
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDesign, synthesis and reactivity of N-adamantyl cyclometalated cyclic(alkyl)(amino)carbene ruthenium complexes in Z-selective olefin metathesis
Clément
Casalta
a,
Fanny
Morvan
 a,
Sophie
Colombel-Rouen
a,
Thierry
Roisnel
a,
Rodolphe
Jazzar
*b,
Angelino
Doppiu
*c and
Marc
Mauduit
*a
a,
Sophie
Colombel-Rouen
a,
Thierry
Roisnel
a,
Rodolphe
Jazzar
*b,
Angelino
Doppiu
*c and
Marc
Mauduit
*a
aUniv Rennes, École Nationale Supérieure de Chimie de Rennes, CNRS, ISCR UMR 6226, 35000, Rennes, France. E-mail: marc.mauduit@ensc-rennes.fr
bDepartment of Chemistry and Biochemistry, San Diego State University, 5500 Campanile Drive, San Diego, California 92182, USA. E-mail: rjazzar@sdsu.edu
cUmicore AG & Co. KG, Rodenbacher Chaussee 4, D-63457, Hanau-Wolfgang, Germany. E-mail: Angelino.Doppiu@eu.umicore.com
First published on 14th January 2026
Abstract
The synthesis of a Z-selective ruthenium complex featuring a cyclic(alkyl)(amino)carbene (CAAC) ligand is reported. Its preparation proceeds through a selective intramolecular C(sp3)–H activation at the N-adamantyl substituent of the CAAC ligand featuring a key spiro-tetraline moiety. The resulting cyclometalated precatalyst was isolated in good yield and fully characterized by X-ray diffraction analysis. This catalyst displays high Z-selectivity (up to 95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 Z/E ratio) in both self-metathesis (SM) and cross-metathesis (CM) reactions.
:5 Z/E ratio) in both self-metathesis (SM) and cross-metathesis (CM) reactions.
Since the introduction of the cyclometalated ruthenium complex Ru1 by Grubbs and co-workers in 2011 (Fig. 1a),1Z-selective olefin metathesis2 has emerged as an efficient and competitive approach to access Z-alkenes, which are widely found in pharmaceuticals,3 fragrances,4 agrochemicals,5 and material sciences.6 In recent years structural refinements of these cyclometalated ruthenium complexes (Ru2–5, Fig. 1b) further enhanced catalytic efficiency and expanded the scope of accessible transformations.7–10 In parallel, the remarkable advances achieved with ruthenium complexes bearing cyclic(alkyl)(amino)carbene (CAAC) ligands11 in olefin metathesis,12 naturally motivated Grubbs and co-workers to merge the CAAC framework with these cyclometalated architectures (Ru6, Fig. 1c).13 Unfortunately, the later displayed modest catalytic performances in the SM of allylbenzene (15% conv., 49% Z-selectivity). We reasoned that the adamantyl group positioned at the quaternary carbon of the CAAC framework, enforces a rigid and congested environment around the metal centre, limiting the ligand's capacity to adapt during key steps of the metathesis cycle. To address this constraint, we envisaged redirecting the adamantyl substituent to the nitrogen atom, a modification expected to reduce proximal steric pressure, introduce conformational flexibility. With this design principle in mind, herein, we report the synthesis of a CAAC iminium salt precursor AdCAAC·BF4 containing the N-adamantyl fragment and a spirotetraline moiety (Fig. 1d). The resulting cyclometalated CAAC Ru-complex Ru7 displays high Z-selectivity (up to 95
:5 Z/E ratio) across a range of self- and cross-olefin metathesis transformations.
