 8-chome, Kita-ku, Sapporo, Hokkaido 060-8628, Japan
eInnovative Catalysis Science Division, Institute for Open and Transdisciplinary Research Initiatives (ICS-OTRI), The University of Osaka, Suita, Osaka 565-0871, Japan
fResearch Center for Solar Energy Chemistry, Graduate School of Engineering Science, The University of Osaka, 1-3 Machikaneyama, Toyonaka, Osaka 560-8531, Japan
First published on 23rd January 2026
Abstract
An effective heterogeneous catalyst for the depolymerization of poly(ε-caprolactone) into ε-caprolactone was demonstrated using a tailored SnO catalyst. The yield of ε-caprolactone reached up to 94% under mild conditions. The catalyst also exhibited excellent durability and scalability. The high catalytic performance was attributed to the formation of SnO nanoparticles generated by calcination, which provided increased surface area and accessible Sn2+ sites.
Plastic pollution has emerged as a major global environmental issue due to its persistence, widespread distribution, and harmful effects on ecosystems and human health.1–6 To address this growing concern, closed-loop chemical recycling, which enables the recovery of monomers from end-of-life plastics, has attracted increasing attention as a sustainable solution that complements or surpasses conventional degradation-based approaches.7–12
Poly(ε-caprolactone) (PCL), typically synthesized via ring-opening polymerization (ROP) of ε-caprolactone (CL), is of particular interest as a green plastic because of its mechanical properties such as flexibility and durability, along with high biodegradability, and potential biomedical applications.13–16 However, unmanaged biodegradation of PCL in the environment can lead to environmental risks, including microbial proliferation and greenhouse gas emissions.17–19 Furthermore, the production of CL from fossil-derived resources relies on carbon-intensive processes that emit substantial greenhouse gases and generate acidic waste.20 Such concerns highlight the need for efficient chemical recycling strategies to depolymerize PCL back into CL. These approaches would promote resource circularity, and reduce the environmental footprint of PCL-based materials.21–23
Ring-closing depolymerization (RCDP) of PCL to CL is in thermodynamic equilibrium with the ROP. RCDP is significantly less favourable than ROP due to the instability of the seven-membered ring structure of CL. The equilibrium conversion of CL at 180 °C is estimated to be 15.4% in toluene at 1 M concentration and significantly lower under bulk conditions.24 Scheme 1 and Table S1 provide a concise summary of previous key research efforts.
 | ||
| Scheme 1 Comparison of previously reported methods and this work for the RCDP of PCL to CL. | ||
Herrera-Kao et al. reported that temperatures exceeding 430 °C are required for effective RCDP of PCL.25 On the other hand, continuous removal of CL by reactive distillation enables the equilibrium shift toward depolymerization. However, at lower temperatures, the depolymerization rate becomes extremely slow, requiring prolonged reaction times. The use of catalysts such as Sn(Oct)2, Bu2Sn(OMe)2, and Mg(HMDS)2 enables high yields of CL within 4.5 hour even at relatively low temperatures below 270 °C under pressures below 20 kPa.22,26,27 Recently, Gallin et al. reported the lower temperature RCDP of PCL at 160 °C by combining ZnCl2 catalyst with ethylene glycol-based additives, achieving a 98% yield of CL under 0.1 torr vacuum.28
However, all these advances rely exclusively on thermally and chemically unstable homogeneous catalysts, which suffer from limited recyclability and the risk of product contamination by catalyst degradation residues. In contrast, heterogeneous catalysts offer advantages in reusability and operational simplicity, making them promising for sustainable depolymerization systems.
In this study, we report that SnO nanoparticles (NPs) serve as a heterogeneous catalyst for RCDP of PCL in the presence of polyethylene glycol (PEG), achieving up to 94% yield of CL under mild conditions. The SnO NPs catalyst also demonstrated excellent durability, including high reusability and scalability.
