
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetrical capacitive deionization battery enabling dual capture of fluoride and copper ions
Qisheng
Huang†
a,
Meiying
Du†
a,
Lei
Huang
 *ab,
Jia
Yan
a,
Xingtong
Liang
a,
Shaojian
Xie
a,
Hongguo
Zhang
*ac,
Meng
Li
a,
Shaocheng
Chen
a and
Hongbo
Zeng
*b
*ab,
Jia
Yan
a,
Xingtong
Liang
a,
Shaojian
Xie
a,
Hongguo
Zhang
*ac,
Meng
Li
a,
Shaocheng
Chen
a and
Hongbo
Zeng
*b
aSchool of Environmental Science and Engineering, Guangzhou University, Guangzhou 510006, China. E-mail: huanglei@gzhu.edu.cn; hgzhang@gzhu.edu.cn
bDepartment of Chemical and Materials Engineering, University of Alberta, Edmonton, Alberta T6G 1H9, Canada. E-mail: hongbo.zeng@ualberta.ca
cGuangzhou University-Linköping University Research Center on Urban Sustainable Development, Guangzhou University, Guangzhou 510006, China
First published on 7th January 2026
Abstract
This work reports a capacitive deionization battery using lithium iron phosphate (LiFePO4) electrodes. A ‘Deintercalation–Adsorption–Desorption–Intercalation’ (DADI) cycle achieves over 95% removal capacity retention after 20 cycles, offering new insights into electrode materials and mechanisms for capacitive deionization technology.
Semiconductor manufacturing processes typically generate wastewater containing substantial quantities of copper ions and fluoride ions.1 Proper treatment of this wastewater and efficient recovery of valuable ions not only helps mitigate environmental pollution but also enables the recycling of resources.
Capacitive Deionization (CDI) is an emerging advanced water treatment method that operates without the need for chemical additives, thereby avoiding secondary pollution.2 Therefore, it is increasingly becoming an important technology in fields such as desalination, purification, and resource recovery.3–5 This technology's primary principle involves applying an electric field to induce ion electro-migration. Subsequently, ions adsorbed on the electrode surface undergo charge transfer and capacitive capture at the electrode–electrolyte interface, enabling low-energy seawater desalination.6 Although capacitive deionization (CDI) has emerged as a cost-effective and eco-friendly technology for ionic contaminant remediation, most reported CDI systems, either symmetrical or asymmetrical configurations, predominantly focus on the removal of a single target ion. This single-ion remediation paradigm renders conventional CDI technologies inefficient for treating actual wastewater where fluoride and copper ions coexist, as the competitive adsorption behaviour between different ions often leads to compromised removal performance and complicated post-treatment procedures. Against this backdrop, the development of a high-performance symmetrical CDI battery capable of simultaneously removing fluoride and copper ions is urgently needed to address this practical bottleneck and advance the applicability of CDI in complex water treatment scenarios.
Lithium iron phosphate (LiFePO4, abbreviated as LFP) is extensively employed in commercial battery applications owing to its exceptional cycle life, high safety profile, and environmental credentials, conferred by its stable olivine structure. Its unique Li+ intercalation–deintercalation mechanism forms a Fe2+/Fe3+ redox pair, enabling efficient ion migration and capacitive capture.7 This mechanism effectively facilitates ion capture and removal during the CDI process. Kang's research confirms that its outstanding lithium-ion mobility supports ultra-fast charging and discharging processes while maintaining energy density.8 Combined with its exceptional specific capacitance characteristics, LFP demonstrates significant application potential as a CDI electrode material.
