
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C7-Sulfonamide functionalization of 7-deazaadenosines: sangivamycin analogues with Haspin inhibitory activity
Milan
Štefek
 ab,
Milan
Dejmek
a,
Michal
Šála
a and
Radim
Nencka
*a
ab,
Milan
Dejmek
a,
Michal
Šála
a and
Radim
Nencka
*a
aInstitute of Organic Chemistry and Biochemistry of the Czech Academy of Sciences, Flemingovo náměstí 2, Prague 6 166 10, Czech Republic. E-mail: radim.nencka@uochb.cas.cz
bDepartment of Organic Chemistry, Faculty of Science, Charles University, Prague 128 00, Czech Republic
First published on 26th December 2025
Abstract
We report a robust method for C7-sulfonamide installation on 7-deazaadenosines, enabled by an unprecedentedly stable sulfonyl chloride. This general transformation introduces a new functionalization site and expands nucleoside chemical space, offering a powerful platform for constructing tailored probes and analogues in synthetic and bioorganic chemistry.
Nucleoside analogues are indispensable tools in medicinal chemistry and chemical biology, serving as both therapeutic agents and molecular probes. Structural modification of nucleosides has yielded numerous antiviral and anticancer drugs.1 Pyrrolo[2,3-d]pyrimidine (7-deazapurine) nucleosides form a class of compounds closely mimicking purine nucleosides, and naturally occurring 7-deazaadenosine (tubercidin) (1), sangivamycin (2) and toyocamycin (3) (Fig. 1) display potent cytotoxic activities, making this scaffold highly attractive for further derivatization.2
 | ||
| Fig. 1 Natural bioactive 7-deazaadenosines. | ||
Replacement of N7 by carbon in 7-deazapurines generates a unique substitution site at C7. Halo, (het)aryl, alkynyl, alkenyl, and alkyl substituents at this position have been extensively explored and shown to modulate the biological properties of the respective molecules.3–5 Our group has recently demonstrated that C7 substitution can be utilized to enhance activity and selectivity against several methyltransferase targets.6–10 In contrast, heteroatom-linked substituents remain underexplored. While a C7-carboxamide is occasionally present in sangivamycin analogues, it has rarely been used as a linker for further functionalization.11
Sangivamycin itself has attracted longstanding interest. Initially studied as an anticancer nucleoside,12,13 it was later shown to possess broad antiviral activity,14–16 and to inhibit protein kinase C.17 More recently, it was identified as a potent inhibitor of Haspin kinase,18 a serine/threonine kinase essential for mitotic chromosome alignment, which is overexpressed in malignant tissue and is responsible for cancer cell proliferation.19 Inhibition of Haspin by sangivamycin has been linked to pronounced anticancer effects, including induction of cell death in pancreatic cancer cell lines and tumor regression in vivo.18
In our exploration of 7-substituted-7-deazaadenosine derivatives, we identified C7-sulfonamides as an unexplored yet desirable modification. Sulfonamides are privileged motifs in drug discovery, valued for their polarity, hydrogen-bonding capacity, and metabolic stability, and they occur in numerous clinical agents.20 Despite their widespread use in medicinal chemistry,21,22 C7-sulfonamide substitution of 7-deazapurines has not been reported. We therefore set out to establish a general method for their synthesis, demonstrate its applicability across diverse nucleoside scaffolds, and assess the biological activity of the resulting analogues.
Sulfonamides have been introduced to other positions of purine nucleosides, notably C2 and C6, yielding derivatives with antitumor and antiviral activities.23–25 However, C7 substitution of 7-deazapurines with sulfonamides has not been described, leaving this modification unexplored.
Despite the availability of numerous approaches to sulfonamide synthesis, the use of sulfonyl chlorides remains to be the preferred method because of their reliable reactivity and broad substrate compatibility.26–28 The C2 and C6 chlorosulfonyl-purines have previously been described as unstable, prone to hydrolysis or substitution by chloride anions. Yet, they are still employed as reactive intermediates in the synthesis of sulfonyl fluorides.29,30 Considering the different electron properties of the 7-deazapurine core, its use was expected to offer improved stability, providing modular access to previously inaccessible derivatives. Herein we report the synthesis and preliminary evaluation of novel sulfonamide analogues of sangivamycin, designed to retain the key pharmacophore features of the parent compound while exploring the impact of this functional group on biological activity.