 | ||
| Fig. 1 State of the art of cyclometalated Ru complexes for Z-selective olefin metathesis (a)–(c). New design of cyclometalated CAAC–Ru complex featuring a N-adamantyl unit and a key spiro-tetraline moiety ((d) this work). | ||
We began our study by investigating the Grubbs’ cyclometalation protocol7–10 on CAAC Ru-complexes Ru8a,b featuring the N-adamantyl unit, which were recently reported by Tuba and co-workers (Scheme 1).14 Unfortunately, in both cases, the intramolecular cyclometalation of the N-Adamantyl fragment using a carboxylate-assisted C–H activation strategy failed. We attribute this behaviour to competing cyclometalation at the phenyl group attached to the quaternary carbon that could led to corresponding species A, which are prone to rapidly decompose (Scheme 1).15
 | ||
| Scheme 1 Failure of the intramolecular cyclometalation process performed on known N-adamantyl CAAC Ru-complex Ru8a,b.14b | ||
To address this limitation, we introduced additional steric constraints by introducing a spiro-cycloalkyl backbone into the CAAC framework. Accordingly, CAAC iminium salts AdCAACa–c·BF4 featuring respectively spiro-indanyle, -tetraline and -fluorenyl fragments was prepared starting from pre-alkylated aldehydes 1a–c (Scheme 2a).16 Condensation of 1-adamantylamine with 1a–c led to the desired imines 2a–c, which were directly subjected to hydroiminiumation,11b followed by anion metathesis. The desired iminium salt AdCAACa·BF4 was obtained with respectable 43% isolated yield (over 3 steps). Unfortunately, despite the accessibility of imines 2b,c, both failed to cyclise under hydroiminiumation precluding access to corresponding iminium salts CAACb,c·BF4. Next, the Hoveyda-type Ruthenium complexes Ru8c was prepared following the standard protocol (Scheme 2b).12b
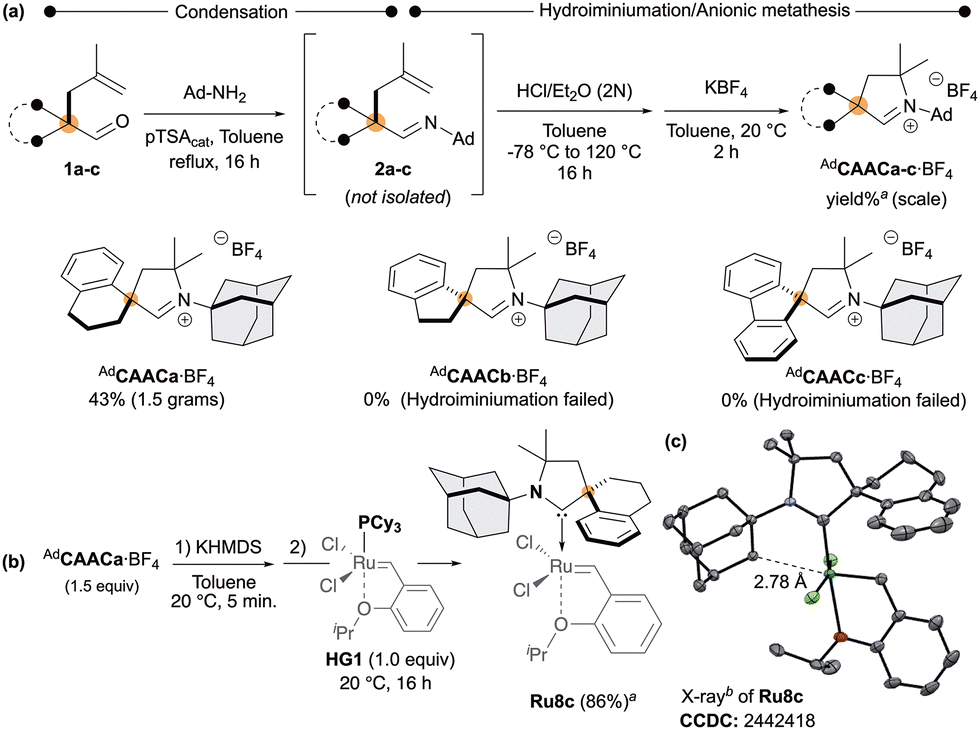 | ||
| Scheme 2 Synthesis of N-adamantyl CAAC precursors containing spiroalkyl fragment (a), the corresponding ruthenium complex Ru8c (b) and its solid-state structure determined by single-crystal X-ray diffraction (c). aIsolated yield. bDisplacement ellipsoids are drawn at 30% probability. Hydrogen atoms have been omitted for clarity. | ||
Deprotonation of AdCAACa·BF4 with potassium hexamethyldisilazide (KHMDS) followed by the addition of the phosphine-based Hoveyda–Grubbs (HG1) afforded the corresponding Ru-complex Ru8c in a good 86% yield. X-ray diffraction analyses from a suitable crystal of Ru8c established the key proximity between the targeted C–H bond and the ruthenium centre with a distance of 2.78 Å (Ru8a) and 2.77–2.78 Å (Ru8b,c) (Scheme 2c).
With this complex in hands, we next examined the intramolecular cyclometalation of the N-Adamantyl fragment (Scheme 3a).7–10 In marked contrast to Ru8a and Ru8b (Scheme 1), Ru8c cleanly delivered the desired cyclometalated complex Ru7 which was isolated in a 35% yield over two steps.