Screening of various metal oxides was conducted using PEG (average molecular weight 400 g mol−1) as the additive in sealed reaction vessels at 180 °C for 16 h (Fig. 1a). The reaction was carried out under autogenous pressure to suppress vaporization effects, and ensure consistent comparison of catalytic activity. Under these conditions, all catalysts predominantly produced PEG-CL rather than free CL, which represents the transesterification step involved in the depolymerization pathway (Scheme 2). Among the metal oxides tested, tin(II) oxide (SnO) and magnesium oxide (MgO) afforded PEG-CL in 12% and 16% yield, respectively, whereas other metal oxides —ZnO, TiO2, Al2O3, and SnO2—resulted in low yields (<10%). Although SnO appeared less effective than MgO on a weight basis, Table S2 suggests that its activity per mole of metal could be superior to that of MgO. Subsequently, calcination effects of SnO and MgO were investigated by pre-calcining the oxides in air at various temperatures for 1 h prior to the reaction. Hereafter, SnO and MgO calcined at XXX °C are denoted as SnO-XXX and MgO-XXX, respectively. Notably, the calcined SnO exhibited a clear volcano-type catalytic trend in the 300–700 °C range, while MgO showed negligible improvement in PEG-CL yield regardless of the calcination temperature (Fig. 1b). The activity of SnO increased with calcination temperature and SnO-500 achieved a maximum PEG-CL yield of 70%. The PEG-CL yield further increased to 83% by extending reaction time to 24 h under the same conditions, indicating that the reaction approached equilibrium (Fig. S3).
 | ||
| Fig. 1 Catalytic performance of SnO catalysts for the depolymerization of PCL with PEG. General reaction conditions: catalyst (0.25 g), PCL (Mw 10 kg mol−1, 0.25 g), PEG (Mw = 400 g mol−1, 3.60 g), Ar (0.3 MPa), 180 °C, 16 h. (a) Comparison of various metal oxides under non-vacuum condition. (b) Effect of calcination temperature under non-vacuum condition. (c) Reuse experiment using SnO-500 under non-vacuum condition (reaction time: 24 h). (d) Product distribution using SnO-500 under non-vacuum and vacuum conditions for 16 h. (e) Gram-scale experiment using SnO-500 under vacuum condition: SnO-500 (0.25 g, 4 mol% Sn to PCL monomer unit), PCL (Mw 10 kg mol−1, 5.00 g), PEG (Mw = 400 g mol−1, 3.6 g), 180 °C, 24 h. | ||
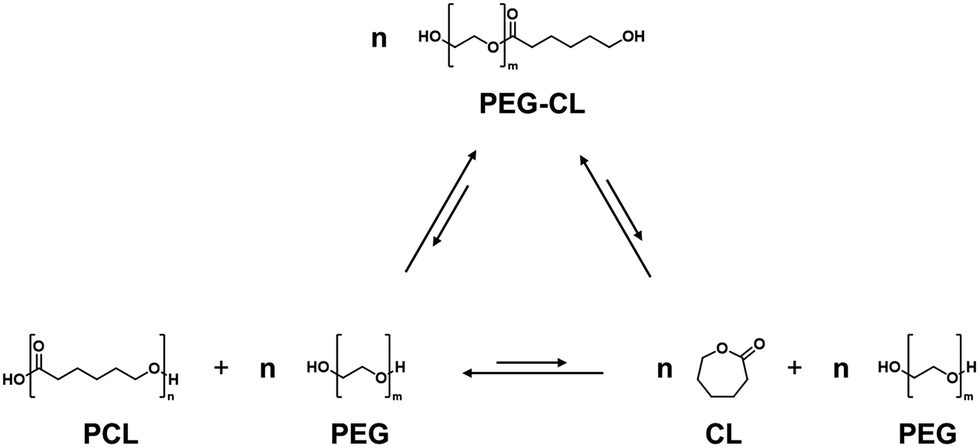 | ||
| Scheme 2 Equilibrium relationships driven by transesterification among PCL, CL, and PEG-CL species in a closed system with excess PEG. | ||
After the reaction, the used SnO-500 catalyst was readily recovered by simple filtration and reused for three consecutive cycles without significant loss of activity, demonstrating high durability (Fig. 1c). A hot filtration test was conducted by terminating the reaction at 6 h and removing the catalyst, after which the reaction was continued for an additional 10 h under identical conditions. The CL yield remained nearly constant at 42% (6 h) and 44% (16 h), supporting that the depolymerization of PCL proceeds under heterogeneous conditions (see Fig. S4).