Our synthesis commenced with benzoyl-protected 6-chloro-7-iodo-7-deazaadenosine (4) (Scheme 1). Conversion of 4 to amine 5 followed by benzylthio substitution furnished intermediate 6, and oxidative chlorination with N-chlorosuccinimide in aqueous acetic acid produced the key sulfonyl chloride 7. Notably, compound 7 could be isolated and stored at room temperature for at least six months without special precautions, in striking contrast to the instability of purine sulfonyl chlorides. This enhanced stability is likely a consequence of the higher electron density of the 7-deazapurine core relative to purine,2,31 which makes the sulfonyl chloride less prone to hydrolysis or to decomposition via chloride displacement—pathways previously observed for purine-derived sulfonyl chlorides.29
 | ||
Scheme 1 Synthesis of key intermediate 7 and preparation of sulfonamide sangivamycin analogues. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Reaction conditions: 9 (1.0 eq.), amine (1.2 eq.), DMAP (1.2 eq.) in DCM (0.1 M) at rt (see SI for more details). bIsolated yields are reported. c7 M ammonia in methanol at 60 °C, overnight. dSulfonamide intermediate was not isolated. e33% MeNH2 in ethanol at rt, 6 h. f40% Me2NH (aq) (5.0 eq.) in DCM at rt, 10 minutes, then 33% MeNH2 in ethanol at rt, 6 h. gt-Butylamine (2.5 eq.) at rt, overnight. hCyclopropylamine (3.0 eq.) at RT, 40 minutes. i2.4 eq. of DMAP were used. jAmine (2.2 eq.) at rt, overnight. k0% TFA in DCM at RT, 90 minutes. lMorpholine (3.0 eq.) at RT, 10 minutes. mAmine (1.3 eq.) at 0 °C, 10 minutes. n1-Boc-4-(aminomethyl)piperidine (1.2 eq.), DMAP (1.2 eq.) in DCM (0.1 M) at rt, 15 minutes, then 10% TFA in DCM at rt, 1 hour. oAniline (3 eq.) and DMAP (1.2 eq.) at rt, 1 hour. pAzetidine-3-carboxylic acid (1.2 eq.), Na2CO3 (6 eq.) in THF/water (1:1) mixture at rt, 1 hour. Reaction conditions: 9 (1.0 eq.), amine (1.2 eq.), DMAP (1.2 eq.) in DCM (0.1 M) at rt (see SI for more details). bIsolated yields are reported. c7 M ammonia in methanol at 60 °C, overnight. dSulfonamide intermediate was not isolated. e33% MeNH2 in ethanol at rt, 6 h. f40% Me2NH (aq) (5.0 eq.) in DCM at rt, 10 minutes, then 33% MeNH2 in ethanol at rt, 6 h. gt-Butylamine (2.5 eq.) at rt, overnight. hCyclopropylamine (3.0 eq.) at RT, 40 minutes. i2.4 eq. of DMAP were used. jAmine (2.2 eq.) at rt, overnight. k0% TFA in DCM at RT, 90 minutes. lMorpholine (3.0 eq.) at RT, 10 minutes. mAmine (1.3 eq.) at 0 °C, 10 minutes. n1-Boc-4-(aminomethyl)piperidine (1.2 eq.), DMAP (1.2 eq.) in DCM (0.1 M) at rt, 15 minutes, then 10% TFA in DCM at rt, 1 hour. oAniline (3 eq.) and DMAP (1.2 eq.) at rt, 1 hour. pAzetidine-3-carboxylic acid (1.2 eq.), Na2CO3 (6 eq.) in THF/water (1:1) mixture at rt, 1 hour. | ||
Sulfonyl chloride 7 reacted smoothly with diverse amines to deliver sulfonamides (8a–p) in moderate to excellent yields (Scheme 1). Ammonia, primary and secondary aliphatic amines, bulky substituents, and electron-deficient amines were all usable. Piperidines bearing hydroxymethyl or aminomethyl substituents furnished exclusively tertiary sulfonamides, showing selectivity for secondary amines.
Application of this methodology to valuable amines or their salts required the identification of a suitable sacrificial base. Optimization (see SI, Table S1) revealed, that N,N-dimethylaminopyridine (DMAP) was the most effective additive, acting as both a proton scavenger and catalyst. By contrast, TEA, DBU, and pyridine promoted side reactions, while K2CO3 in DMF led exclusively to sulfonic acid, most likely via reaction of the sulfonyl chloride with the solvent to form a transient iminium salt.32 A biphasic DCM/water system with K2CO3 gave acceptable yields only after prolonged stirring, underscoring the hydrolytic stability of the sulfonyl chloride.