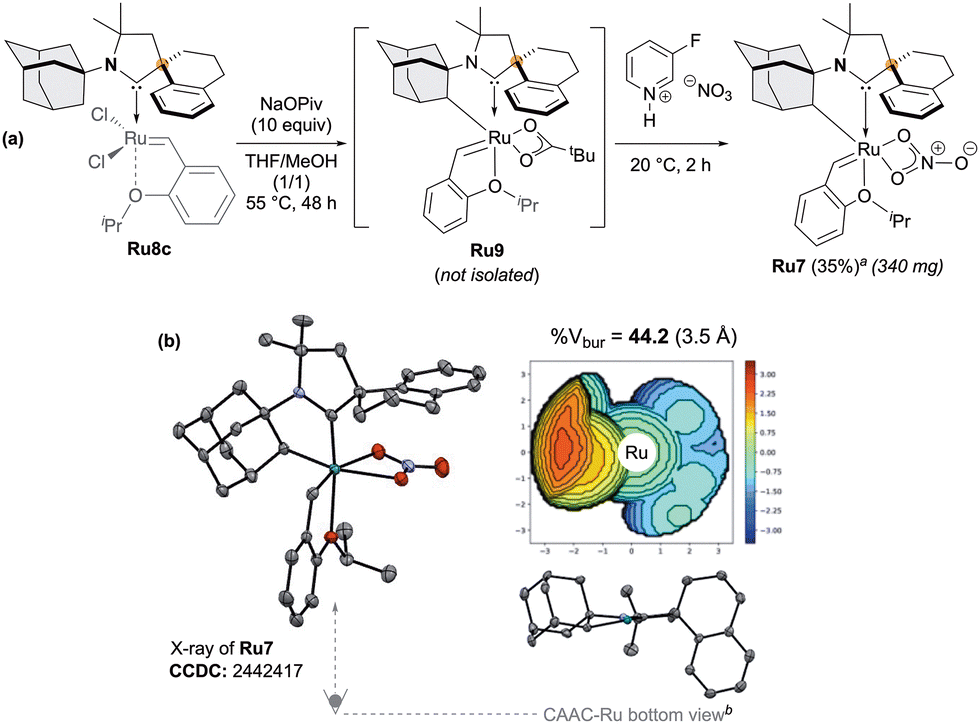 | ||
| Scheme 3 Synthesis of the cyclometalated CAAC–Ru complex Ru7 (a) and its solid-state structure with corresponding steric map and buried volume analysis (b). Displacement ellipsoids are shown at 30% probability; hydrogen atoms are omitted for clarity. Buried volumes (%Vbur) and steric maps (3.5 Å radius) were calculated using SambVca 2.1 (ref. 17). aIsolated yield. bStyrenyl ether and nitrato ligands omitted for clarity. | ||
Single-crystal X-ray analysis of the nitrato derivative Ru7 confirmed the expected chelating architecture. We thus confirm the divergent outcome occurring with Ru8a and Ru8b (Scheme 1), as the competitive cyclometalation pathway is not accessible in Ru8c due to the steric constraints imposed by the spiro-tetraline backbone.
Using the novel cyclometalated complex Ru7, we next evaluated its catalytic performance in representative self- and cross-metathesis reactions (Table 1 and Scheme 3). We first examined the self-metathesis of allylbenzene S1a (Table 1). Using 1 mol% of Ru7 at 25 °C in THF (2.1 M), the reaction reached 76% conversion and 68% isolated yield after 28 h. Notably, the catalyst delivered excellent Z-selectivity throughout the reaction, with only a minor erosion over time (97:3 Z/E at 6.5 h; 95:5 Z/E at 28 h; entries 2–3). This stereocontrol significantly outperforms the CAAC cyclometalated catalyst Ru6 which has been reported by Grubbs and colleagues to provide only 49% Z-selectivity under analogous conditions (Fig. 1).13 Increasing the catalyst loading to 1.5 mol% enhanced productivity, affording up to 74% yield after 28 h, while maintaining good Z-selectivity (94:6 Z/E ratio; entry 10).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ru7 (mol%) | Temp. (°C) | Time (h) | Conv.b (yield)c (%) | Z/E ratiod |
| a Reaction conditions: S1 (0.45 mmol), Ru7 (0.0045 or 0.00675 mmol), THF (0.215 mL) in Glove-box. b Conversions were determined by 1H NMR spectroscopy. c Yields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. d E/Z ratio was determined by GC analysis (see SI for details). | |||||
| 1 | 3.5 | 5 (2) | 97:3 |
||
| 2 | 6.5 | 19 (14) | 97:3 |
||
| 3 | 1 | 25 | 21 | 65 (60) | 95:5 |
| 4 | 24 | 69 (64) | 95:5 |
||
| 5 | 28 | 76 (68) |
95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) :5 :5
|
||
| 6 | 3.5 | 12 (8) | 96:4 |
||
| 7 | 6.5 | 32 (30) | 96:4 |
||
| 8 | 1.5 | 25 | 21 | 69 (68) | 95:5 |
| 9 | 24 | 74 (72) | 94:6 |
||
| 10 | 28 | 79 (74) |
94:6
|
||
| 11 | 1 | 35 | 16 | 74 (66) | 87:13 |
| 12 | 1.5 | 35 | 16 | 74 (70) | 82:18 |
Raising the reaction temperature to 35 °C accelerated the reaction (66–70% yield after 16 h, entries 11 and 12), but at the expense of stereocontrol decreasing to 87:13 to 82:18 Z/E ratio.