This paper presents a low-cost and high-efficiency capacitive deionization battery. By moving beyond the conventional single-ion removal paradigm, the proposed symmetrical CDI battery not only fills the research gap in multi-ion co-remediation but also provides a more practical solution for the treatment of complex industrial wastewater containing both fluoride and copper pollutants.9
It mainly presents a spherical shape at the nanoscale and a strip-like shape at the micrometre scale (Fig. 1(a) and Fig. S1). Energy dispersive X-ray spectroscopy (EDS) scanning reveals that the various elements show almost no agglomeration and present a highly uniform distribution. The X-ray diffraction analysis (PDF #998-000-0443) reveals that the characteristic peaks of LFP (Fig. 1b) correspond to those of the trihydroxy phosphate phase. However, following desalination, the crystallographic peaks of LFP-F exhibit a blue shift (Fig. S2), confirming lattice distortion induced by Li+ extraction and F− insertion. The attenuation of the LFP characteristic peak comes from the partial escape of Li ions, while fluoride ions form amorphous fluoride bonds on the surface, resulting in a decrease in the intensity of its diffraction peak. The weak interaction between the crystal plane LFP and Cu leads to a more pronounced characteristic peak.
 | ||
| Fig. 1 (a) SEM image of LFP at 10 µm resolution, with colours on the right representing the intensity of different atoms. The inset shows a magnified view at 1 µm resolution. (b) and (c) XRD patterns and FT-IR spectra of LFP and the post-reaction products LFP-F and LFP-Cu. (d) N2 adsorption–desorption isotherms (inset image shows BJH pore size distribution profiles). (e) Schematic of the deintercalation–adsorption–desorption–intercalation cycle for LFP and the capacitive deionisation of copper ions. | ||
In the FT-IR image (Fig. 1c), the stretching vibration at 970 cm−1 primarily originates from the [PO4] tetrahedron. Compared to the calculated standard, a red shift has occurred, primarily due to the Li-Fe anti-site defect.10 The 1075 cm−1 peak of LFP-Cu exhibits a blue shift, indicating that partial replacement of Fe2+ by Cu2+ results in a slight increase in the bond length of the Fe–O–P framework, thereby lowering the vibrational frequency. Furthermore, the peak shape in the LFP-F sample is sharper, suggesting higher crystallinity and a more stable [PO4] framework in its structure. After the capacitive deionization process, a blue shift occurs, indicating that the positively charged Li-Fe defects are successfully replaced by fluoride ions, thereby reducing the defect extent.11
The nitrogen adsorption–desorption isotherm of LFP exhibits a typical Type IV adsorption curve (Fig. 1d), with a pronounced hysteresis loop emerging at a relative pressure P/P0 approaching 1.0. Its specific surface area is approximately 9.6785 m2 g−1. This relatively small specific surface area implies that electrode removal primarily relies on pseudocapacitive effects. Meanwhile, LFP exhibits a typical mesoporous morphology. This mesoporous morphology facilitates electrolyte permeation and rapid Li+ diffusion, thereby reducing electrochemical polarization and enhancing the battery's rate performance and cycling stability.
As shown in Fig. 2a, cyclic voltammetry (CV) curves at different scan rates exhibit near-rectangular shapes without distinct redox peaks, indicating the absence of strong faradaic reactions. This suggests that the entire charge storage process is dominated by capacitive behavior, accompanied by reversible ion adsorption/desorption at the electrode–electrolyte interface. At a scan rate of 1 mV s−1, the specific capacitance reaches a maximum value of 140.23 F g−1, confirming highly efficient interfacial charge accumulation.
 | ||
| Fig. 2 (a) CV curves of LFP at scan rates from 1 mV s−1 to 100 mV s−1. (b) Relative contribution ratios of capacitive (blue) and diffusive (red) behaviour to removal at scan rates of 1 mV s−1, 5 mV s−1, and 10 mV s−1. (c) Nyquist plots. (d) GCD curve of LFP after 100 cycles at a current density of 0.2 A g−1. | ||
The contributions of capacitive and diffusion-controlled processes are quantified using the Dunn method (Fig. 2b). At low scan rates, ion storage is predominantly governed by diffusion-controlled processes, whereas at 10 mV s−1, the capacitive contribution becomes dominant.
