
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Bridging the hydrophobic gap: visible-light-driven photocatalysis of organic reactions in water with soft-matter nanoreactors
Davide
Arena†
ab and
José A.
Pomposo
 *abc
*abc
aMaterials Physics Center (CFM-MPC), CSIC-UPV/EHU, 20018 Donostia, Spain. E-mail: Josetxo.pomposo@ehu.eus
bDepartment of Polymers and Advanced Materials: Physics, Chemistry and Technology, University of the Basque Country (UPV/EHU), 20800 Donostia, Spain
cIKERBASQUE−Basque Foundation for Science, 48009 Bilbao, Spain
First published on 8th January 2026
Abstract
Visible-light-driven photocatalysis of organic reactions in water represents a cornerstone of green chemistry, yet its application is often hampered by the poor solubility and stability of organic substrates and catalysts in aqueous media. In this review article we highlight recent advances in overcoming these challenges using state-of-the-art self-assembling soft-matter platforms—including micelles, supramolecular nanocapsules, and single-chain polymer nanoparticles (SCNPs). When careful designed, these constructs create confined, hydrophobic environments that enhance catalyst stability, substrate solubilization, and reaction efficiency under mild conditions. We show key applications such as amide synthesis and C–X bond activation by photocatalytic micellar systems, reductive cleavages by supramolecular capsules stabilizing photocatalysts, and diverse visible-light-driven photocatalytic transformations by SCNPs serving as tunable enzyme-mimetic nanoreactors. Collectively, these selected examples demonstrate the immense promise of soft-matter nanoreactors for shaping the future of aqueous visible-light-driven photocatalysis.
 Davide Arena | Davide Arena completed his Bachelor's degree in Chemistry (2017) and his Master's degree in Organic & Biomolecular Chemistry (2019) at the Sapienza University of Rome. He then joined the Materials Physics Center, Polymers & Soft Matter Group, under the supervision of Prof. Pomposo, to pursue his PhD in Physics of Nanostructures and Advanced Materials at the University of the Basque Country, which he completed in 2024. He is currently a postdoctoral researcher in the group of Prof. Salmeron-Sanchez, working on the design of advanced biomaterials to engineer the cellular microenvironment. |
 José A. Pomposo | José A. Pomposo received his PhD from the University of the Basque Country (UPV/EHU) in 1994. After more than 30 years of research experience in Materials Science, more than 200 scientific publications and 9 international patents, currently, he is IKERBASQUE Research Professor at the Materials Physics Center, Polymers & Soft Matter Group, and Distinguished Professor at UPV/EHU. His research interests include the synthesis of uniform soft nano-objects as artificial enzymes, research in the structure, dynamics & self-assembly behaviour of complex single-chain nano-objects for nanomedicine and (photo) catalysis applications, hybrid organic nanostructures, and polymer networks based on reversible covalent bonds. |
1. Introduction
Photocatalysis, the acceleration of chemical reactions through the absorption of light by a catalyst, has its roots in early studies of photochemical processes dating back to the 19th century.1 In this sense, light is an abundant, therefore cheap, source of energy we have access to almost without any time restriction and readily form our planet. Life itself evolved through precise and intricate light-harvesting strategies to sustain its long-lasting existence in a sustainable fashion. For these simple and intuitive reasons, its use as a reactant to manipulate and control our environment fascinated humanity since ancient times.2However, it was not until the late 20th century that photocatalysis began to gain traction in synthetic organic chemistry, particularly with the development of transition-metal complexes and semiconductors capable of harnessing visible light. Landmark contributions, such as the use of ruthenium and iridium polypyridyl complexes, laid the foundation for photoredox catalysis—a subfield that enables single-electron transfer processes under mild conditions and has revolutionized the way chemists approach bond formation and activation.3–5 Over the past two decades, visible-light photoredox catalysis has evolved into a versatile and widely adopted strategy for organic synthesis. It has enabled a broad range of transformations, including C–C and C–X bond formation, oxidation and reduction reactions, and functionalization of complex molecules. Despite its success, most photoredox reactions have been conducted in organic solvents, which pose environmental and safety concerns and limit the scalability and sustainability of these methods.6
The shift toward aqueous photocatalysis represents a significant advancement in the pursuit of greener chemical processes.7 Water is an ideal solvent from an environmental standpoint: it is non-toxic, abundant, inexpensive, and inherently safe. However, its use in photocatalysis introduces several challenges. Many organic substrates and photocatalysts are poorly soluble or unstable in water, and the reactivity can be hindered by quenching effects or competing side reactions.