While most tested amines provided the corresponding sulfonamides in good to excellent yields, several substrates proved incompatible with sulfonyl chloride 7. Electron-poor amines such as 3-aminopyridine, 4-aminopyridine, and 2-aminothiazole afforded only complex mixtures (data not shown), whereas diphenylamine was insufficiently reactive and did not yield any detectable sulfonamide product (see SI, Table S1). These observations indicate that strongly electron-deficient or weakly nucleophilic amines represent practical limitations of the current methodology.
Prepared secondary sulfonamides were amenable to further derivatization, as demonstrated with compound 8p (Scheme 2). Alkylation under Mitsunobu conditions furnished benzylated compound 10 in excellent yield, while 12, unavailable via direct sulfonation of diphenylamine, was obtained through Cu-catalysed coupling with Ph2IOTf.33 Final deprotection using MeNH2 provided the free nucleosides.
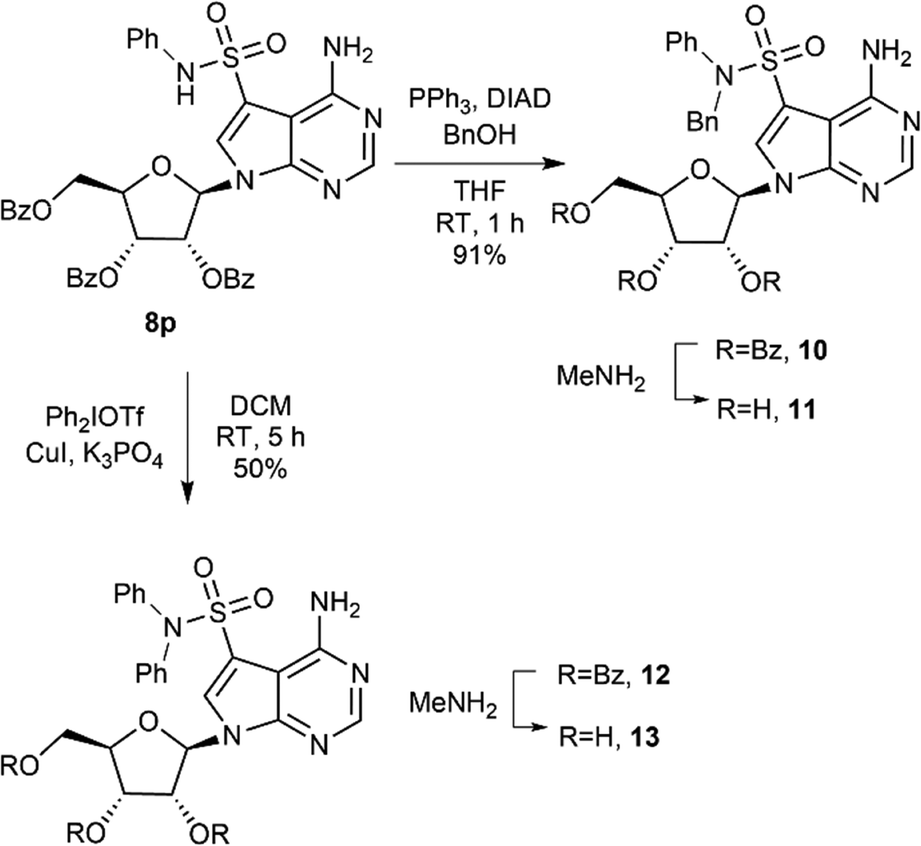 | ||
| Scheme 2 N-Functionalization of sulfonamide 8p. | ||
Importantly, the methodology proved highly general. Beyond the sangivamycin analogue, it was successfully extended to multiple nucleoside scaffolds, including 2-substituted-7-deazaadenosines (14, 15), pseudoadenosine with a pyrrolo[2,1-f]triazin core (16), 2′-modified nucleosides (17, 18), and a carbocyclic nucleoside (19) (Fig. 2). This highlights the robustness of the transformation and establishes C7-sulfonamides as a versatile platform for nucleoside diversification.