We then evaluated Ru7 (1 mol%) in the self-metathesis of 1-dodecene S1b, 9-decenyl acetate S1c and methyl 1-decenoate S1d conduced at 25 and 35 °C (Scheme 4a). In all cases, the corresponding homo-metathesis products P1b-d were formed in modest yields (52 to 65%) but high Z-selectivity, with Z/E ratio ranging from 93:7 to 90:10. Moving to more challenging cross-metathesis reactions, CAAC-Ru7c delivered comparable catalytic performance, albeit requiring higher catalyst loading (2–3 mol%, Scheme 4b). The expected cross-metathesis products P2a–c were obtained in modest yields (22 to 50%) while maintaining consistently good to high Z-selectivity (92:8 to 94:6 Z/E ratio).18 It should be noted that for P2c, the increase in reaction time led to a slight improvement in yield but at the expense of Z-selectivity (88:12 to 85:15 Z/E ratio).
 | ||
| Scheme 4 Scope of Z-selective SM (a) and CM (b) reactions catalysed by Ru7. aConversions were determined by 1H NMR spectroscopy. bYields were determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. cE/Z ratio was determined by GC analysis. dE/Z ratio was determined by 13C NMR analysis. eNMR yield of the Z isomer determined by 1H NMR spectroscopy using 1,3,5-trimethoxybenzene as internal standard. | ||
In summary, we have developed the first Z-selective cyclometalated ruthenium catalyst Ru7 bearing an N-adamantyl CAAC ligand. Thanks to the introduction of a spiro-tetraline moiety at the quaternary carbon center, the cyclometalation process successfully delivered the targeted complex, which was isolated in 35% yield over two steps and fully characterized, including by single-crystal X-ray diffraction. This catalyst delivers moderate to good yields across a range of self- and cross-metathesis reactions while maintaining consistently high Z-selectivity (90:10 to 95:5 Z/E). Building on these results and our recent access to enantiopure CAAC ligands, efforts toward new chiral cyclometalated CAAC–Ru catalysts for asymmetric Z-selective olefin metathesis are currently underway in our laboratories.19
R. J., A. D. and M. M. conceptualized and supervised this work. C. C., F. M and S. C.-R. conducted all the experiments. T. R. accomplished the X-Ray diffraction analysis. The manuscript was written by R. J. and M. M. and was reviewed by all the authors.
Conflicts of interest
There are no conflicts to declare.Data availability
All experimental and crystallographic data associated with this work are available in the supplementary information (SI). Supplementary information: experimental procedures, NMR spectra, and GC analysis. See DOI: https://doi.org/10.1039/d5cc07127e.CCDC 2442417 and 2442418 contain the supplementary crystallographic data for this paper.20a,b
Acknowledgements
We are grateful to the CNRS, the ENSC de Rennes and SDSU. This work was supported by Umicore AG & Co (grant to C. C.). and the Region Bretagne (ARED 2022 No 2671, grant to F. M.)References
- (a) K. Endo and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 8525 CrossRef CAS PubMed; (b) B. K. Keitz, K. Endo, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2011, 133, 9686 CrossRef CAS PubMed.