The SnO-500 catalyst was then employed for PCL depolymerization under vacuum to obtain free CL. Remarkably, CL was obtained as a distillate in an excellent yield of 94% (Fig. 1d). To the best of our knowledge, SnO-500 represents the first example of a heterogeneous catalyst capable of promoting the selective depolymerization of PCL into CL. Moreover, this catalyst system was 20-fold scalable: 5 g of PCL efficiently transformed to CL with an 83% yield, confirming the robustness of SnO-500 (Fig. 1e). These results demonstrate that SnO-500 catalyzed depolymerization of PCL to CL is a highly active, durable, and scalable heterogeneous catalyst system under mild conditions.
To gain insight into the calcination effect on SnO, its structural evolution was monitored by in-situ synchrotron X-ray diffraction (SXRD) during heating at 80 °C min−1 from 60 to 800 °C in air (Fig. 2a). The left figure shows SXRD patterns at 100 °C intervals, while the right figure presents a heatmap of diffraction intensity changes as a function of temperature and diffraction angle. Below 500 °C, peaks corresponding exclusively to tetragonal SnO were observed (see details in Fig. S5). Peaks attributed to rutile-type tetragonal SnO2 appeared above 440 °C and became dominant above 700 °C, SnO undergoes disproportionation upon heating, producing metallic Sn and SnO2 (see details in Fig. S6).29,30 These results indicate that SnO is the catalytically active species, while the formation of SnO2 leads to a significant loss of activity in the PCL depolymerization.
 | ||
| Fig. 2 Structural and textural changes of SnO induced by calcination investigated using in situ SXRD, nitrogen physisorption, and crystal structure analysis. (a) In situ SXRD patterns (left), and heatmap of diffraction intensity changes (right). (b) BJH pore size distribution curves of SnO at different calcination temperatures. (c) BET surface area of SnO at different calcination temperature, (d) Crystal structure of tetragonal SnO (P4/nmm). | ||
Although SXRD showed that both uncalcined SnO and SnO-500 samples consisted of SnO, their catalytic activities were markedly different. To investigate the origin of this difference, Fig. 2b and c compare the nitrogen adsorption–desorption results including BJH pore size distribution curves and BET surface area values for uncalcined and calcined SnO. The calcination induced the formation of small pores (<5 nm). It is noted that a significant increase in mesoporous structures (2–100 nm) was observed at temperatures above 500 °C (highlighted yellow area in Fig. 2b). Correspondingly, the BET surface area increased between 400 and 500 °C (Fig. 2c). These results indicate that calcination produces SnO nanoparticles with micro- and mesoporous structures, leading to the enhanced catalytic activity in PCL depolymerization.31
SnO nanoparticles generated from calcination exhibited high activity for PCL depolymerization in PEG solvent, which can be ascribed to their crystal structure (Fig. 2d). The lone pair on Sn2+ induces a pseudo-layered arrangement that exposes Sn layers on the surface.32,33 These surface-exposed Sn sites are accessible to the bulky PCL molecules and can act as Lewis acid centers that activate the carbonyl or hydroxyl moieties of PCL and PEG-CL, thereby facilitating the depolymerization.34–37
In conclusion, this study demonstrates the first heterogeneous catalyst system capable of depolymerizing PCL into CL under mild conditions. The use of calcined SnO with PEG promotes PEG-CL intermediate formation, offering alternative to conventional high-temperature, energy-intensive methods, while also providing advantages in reusability and operational simplicity. These results highlight a promising direction toward practical, scalable, and environmentally responsible recycling of biodegradable polymers.