 | ||
| Fig. 2 Scope of C7-sulfonamide nucleosides. The transformation is compatible with diverse scaffolds. | ||
Selected compounds were evaluated for inhibition of Haspin kinase (Table 1). Four analogues showed double-digit nanomolar IC50 values. The most potent, 9a, inhibited Haspin at 21 nM, which is comparable to 7 nM value for sangivamycin. The IC50 data also allowed a basic SAR analysis. Changing the nucleobase core (compound 16) afforded an analogue with similar potency, suggesting that the central heterocycle can tolerate certain modifications. Introduction of substituents on the sulfonamide nitrogen (9b, 9c) resulted in only a minor loss of activity, indicating that this moiety does not engage in crucial interactions with the protein. In contrast, modifications of the ribose portion of the nucleoside (17, 18, 19) led to substantially reduced inhibitory activity, and substitution at the C2 position produced compounds (14, 15) with no detectable inhibition of Haspin kinase. These observations imply that the binding site is highly constrained around the C2 region and cannot accommodate additional substituents. To assess its selectivity, 9a was profiled in a 63-kinase panel. Comparison with sangivamycin revealed that 9a possesses an altered selectivity profile. Whereas sangivamycin inhibited several kinases in the panel, including PKCδ, YSK4, KDR, DYRK1A, and DYRK2, compound 9a showed reduced activity toward these off-targets but displayed measurable inhibition of AMPKα1 and RSK1 instead (SI, Table S2). Overall, 9a retains potent Haspin inhibition while exhibiting a distinct and somewhat narrower pattern of off-target interactions (see SI, Table S2).
| Compound | Haspin IC50 (nM) |
|---|---|
| Sangivamycin | 7 |
| 9a | 21 |
| 9b | 27 |
| 9c | 55 |
| 14 | >10000 |
| 15 | >10000 |
| 16 | 30 |
| 17 | 354 |
| 18 | 344 |
| 19 | 1708 |
In summary, we report the first synthesis of C7-sulfonamide-7-deazaadenosines enabled by a uniquely stable sulfonyl chloride intermediate. The transformation is general, applicable to a wide range of amines and extending across multiple nucleoside scaffolds to provide derivatives readily suited for further modification. Several of the synthesized compounds exhibited potent low-nanomolar Haspin inhibition with high selectivity against other kinases. These findings establish C7-sulfonamides as a new and versatile entry point for nucleoside diversification with direct relevance to medicinal chemistry.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc06746d.Acknowledgements
This research was funded by the project New Technologies for Translational Research in Pharmaceutical Sciences/NETPHARM, project ID CZ.02.01.01/00/22_008/0004607, which is co-funded by the European Union. Also, the project was supported by the Academy of Sciences of the Czech Republic as part of the Strategy AV 21 Virology and Antiviral Therapy programme; the Czech Academy of Sciences (RVO: 61388963).References
- K. L. Seley-Radtke and M. K. Yates, Antiviral Res., 2018, 154, 66–86 CrossRef.
- P. Perlikova and M. Hocek, Med. Res. Rev., 2017, 37, 1429–1460 CrossRef.
- A. Bourderioux, P. Nauš, P. Perlíková, R. Pohl, I. Pichová, I. Votruba, P. Džubák, P. Konečný, M. Hajdúch, K. M. Stray, T. Wang, A. S. Ray, J. Y. Feng, G. Birkus, T. Cihlar and M. Hocek, J. Med. Chem., 2011, 54, 5498–5507 CrossRef.
- M. Ondruš, V. Sýkorová, L. Bednárová, R. Pohl and M. Hocek, Nucleic Acids Res., 2020, 48, 11982–11993 CrossRef PubMed.
- F. Hulpia, G. D. Campagnaro, K. J. Alzahrani, I. A. Alfayez, M. A. Ungogo, D. Mabille, L. Maes, H. P. de Koning, G. Caljon and S. Van Calenbergh, ACS Infect. Dis., 2020, 6, 2045–2056 Search PubMed.
- T. Otava, M. Šála, F. Li, J. Fanfrlík, K. Devkota, S. Perveen, I. Chau, P. Pakarian, P. Hobza, M. Vedadi, E. Boura and R. Nencka, ACS Infect. Dis., 2021, 7, 2214–2220 CrossRef.
- M. Štefek, D. Chalupská, K. Chalupský, M. Zgarbová, A. Dvořáková, P. Krafčíková, A. S. M. Li, M. Šála, M. Dejmek, T. Otava, E. Chaloupecká, J. Kozák, J. Kozic, M. Vedadi, J. Weber, H. Mertlíková-Kaiserová and R. Nencka, ACS Omega, 2023, 8, 27410–27418 CrossRef.
- M. Zgarbová, T. Otava, J. Silhan, R. Nencka, J. Weber and E. Boura, Antiviral Res., 2023, 218, 105714 CrossRef PubMed.
- J. Silhan, M. Klima, T. Otava, P. Skvara, D. Chalupska, K. Chalupsky, J. Kozic, R. Nencka and E. Boura, Nat. Commun., 2023, 14, 2259 CrossRef PubMed.