These limitations have prompted researchers to explore innovative strategies to enable efficient photocatalysis in aqueous environments. One of the most promising approaches involves the use of soft-matter assemblies—dynamic, self-organizing systems that create hydrophobic microenvironments within water. These assemblies mimic biological compartmentalization and offer confined spaces where photocatalysts and substrates can interact effectively. Among the most studied soft-matter systems are micelles, supramolecular nanocapsules, and single-chain nanoparticles (SCNPs).
Micellar systems, formed by surfactants in water, have been shown to facilitate a variety of photocatalytic transformations by concentrating hydrophobic reactants and catalysts within their cores. Recent studies by Giustiniano et al.8 and Giedyk et al.9 have demonstrated the utility of micellar photocatalysis in amide synthesis and C–X bond activation, respectively, under mild and recyclable conditions.
Supramolecular nanocapsules, such as those developed by Akita et al.,10 provide tailored cavities for hosting photocatalysts, enhancing their stability and reactivity in water. These systems have enabled complex transformations like pinacol coupling and N–O bond cleavage, highlighting the potential of molecular design in aqueous photocatalysis.
SCNPs represent a more recent and sophisticated strategy, where polymer chains fold into defined nanostructures with internal hydrophobic pockets.11 These systems combine the robustness of polymeric materials with the precision of molecular engineering. Palmans et al.12,13 and Barner-Kowollik et al.14 have reported SCNPs capable of catalysing reductions, cross-couplings, and photo-oxidations in water, with enhanced efficiency and selectivity compared to free photocatalysts. Furthermore, we have very recently developed artificial photo-synthases (APS) in the form of SCNPs with multifaceted visible-light photocatalytic activity for challenging organic transformations in aqueous media.15
This feature article aims to highlight the conceptual and practical innovations that have enabled the use of soft-matter assemblies in aqueous photocatalysis. By examining representative examples across micellar, supramolecular, and single-chain polymeric nanoparticle systems, we seek to illustrate how these strategies are reshaping the landscape of photoredox chemistry, offering new opportunities for sustainable synthesis and expanding the scope of reactions that can be performed in water.
We begin with a concise definition of photocatalysis, followed by a historical overview of the concept and a summary of its foundational principles, with particular emphasis on its application in organic synthesis. Next, recent developments in organic photocatalysis in water are examined, with emphasis on soft-matter nanoreactors that facilitate light-driven transformations in aqueous environments. Finally, we present a forward-looking perspective on future directions in this emerging field.
2. Photocatalysis: fundamentals and historical development
The alteration in the rate of a chemical reaction due to electromagnetic radiation effectively summarizes how most authoritative English dictionaries define the term photocatalysis.16–18 According to the Oxford English Dictionary, the earliest recorded use of the noun photocatalysis dates back to the 1890s, with its first known appearance in the writings of C. H. Bothamley on photography.18 In its Compendium of Chemical Terminology, the International Union of Pure and Applied Chemistry (IUPAC) broadens the definition of photocatalysis to include the change in rate of a chemical reaction or its initiation. It also limits the relevant electromagnetic radiation to ultraviolet, visible, or infrared wavelengths, and introduces the concept of a photocatalyst—a substance that absorbs light and is involved in the chemical transformation of the reaction partners.19 Both the linguistic and scientific points of view agree on the vastness of the term photocatalysis, which indeed is, at the date, declined in a variety of applications, spanning through light-dependent conductive materials, water purification, solar energy conversion, optoelectronics, water splitting, and carbon dioxide reduction, just to cite a few, all sharing the manipulation of matter through its interaction with either ultraviolet (UV), visible or infrared (IR) electromagnetic radiation.The curiosity for the chemistry induced by light accompanies the history of modern science since its very beginning. The experiment of the combustion of diamond employing solar light and the apparatus shown in Fig. 1, performed in Florence by Sir Humphry Davy and Michael Faraday in 1814, legendarily exemplifies the scientific need of thinking about light as a proper reagent.20–22
 | ||
| Fig. 1 (a) Lens worked by Benedict Bregans and later used by Sir Humphry Davy and Michael Faraday in their experiments on the combustion of diamond.21,22 (b) Complete apparatus used in these experiments which is preserved at the Museo Galileo in Florence, Italy.22Fig. 1 reproduced from ref. 21 with permission from IOP Publishing, copyright 1931. | ||
As is often the case in many scientific disciplines, establishing a definitive chronological starting point can be challenging and prone to error. This difficulty applies not only to photocatalysis, but to photochemistry as a whole. As the name implies, photochemistry is the branch of chemistry concerned with the chemical effects of light—ranging from far UV to IR radiation.23 This broad definition makes it difficult to pinpoint a universally accepted starting point for the field without narrowing its scope. A first account of a photochemical organic reaction was documented in 1834 by Hermann Trommsdorff, in his research on anthelmintic substances, in which reported that solid α-santonin underwent a change in colour and crystalline structure upon exposure to sunlight24 (Fig. 2).