T. N. and K. M. designed the experiments, conducted the catalytic activity tests, and characterized the catalysts. A. M. performed SXRD analysis. T. Miz. discussed the experiments and results. T. N. and T. Mit. directed the project and wrote the manuscript with input from all the authors. All authors commented critically on the manuscript and approved the final manuscript.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: the optimization process of experimental conditions, specific procedures for the reaction, relevant characterization data of products, and data from the calculation section are all included in the supplementary information files. See DOI: https://doi.org/10.1039/d5cc06942d.Acknowledgements
This research work was supported by the Daicel-Engineering Science Collaborative Laboratory. This work was also supported by JST-CREST (G. No. JPMJCR21L5) and JSPS KAKENHI grants (G. No. 23H01761 and 24K01255). Part of the experimental analysis was supported by the “Advanced Research Infrastructure for Materials and Nanotechnology in Japan (ARIM)” (Program for supporting construction of core facilities) (G. No. JPMXS0441200025) of the Ministry of Education, Culture, Sports, Science, and Technology (MEXT), Japan.References
- M. MacLeod, H. P. H. Arp, M. B. Tekman and A. Jahnke, Science, 2021, 373, 61–65 CrossRef CAS PubMed.
- R. Meys, A. Kätelhön, M. Bachmann, B. Winter, C. Zibunas, S. Suh and A. Bardow, Science, 2021, 374, 71–76 CrossRef CAS PubMed.
- R. A. Sheldon and M. Norton, Green Chem., 2020, 22, 6310–6322 RSC.
- R. Geyer, J. R. Jambeck and K. L. Law, Sci. Adv., 2017, 3, e1700782 CrossRef PubMed.
- S. J. Borah, A. K. Gupta, A. Gupta, B. Bhawna, S. Kumar, R. Sharma, R. Kumar, P. Kumar, K. K. Dubey, S. Kaushik, A. K. Mishra and V. Kumar, J. Mater. Sci., 2023, 58, 12899–12928 CrossRef CAS.
- Y. Li, L. Tao, Q. Wang, F. Wang, G. Li and M. Song, Environ. Health, 2023, 1, 249–257 CAS.
- H. A. Alrazen, S. M. Aminossadati, H. A. Mahmood, A. K. Hussein, K. A. Ahmad, S. S. Dol, S. Jabbar, S. J. M. Algayyim, M. Konarova and I. M. R. Fattah, Mater. Renew. Sustainable Energy, 2025, 14, 50 CrossRef.
- X. Zhang, M. Fevre, G. O. Jones and R. M. Waymouth, Chem. Rev., 2018, 118, 839–885 CrossRef CAS PubMed.
- K. L. Law and R. Narayan, Nat. Rev. Mater., 2022, 7, 104–115 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- M. Hong and E. Y.-X. Chen, Green Chem., 2017, 19, 3692–3706 RSC.
- T. Uekert, A. Singh, J. S. DesVeaux, T. Ghosh, A. Bhatt, G. Yadav, S. Afzal, J. Walzberg, K. M. Knauer, S. R. Nicholson, G. T. Beckham and A. C. Carpenter, ACS Sustainable Chem. Eng., 2023, 11, 965–978 Search PubMed.
- R. A. Gross and B. Kalra, Science, 2002, 297, 803–807 CrossRef CAS PubMed.
- M. A. Ntrivala, A. C. Pitsavas, K. Lazaridou, A. Baziakou, D. Karavasili, M. Papadimitriou, G. Ntagkopoulou, E. Balla and D. N. Biliaris, Eur. Polym. J., 2025, 234, 114033 CrossRef CAS.
- M. Suzuki, Y. Tachibana and K. Kasuya, Polymer J., 2021, 53, 47–66 Search PubMed.