As the scan rate increases, the capacitive contribution gradually transitions from bulk ion diffusion to surface-controlled pseudocapacitive behavior.12 Electrochemical impedance spectroscopy (EIS) curves (Fig. 2c) exhibit a small semicircle in the high-frequency region and a steep line in the low-frequency region, indicating fast charge transfer dynamics and enhanced ion diffusion within the electrode structure.
The charge–discharge (GCD) curve (Fig. S3) exhibits distinct potential plateaus and a short cycle duration, further indicating that the total capacitance is not primarily generated by faradaic desalination mechanisms. It exhibits a maximum specific capacitance of 30.212 F g−1 at a current density of 0.2 A g−1. Notably, the LFP electrode maintains excellent electrochemical stability after 500 consecutive charge–discharge cycles (Fig. 2d), confirming its robust structural integrity and outstanding reversibility in the fluoride aqueous system.
An intermittent electrochemical system based on symmetrical LFP is established (Fig. S4) to evaluate dual copper removal and fluoride ion storage performance. The fluoride ion removal capacity gradually decreases within the 0–0.6 V range but abruptly increases to 62.7 mg g−1 at 1.2 V, representing a 132.4% improvement over open-circuit conditions. Cu2+ removal exhibits analogous voltage dependence, reaching 182 mg g−1 at 1.2 V (Fig. 3a). Enhanced performance at elevated voltages comes from intensified interfacial polarization, which facilitates ion migration and electrostatic adsorption. Fluoride ions are more prone to forming hydrogen-bonded complexes under acidic conditions and exhibit higher dehydration energies. This renders fluoride ion removal comparatively more challenging than copper ion removal. Consequently, this explains why LFP demonstrates significantly greater removal capacity for copper ions than fluoride ions, as illustrated in Fig. 3a.13 The battery exhibits outstanding activity under weakly acidic conditions, as surface protonation strengthens the Fe–O–P–F bond (Fig. S5). However, excessively high flow rates disrupt the double layer, with 13.8 mm s−1 identified as the optimal flow rate (Fig. S6).14 Kinetic analysis aligns well with the pseudo-second-order model, confirming chemisorption-controlled processes (Fig. S7). SAC increases proportionally with fluoride ion concentration, indicating substantial application potential in high-salinity environments.
 | ||
| Fig. 3 (a) SAC values measured at different voltages. (b) SAC values at different fluoride concentrations. The ion competition effects for anions (c) and cations (d) are shown, respectively. (e) Ragone plots for LFP-F and LFP-Cu. | ||
Fig. 3c illustrates fluoride ion removal capacity under anion competition. Only PO43− significantly suppresses adsorption by preferentially coordinating with Fe sites (Fig. 3c). The addition of other anions has a negligible impact on fluoride removal performance. Concurrently, it demonstrates LFP's removal efficiency under cation competition (Fig. 3d). Cations scarcely affect copper ion removal, maintaining exceptionally high removal efficacy. Ragone plots reveal exceptionally rapid q ion adsorption rates (SARF− = 31.98 mg g−1 h−1; SARCu2+ = 11.65 mg g−1 h−1), with the system retaining 95% capacity after 20 cycles, demonstrating outstanding regeneration stability. LFP shows a superior SAC compared to other electrodes (Fig. S8, S9 and Table S2).