- H. Kocek, D. Chalupská, M. Dejmek, A. Dvořáková, M. Zgarbová, M. Šála, K. Chalupský, P. Krafčíková, T. Otava, M. Drexler, E. Procházková, B. Klepetářová, M. Štefek, J. Kozic, H. Mertlíková-Kaiserová, E. Boura, J. Weber and R. Nencka, RSC Med. Chem., 2024, 15, 3469–3476 RSC.
- Y. Saito, K. Kato and K. Umezawa, Tetrahedron, 1998, 54, 4251–4266 Search PubMed.
- R. L. Tolman, R. K. Robins and L. B. Townsend, J. Am. Chem. Soc., 1968, 90, 524–526 CrossRef PubMed.
- J. A. Cavins, T. C. Hall, K. B. Olson, C. L. Khung, J. Horton, J. Colsky and R. K. Shadduck, Cancer Chemother. Rep., 1967, 51, 197–200 Search PubMed.
- E. De Clercq, R. Bernaerts, D. E. Bergstrom, M. J. Robins, J. A. Montgomery and A. Holy, Antimicrob. Agents Chemother., 1986, 29, 482–487 CrossRef PubMed.
- N. Kučić, H. Mahmutefendić and P. Lučin, J. Gen. Virol., 2005, 86, 2153–2161 Search PubMed.
- R. P. Bennett, E. N. Postnikova, B. P. Eaton, Y. Cai, S. Yu, C. O. Smith, J. Liang, H. Zhou, G. A. Kocher, M. J. Murphy, H. C. Smith and J. H. Kuhn, JCI Insight, 2022, 7, 105716 Search PubMed.
- C. R. Loomis and R. M. Bell, J. Biol. Chem., 1988, 263, 1682–1692 Search PubMed.
- L. I. Bastea, L. M. A. Hollant, H. R. Döppler, E. M. Reid and P. Storz, Sci. Rep., 2019, 9, 16588 Search PubMed.
- Y. Liu, H. Yang, Y. Fang, Y. Xing, X. Pang, Y. Li, Y. Zhang and Y. Liu, J. Pharm. Pharmacol., 2023, 75, 445–465 Search PubMed.
- C. Giulia, T. Marco, Z. Maria and A. Anna Maria, Curr. Med. Chem., 2023, 30, 128–163 Search PubMed.
- N. A. McGrath, M. Brichacek and J. T. Njardarson, J. Chem. Educ., 2010, 87, 1348–1349 Search PubMed.
- S. Apaydın and M. Török, Bioorg. Med. Chem. Lett., 2019, 29, 2042–2050 Search PubMed.
- K. Ramasamy, N. Imamura, N. B. Hanna, R. A. Finch, T. L. Avery, R. K. Robins and G. R. Revankar, J. Med. Chem., 1990, 33, 1220–1225 CrossRef PubMed.
- G. R. Revankar, N. B. Hanna, N. Imamura, A. F. Lewis, S. B. Larson, R. A. Finch, T. L. Avery and R. K. Robins, J. Med. Chem., 1990, 33, 121–128 CrossRef PubMed.
- G. R. Revankar, N. B. Hanna, K. Ramasamy, S. B. Larson, D. F. Smee, R. A. Finch, T. L. Avery and R. K. Robins, J. Heterocycl. Chem., 1990, 27, 909–918 Search PubMed.
- W. Liu, J. Chen and W. Su, Pharm. Fronts, 2024, 06, e355–e381 Search PubMed.
- A. Muhammad, S. Syed Shoaib Ahmad, N. Tayyaba, S. Salma and R. Gildardo, Mini-Rev. Org. Chem., 2013, 10, 160–170 CrossRef.
- T. K. Pati, S. A. Molla, N. N. Ghosh, M. Kundu, S. Ajarul, P. Maity, U. Khamrai and D. K. Maiti, J. Org. Chem., 2024, 89, 6798–6812 Search PubMed.
- R. K. Robins, J. Org. Chem., 1961, 26, 447–451 CrossRef.
- A. G. Beaman and R. K. Robins, J. Am. Chem. Soc., 1961, 83, 4038–4044 Search PubMed.
- K. E. Richardson and B. M. Znosko, RNA, 2016, 22, 934–942 Search PubMed.
- J. D. Albright, E. Benz, A. E. Lanzilotti and L. Goldman, Chem. Commun., 1965, 413–414, 10.1039/C19650000413.
- X. Geng, S. Mao, L. Chen, J. Yu, J. Han, J. Hua and L. Wang, Tetrahedron Lett., 2014, 55, 3856–3859 CrossRef.
| This journal is © The Royal Society of Chemistry 2026 |