- N. Shin, S. H. Kim, J. Y. Cho, J. H. Hwang, H. J. Kim, S. J. Oh, S.-H. Park, K. Park, S. K. Bhatia and Y.-H. Yang, J. Polym. Environ., 2024, 32, 898–912 CrossRef CAS.
- Z. Piao, A. A. A. Boakye and Y. Yao, Nat. Chem. Eng., 2024, 1, 661–669 CrossRef.
- A. Keyes, C. M. Saffron, S. Manjure and R. Narayan, Polymers, 2024, 16, 3073 Search PubMed.
- K. Akash, R. Parthasarathi, J. K. Bharath and R. Elango, Water Air Soil Pollut., 2025, 236, 786 CrossRef CAS.
- L. M. Bernhard and H. Gröger, ChemSusChem, 2024, 17, e202400073 CrossRef CAS PubMed.
- G. W. Coates and Y. D. Y. L. Getzler, Nat. Rev. Mater., 2020, 5, 501–516 CrossRef CAS.
- C. Cai, J. Ma, X. Liang, S. Zhang, H. Zhang, C. Zhang and S. Zhang, Polym. Chem., 2025, 16, 1568–1577 RSC.
- R. Yang, W. Wei, G. Xu, X. Guo and Q. Wang, Angew. Chem., Int. Ed., 2025, 64, e202504819 CrossRef CAS PubMed.
- P. Olsén, K. Odelius and A.-C. Albertsson, Biomacromolecules, 2016, 17, 699–709 CrossRef PubMed.
- W. A. Herrera-Kao, M. I. Loría-Bastarrachea, Y. Pérez-Padilla, J. V. Cauich-Rodríguez, H. Vázquez-Torres and J. M. Cervantes-Uc, Polym. Bull., 2018, 75, 4191–4205 CrossRef CAS.
- M. Nelißen, H. Keul and H. Höcker, Macromol. Chem. Phys., 1995, 196, 1645–1661 CrossRef.
- J. Sun, G. Xu, B. Du, R. Yu, H. Sun and Q. Wang, Polym. Chem., 2022, 13, 5897–5904 RSC.
- C. F. Gallin, W.-W. Lee and J. A. Byers, Angew. Chem., Int. Ed., 2023, 62, e202303762 Search PubMed.
- H. Giefers, F. Porsch and G. Wortmann, Solid State Ionics, 2005, 176, 199–207 CrossRef CAS.
- C. M. Campo, J. E. Rodríguez and A. E. Ramírez, Heliyon, 2016, 2, e00112 CrossRef PubMed.
- L. Interrante, S. Bensaid, C. Galletti, R. Pirone, B. Schiavo, O. Scialdone and A. Galia, Fuel Process. Technol., 2018, 177, 336–344 CrossRef CAS.
- S. Jaśkaniec, S. R. Kavanagh, J. Coelho, S. Ryan, C. Hobbs, A. Aalsh, D. O. Scanlon and V. Nicolosi, npj 2D Mater. Appl., 2021, 5, 27 Search PubMed.
- C. S. Lim, Z. Sofer, O. Jankovský, H. Wang and M. Pumera, RSC Adv., 2016, 6, 77538–77547 Search PubMed.
- D. K. Yoo, D. Kim and D. S. Lee, Macromol. Res., 2006, 14, 510–516 Search PubMed.
- M. Ryner, K. Stridsberg, A.-C. Albertsson, H. von Schenck and M. Svensson, Macromolecules, 2001, 34, 3877–3881 Search PubMed.
- Z. Liu, S. Hu, Q. Hao, R. Ma, Y. Zhang, B. Wu and J. Zhu, Microporous Mesoporous Mater., 2025, 397, 113787 CrossRef CAS.
- H. Su, Q. Zhao, C. Jiang, Y. Wang, Y. Niu, X. Li, W. Lou, Y. Qi and X. Wang, RSC Adv., 2023, 13, 8934–8941 RSC.
| This journal is © The Royal Society of Chemistry 2026 |