As shown in Fig. 4a, the P 2p peak shifts to lower binding energies with negligible change in peak area, indicating that F−/HF intercalation alters the local chemical environment while the phosphate backbone remains intact. In pristine LFP, Fe exists mainly as Fe3+ with trace Fe2+ (Fig. 4b); after reaction, the enhanced Fe2+ signal confirms that Fe active sites are reduced during F−/HF adsorption, forming stable Fe-F and Fe-HF coordination bonds. This mechanism effectively mitigates the cycling degradation caused by surface deposition in conventional CDI electrodes. The reaction equation is as follows:
 | (1) |
| LiPO4Fe + F− → (LiPO4Fe − F)surface | (2) |
| Fe3+ + F− → Fe − F | (3) |
| Fe3+ + HF → Fe − HF | (4) |
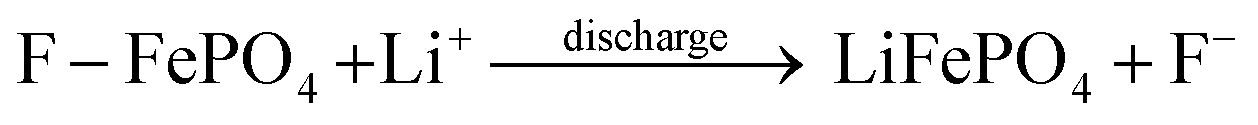 | (5) |
 | ||
| Fig. 4 High-resolution XPS spectra of LFP before and after reaction: (a) P 2p, (b) Fe 2p, (c) O 1s. (d)–(f) Adsorption energies and projected density of states for LFP and the post-reaction products LFP-F and LFP-HF. | ||
In the XPS peak of oxygen (Fig. 4c), the defect oxygen (Ov) peak can also be clearly observed to shift towards lower binding energy, accompanied by a significant increase in its relative concentration.15 The oxygen vacancy releases local stress and electron–ion coupling constraints in one step. This indicates that the removal of fluoride also depends on the intercalation effect of F− and HF.
The removal process of copper ions may be primarily influenced by the double layer effect, accompanied by partial substitution of Fe by Cu (Fig. S10). Electron microscopy of the captured LFP-Cu (Fig. S11) reveals no new grafting in the morphology of either LFP or LFP-Cu, while mapping did not detect extensive copper deposition (elemental copper). Furthermore, no copper lattice is observed in the TEM images of either LFP or LFP-Cu. The reaction equation is as follows:
| LiPO4Fe + Cu2+ → (LiPO4Fe − Cu)surface | (6) |
DFT calculations (Fig. 4d–f and Fig. S12, S13) reveal that the primary adsorption sites for F− and Cu2+ are both located at the Fe site. The corresponding binding energies indicate that the adsorption processes are thermodynamically spontaneous. Projected Density of States (PDOS) analysis reveals new electronic states emerging within the −10 to 0 eV range post-reaction, attributed to strong hybridization between F 2p and Fe 3d orbitals. This reflects significant charge redistribution and Fe3+ to Fe2+ reduction behavior. Cu PDOS analysis further reveals strong coupling between Cu 3d and Fe 3d orbitals alongside adsorption-exchange interactions, consistent with the coexistence of Cu+/Cu2+ detected by XPS.
For the removal of fluoride and copper ions in this system, the overall process involves three key behaviours: double-layer behaviour, Li+-ion intercalation behaviour, and specific adsorption behaviour. This process may be represented as follows: Under the driving force of an applied electric field, F− migrates towards the anode while Cu2+ migrates towards the cathode. Both ions are initially captured at the electrode–electrolyte interface via the electrostatic double layer effect. Cyclic voltammetry confirms this process constitutes a non-faradaic capacitive phenomenon. F− forms stable coordination bonds with active Fe sites on the LFP surface, while some ions, possessing an ionic radius compatible with lattice vacancies, can enter the LFP lattice. Cu2+, due to size mismatch, is excluded from the LFP lattice and adsorbed solely on the material surface via a capacitive process. XPS detected no Cu0 signal, and SEM revealed no extensive copper dendrites, ruling out the possibility of reduction deposition. The Fe2+/Fe3+ redox pair within LFP prevents irreversible reactions that could damage the electrode structure. Applying a reverse voltage releases the captured F− and Cu2+ ions for recovery, whilst Li+ re-intercalates into the LFP lattice to restore its original state, ensuring the cycling stability of the CDI system.