 | ||
| Fig. 2 Schematic illustration of the structural transformation associated to the sunlight-induced color change in α-santonin crystals—a photochemical phenomenon first reported by Trommsdorff in 1834.24 | ||
Although Edmond Becquerel reported the discovery of the photovoltaic effect25 just a few years later—an effect closely related in principle to photocatalysis—the term photocatalysis itself was first explicitly mentioned in scientific literature in 1911, in a study on the photobleaching of Prussian blue by Alexander Eibner.26 By the early 20th century, photochemistry and photocatalysis began to consolidate through the development of a more systematic approach. This evolution was strongly influenced by the pioneering work and visionary ideas of Giacomo Ciamician, brilliantly summarized in his 1912 Science publication.27 His insights sparked growing interest among experimentalists28 and more than a century later, the scientific community still agree with most of the principles stated in these seminal works.
For the contemporary understanding of photocatalytic processes, the acceleration of a certain transformation upon absorption of light by the photocatalyst can be interpreted based on molecular electronic states of the substances involved in the process.29 Considering the simple, thermal reaction (a):
| A + B → C | (a) |
| A + hν → A* | (b) |
| A* + B → C | (c) |
 | ||
| Fig. 3 (a) A schematic representation illustrating the increase in both reduction and oxidation potential of a molecule upon electronic excitation. (b) Thermodynamic diagram schematically displaying a thermally-allowed reaction between A and B, in presence of the photocatalyst, PC, and the effect on the free Gibbs energy of the system upon photo-excitation of PC to PC*. (c) Thermodynamic diagram illustrating the principle of light conversion into energy: a thermally-prohibited process becomes allowed upon photo-excitation of PC to PC*. | ||
In presence of a third substance, the photocatalyst (PC), capable to absorb light and to take place in a certain process without being chemically altered, the photochemical transformation can occur even between reactants which either tend not to react or do not react at all thermally. In this case, the process is said to be photocatalysed by PC.