- For recent review on Z-selective metathesis, see: (a) T. P. Montgomery, A. M. Johns and R. H. Grubbs, Catalysts, 2017, 7, 87 CrossRef; (b) S. Shahane, C. Bruneau and C. Fischmeister, ChemCatChem, 2013, 5, 3436 CrossRef CAS . For general books on olefin metathesis, see: ; (c) Handbook of Metathesis, ed. R. H. Grubbs, A. G. Wenzel, D. J. O’Leary and E. Khosravi, Wiley-VCH, Weinheim, Germany, 2nd edn, 2015 Search PubMed; (d) Olefin Metathesis: Theory and Practice, ed. K. Grela, John Wiley & Sons, Hoboken, N. J., 2014 Search PubMed.
- (a) J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2013, 30, 237 RSC; (b) S. Werrel, J. C. L. Walker and T. J. Donohoe, Tetrahedron Lett., 2015, 56, 5261 CrossRef CAS.
- V. M. Marx, M. B. Herbert, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 94 Search PubMed.
- (a) M. B. Herbert, V. M. Pederson, R. L. Marx and R. H. Grubbs, Angew. Chem., Int. Ed., 2013, 52, 310 CrossRef CAS PubMed; (b) J. Morvan, T. McBride, I. Curbet, S. Colombel-Rouen, T. Roisnel, C. Crévisy, D. L. Browne and M. Mauduit, Angew. Chem., Int. Ed., 2021, 60, 19685 Search PubMed.
- (a) B. K. Keitz, A. Fedorov and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 2040 Search PubMed; (b) L. E. Rosebrugh, V. M. Marx, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 10032 CrossRef CAS PubMed; (c) L. E. Rosebrugh, T. S. Ahmed, V. M. Marx, J. Hartung, P. Liu, J. G. Lopez, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2016, 138, 1394 CrossRef CAS PubMed.
- B. K. Keitz, K. Endo, P. R. Patel, M. B. Herbert and R. H. Grubbs, J. Am. Chem. Soc., 2012, 134, 693 CrossRef CAS PubMed.
- (a) L. E. Rose-brugh, M. B. Herbert, V. M. Marx, B. K. Keitz and R. H. Grubbs, J. Am. Chem. Soc., 2013, 135, 1276 CrossRef CAS PubMed; (b) M. B. Herbert and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 5018 CrossRef CAS PubMed.
- A. Dumas, R. Tarrieu, T. Vives, T. Roisnel, V. Dorcet, O. Baslé and M. Mauduit, ACS Catal., 2018, 8, 3257 CrossRef CAS.
- Y. Xu, J. J. Wong, A. E. Samkian, J. Hoon Ko, S. Chen, K. N. Houk and R. H. Grubbs, J. Am. Chem. Soc., 2020, 142, 20987 CrossRef CAS PubMed.
-
(a) V. Lavallo, Y. Canac, C. Pra
![[s with combining umlaut]](https://www.rsc.org/images/entities/char_0073_0308.gif) ang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 Search PubMed;
(b) R. Jazzar, R. D. Dewhurst, J.-B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899 Search PubMed;
(c) F. Vermersch, L. Oliveira, J. Huner, M. Soleilhavoup, R. Jazzar and G. Bertrand, J. Org. Chem., 2022, 87, 3511 CrossRef CAS PubMed . For recent reviews, see: ;
(d) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed;
(e) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 Search PubMed;
(f) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362 CrossRef CAS;
(g) R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141 CrossRef CAS PubMed;
(h) R. K. Singh, T. K. Khan, S. Misra and A. K. Singh, J. Organomet. Chem., 2021, 956, 122133 CrossRef CAS.