In summary, we have designed a novel capacitive deionization battery. Driven by an external electric field, this battery achieves efficient simultaneous removal of fluoride and copper ions through a synergistic double-layer adsorption-diffusion-intercalation (DADI) mechanism. The symmetrical LFP electrode structure not only maintains a stable and balanced electric field distribution but also effectively reduces polarization effects and interfacial impedance. This provides an innovative design approach and theoretical foundation for capacitive deionization technology in multi-ion synergistic purification applications.
This research is supported by Guangzhou University Large Equipment Open Sharing Fund, the Guangdong Natural Science Foundation General Project (2023A1515010038) and H. Zeng acknowledges the support from the Canada Research Chairs Program.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this study are available in the article and its supplementary information (SI). Supplementary information: Experimental section, SEM, TEM, FT-IR spectra, GCD curves, schematic diagram of capacitive deionization system, the performance of materials, density of states, and additional references. See DOI: https://doi.org/10.1039/d5cc06728f.References
- M. Ahmed, M. O. Mavukkandy, A. Giwa, M. Elektorowicz, E. Katsou, O. Khelifi, V. Naddeo and S. W. Hasan, npj Clean Water, 2022, 5, 12 CrossRef.
- L. Xiang, X. Xu, Y. Liu, H. Zhang, R. Xu, C. Li, F. Xu, Y. Yamauchi and Y. Mai, Nat. Water, 2024, 2, 1195–1206 CrossRef.
- J. Lee, Y. Shin, H. Ryu, C. Boo and S. Hong, Water Res., 2025, 279, 123410 CrossRef PubMed.
- X. Kong, C. Zhang, C.-H. Hou, T. D. Waite and J. Ma, Environ. Sci. Technol., 2025, 59, 13566–13584 Search PubMed.
- M. He, J. Chen, Z. Wang, Z. Chen and L. Deng, Chem. Commun., 2025, 61, 713–716 Search PubMed.
- N. M. Aldaqqa, S. Kumar, J. I. Martínez, N. Elmerhi, E. Alhseinat and D. Shetty, Angew. Chem., Int. Ed., 2025, 137, e202510345 CrossRef.
- C. Delmas, M. Maccario, L. Croguennec, F. Le Cras and F. Weill, Nat. Mater., 2008, 7, 665–671 Search PubMed.
- B. Kang and G. Ceder, Nature, 2009, 458, 190–193 Search PubMed.
- X. Zeng, M. Li, D. Abd El-Hady, W. Alshitari, A. S. Al-Bogami, J. Lu and K. Amine, Adv. Energy Mater., 2019, 9, 1900161 Search PubMed.
- B. Zhang, Y. Xu, J. Wang, X. Ma, W. Hou and X. Xue, Electrochim. Acta, 2021, 368, 137575 Search PubMed.
- Z. Zeng, H. Lei, X. Lu, C. Zhu, Y. Wen, J. Zhu, X. Ji, W. Sun, Y. Yang and P. Ge, Energy Storage Mater., 2025, 74, 103947 Search PubMed.
- D. Wang, Z. Fu, X. Liu, Y. Yao, Y. Ji, X. Gao, J. Wang, Z. Hou, Y. Li, S. Yao, S. Wang, J. Xie, Z. Yang and Y. M. Yan, Small, 2023, 19, 202301717 Search PubMed.
- T. Pang, F. Marken, D. Zhang, J. Shen and M. Xie, ACS ES&T Water, 2024, 4, 5142–5149 Search PubMed.
- Z. Li, M. Xu, Y. Xia, Z. Yan, J. Dai, B. Hu, H. Feng, S. Xu and X. Wang, Nat. Commun., 2025, 16, 3704 CrossRef CAS PubMed.
- Y. Shen, J. Deng, X. Hu, X. Chen, H. Yang, D. Cheng and D. Zhang, Environ. Sci. Technol., 2023, 57, 1797–1806 CrossRef CAS PubMed.
Footnote |
| † These authors have equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2026 |