From a thermodynamic point of view, photocatalysed transformations can occur via two different modalities, which are depicted in Fig. 3: panel (b) shows a photosensitized process, while panel (c) illustrates the conversion of light into chemical energy. In a photosensitized process, a thermodynamically allowed, yet slow, reaction between A and B to yield C and D, is sped up by the presence of PC, whose electronic excited state PC* generated upon absorption of light augment the total free energy of the system. In the case of the conversion of light into chemical energy, an initially thermodynamically impossible reaction becomes allowed by the presence of the photo-excited species PC*. Both modes of action result from the contribution of the electronically excited state of PC to the overall Gibbs free energy of the system.29
It is worth noting that all photocatalysed reactions can be grouped into two main classes, according to the mechanism with which the PC enables the catalytic cycle: (i) energy transfer and (ii) photoredox catalysis. The main process of photosensitized reactions by energy transfer is shown in reaction (d):
| PC* + A → PC + A* → PC + products | (d) |
 | ||
| Fig. 4 Schematic representation (Jablonski diagram) of the main electronic transitions involved in energy transfer photocatalytic processes.30 The fine vibronic structures of the singlet excited states 1Sn with internal relaxation thereof were omitted for pictorial clarity. The reactive triplet state of the acceptor substrate 3A*, the crucial intermediate in the mechanistic pathway to products, is highlighted in yellow. The spectroscopic triplet energies ET and the associated transitions are represented in grey. | ||
3. Photocatalysis in organic solvents
In general, accessing the triplet states of common organic substrates through direct excitation in organic solvents requires short-wavelength irradiation (280–315 nm) and high light intensities. These conditions often lead to unselective absorption and undesired side reactions.To function efficiently, a photocatalyst must exhibit high extinction coefficient and rapid intersystem crossing (ISC), ideally combined with a long-lived triplet state (greater than 100 ns). These characteristics explain the widespread use of heavy-atom Ru/Ir complexes and carbonyl compounds as effective triplet sensitizers.31 Photocycloadditions, heterolytic bond dissociations, photoisomerizations of carbon–carbon double bonds, and reactions involving the formation of photosensitized singlet oxygen represent the four most commonly employed classes of transformations driven by the energy transfer (EnT) mechanism. Fig. 5 illustrates selected applications of these reactions.32–35
 | ||
| Fig. 5 On top, four examples of recently reported photocatalysed reactions via energy transfer (EnT) mechanism: heterolytic bond dissociation of carbazides,32 regioselective photo Diels–Alder cycloadditions of anthracenes to olefins,33 singlet oxygen cycloaddition to dienes,34 and isomerization of carbon–carbon double bonds.35 On the bottom, the structure of commonly used photosensitizers with their respective EnT values (in kcal mol−1) shown in parenthesis.31 | ||
Concerning the second mode of action of a photocatalysed reaction, the photoredox catalysis, the reaction mechanism involves the electron transfer between the excited photocatalyst and substrates, facilitated by the principle discussed above by which the excited states of a photocatalyst are usually both better reductants and better oxidizers.29 If PC's excited state transfers an electron to one substrate to initiate the catalytic cycle, the mechanistic route is called reductive quenching cycle (Scheme 1), which is usually completed by the reduction of the oxidized intermediate of the photocatalyst via electron transfer form a second, electron donating reagent. In some cases, the redox potentials of the species involved in the cycle can undergo the opposite process, an oxidative quenching cycle, in which the photoexcited state of PC abstract an electron from a donor to initiate the catalysis (Scheme 1). As a rule, it is possible to tune the oxidation potentials of the photocatalyst to switch from one mechanism to another, opening new synthetically valuable possibilities.36
 | ||
| Scheme 1 Possible mechanisms in photoredox catalysis. On the right, the photocatalyst PC is reduced by a donor reactant D via single-electron transfer (SET) after being excited upon light absorption, the reduced form of PC is oxidized by an acceptor reactant A to finally yield the regenerated PC. On the left, after being promoted to the reactive excited state upon light absorption, PC* transfers an electron via SET to an accepting reactant A. The ground state photocatalyst is then regenerated by a second SET step in which a donating reactant D is oxidized by the intermediate oxidized PC. | ||
Photoredox catalysis, particularly under visible light, has become a powerful and increasingly prominent strategy for the synthesis of organic molecules. Although the field has seen rapid development in recent years, its origins trace back to 1979, when Kellogg37 first reported the use of photoredox systems. Subsequent foundational contributions by Fukuzumi and Tanaka,38 as well as Deronzier,39 further demonstrated the potential of ruthenium polypyridyl complexes to facilitate a wide range of organic transformations. Fig. 6 presents several representative examples that underscore the versatility of these systems.