ang, B. Donnadieu and G. Bertrand, Angew. Chem., Int. Ed., 2005, 44, 5705 Search PubMed;
(b) R. Jazzar, R. D. Dewhurst, J.-B. Bourg, B. Donnadieu, Y. Canac and G. Bertrand, Angew. Chem., Int. Ed., 2007, 46, 2899 Search PubMed;
(c) F. Vermersch, L. Oliveira, J. Huner, M. Soleilhavoup, R. Jazzar and G. Bertrand, J. Org. Chem., 2022, 87, 3511 CrossRef CAS PubMed . For recent reviews, see: ;
(d) M. Soleilhavoup and G. Bertrand, Acc. Chem. Res., 2015, 48, 256 CrossRef CAS PubMed;
(e) M. Melaimi, R. Jazzar, M. Soleilhavoup and G. Bertrand, Angew. Chem., Int. Ed., 2017, 56, 10046 Search PubMed;
(f) U. S. D. Paul and U. Radius, Eur. J. Inorg. Chem., 2017, 3362 CrossRef CAS;
(g) R. Jazzar, M. Soleilhavoup and G. Bertrand, Chem. Rev., 2020, 120, 4141 CrossRef CAS PubMed;
(h) R. K. Singh, T. K. Khan, S. Misra and A. K. Singh, J. Organomet. Chem., 2021, 956, 122133 CrossRef CAS. - For a recent review on CAAC–Ru-complexes, see: (a) J. Morvan, M. Mauduit, G. Bertrand and R. Jazzar, ACS Catal., 2021, 11, 1714 CrossRef CAS . For pioneer works on CAAC–Ru complexes, see: ; (b) D. R. Anderson, V. Lavallo, D. J. O’Leary, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2007, 46, 7262 CrossRef CAS PubMed; (c) V. M. Marx, A. H. Sullivan, M. Melaimi, S. C. Virgil, B. K. Keitz, D. S. Weinberger, G. Bertrand and R. H. Grubbs, Angew. Chem., Int. Ed., 2015, 54, 1919 CrossRef CAS PubMed.
- S. M. Bronner, M. B. Herbert, P. R. Patel, V. M. Marx and R. H. Grubbs, Chem. Sci., 2014, 5, 4091 Search PubMed.
- For a recent synthesis of N-adamentyl CAAC Ru8a,b, see: (a) V. Farkas, D. Csókás, Á. Erdélyi, G. Turczel, A. Bényei, T. Nagy, S. Kéki, I. Pápai and R. Tuba, Adv. Sci., 2024, 11, 2400118 CrossRef CAS PubMed; (b) for this study, we used homemade complexes Ru8a,b (see SI).
- T. S. Ahmed, J. M. Grandner, B. L. H. Taylor, M. B. Herbert, K. N. Houk and R. H. Grubbs, Organometallics, 2018, 37, 2212 CrossRef CAS.
- For previous synthesis of prealkylated aldehydes 1a–c, see: (a) F. Vermersch, L. Oliveira, J. Hunter, M. Soleilhavoup, R. Jazzar and G. Bertrand, J. Org. Chem., 2022, 87, 3511 CrossRef CAS PubMed; (b) A. Madron du Vigné and N. Cramer, Organometallics, 2022, 41, 2731 CrossRef; (c) R. Gawin, A. Tracz, P. Krajczy, A. Kozakiewicz-Piekarz, J. P. Martínez and B. Trzaskowski, J. Am. Chem. Soc., 2023, 145, 25010 CAS; (d) Á. Erdélyi, V. Farkas, G. Turczel, M. Nagyházi, A. Bényei, M. L. L. Recta, T. Nagy, S. Kéki, O. Osterthun, J. Klankermayer and R. Tuba, Chem. – Eur. J., 2024, 30, e202401918 CrossRef PubMed.
- L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872 CrossRef CAS PubMed.
- Some amounts of self-metathesis products were also detected by GC (see SI for details).
- (a) J. Morvan, F. Vermersch, Z. Zhang, L. Falivene, T. Vives, V. Dorcet, T. Roisnel, C. Crévisy, L. Cavallo, N. Vanthuyne, G. Bertrand, R. Jazzar and M. Mauduit, J. Am. Chem. Soc., 2020, 142, 19895 CrossRef CAS PubMed; (b) J. Lorkowski, P. Yorkgitis, J. Morvan, F. Morvan, N. Vanthuyne, T. Roisnel, M. Gembicki, G. Bertrand, M. Mauduit and R. Jazzar, J. Am. Chem. Soc., 2025, 147, 14777 CrossRef CAS PubMed; (c) M. Liu, D. Bouëtard, J. Talcik, J. Lorkowski, Z. Zhang, T. Vives, T. Roisnel, M. Serradeil-Albalat, N. Vanthuyne, L. Cavallo, L. Falivene, R. Jazzar and M. Mauduit, JACS Au, 2025, 5, 5170 Search PubMed.
- (a) C. Casalta, F. Morvan, S. Colombel-Rouen, T. Roisnel, R. Jazzar, A. Doppiu and M. Mauduit, CCDC 2442417: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2mzjnt; (b) C. Casalta, F. Morvan, S. Colombel-Rouen, T. Roisnel, R. Jazzar, A. Doppiu and M. Mauduit, CCDC 2442418: Experimental Crystal Structure Determination, 2026 DOI:10.5517/ccdc.csd.cc2mzjpv.
| This journal is © The Royal Society of Chemistry 2026 |