 | ||
| Fig. 6 Three representative examples of historically relevant photoredox organic reactions. From top to bottom: the light-induced acceleration of the reductive desulfuration of trialkyl sulfonium salts in presence of catalytic amounts of [Ru(bpy)3]Cl2;37 the light-dependent reductive coupling of benzyl bromide in presence of 1-benzyl-1.4-dihydronicotinamide and catalytic amount of [Ru(bpy)3]Cl2;38 and the photo-oxidation of benzylic alcohols to the corresponding aldehydes, mediated by polypyridyl Ru(II) complexes and using diazonium salts as sacrificial electron donors.39 | ||
At the turn of the century, photoredox catalysis entered a period of rapid expansion, catalysed by seminal contributions from the groups of Nobel Laureate David MacMillan and Yoon, who demonstrated the transformative effect of visible light on organic reactions mediated by metal complexes.7 Since then, the field has grown to encompass a wide array of applications and reaction types, many of which proceed under significantly milder conditions than their thermal counterparts.29 The ability to introduce structurally complex moieties under such gentle conditions has attracted increasing interest from the pharmaceutical and fine chemical industries. As a result, photoredox catalysis is now being integrated into synthetic routes and late-stage functionalization strategies for the production of high-value molecules.35
In particular, experimental setups for visible-light photocatalysis—which traditionally relied on sunlight or household light bulbs—have greatly benefited from the commercialization of standardized light-emitting diodes (LEDs). These systems offer enhanced reaction reproducibility due to their narrow spectral emission profiles, as well as improved overall performance.40 This exponential growth is visually illustrated in Fig. 7, which presents a timeline of publication volume in the fields of visible-light photocatalysis and photochemistry, overlaid with three milestone optoelectronic innovations that have undeniably contributed to the recent transformation of the scientific landscape.41
 | ||
| Fig. 7 Trends in scientific output over time, represented by the number of publications containing the terms “visible-light photocatalysis,” “photochem,” and “photocatalysis,” aggregated per quinquennium from the 1910s to the 2020s (data from the digital search portal Google Scholar). | ||
4. Photocatalysis in water
This section opens with a concise overview of the limited examples of naked photocatalysts that operate in water, followed by a discussion of recent progress in soft-matter-based photocatalytic nanoreactors tailored for organic transformations in aqueous environments. Among contemporary strategies in photocatalysis, supramolecular scaffolds42 emerge as a particularly promising solution to critical challenges such as poor solubility of catalysts and products—often achieving performance that rivals natural systems.Supramolecular systems rely on discrete molecules that self-assemble into well-defined architectures through noncovalent interactions. Inspired by biological processes, which often occur in the confined pockets of enzymes, these assemblies create nanocavities that host reactant molecules, lowering transition-state energies and facilitating reactions within a tailored microenvironment.43,44 Numerous supramolecular hosts with precise cavities have been developed to mimic enzymatic activity, and the literature offers outstanding examples of how weak interactions can be harnessed to construct functional catalytic architectures.45–49 Notably, supramolecular hosts often surpass traditional molecular catalysts, delivering higher yields and improved selectivity.50–52
Despite these advances, the application of soft-matter nanoreactors for visible-light photocatalysis of organic transformations in water remains in its early stages. While several representative studies have demonstrated the remarkable potential of this approach, significant challenges and unresolved questions persist, leaving ample room for future innovation.
4.1. Nacked photocatalysts
Photocatalysis has long drawn inspiration from nature, given its central role in ecological balance and life processes. The Earth itself can be viewed as a vast, complex photoreactor. Historical milestones include Priestley's 1772 discovery of oxygen production by plants and Ingen-Housz's demonstration that this process depends on light.53 These findings sparked centuries of research into photosynthesis, culminating in modern biochemical insights. Early attempts to mimic these natural processes appeared in 1921 when Barker54 reported photo-induced synthesis of formaldehyde and carbohydrates from carbon dioxide in water using uranium salts and iron oxides. Despite these pioneering efforts, photocatalysis in aqueous media remained largely unexplored until recent years.The use of water as a reaction medium has deep roots in synthetic chemistry, dating back to Wöhler's urea synthesis in the 19th century.55 While water was common in early reactions, its use declined with the rise of Grignard reagents and petrochemical solvents in the 20th century. Today, sustainability concerns have renewed interest in water as a green solvent. Organic solvents pose significant hazards—flammability, toxicity, carcinogenicity, and environmental impact—making the shift toward safer, renewable alternatives imperative. Water meets all criteria for a green solvent, reducing health and environmental risks while offering unique polarity-tuning capabilities for diverse reactions.
Combining photocatalysis with water as a solvent aligns perfectly with green chemistry principles, yet remains challenging. Purely aqueous photocatalytic organic reactions are still rare due to solubility limitations of potent photocatalysts and substrates. Nevertheless, recent reviews highlight the potential of this approach to unlock new reactivities and sustainable pathways.42,56–59 Merging the intrinsic benefits of photocatalysis with water's environmental advantages represents a promising frontier for both academic research and industrial applications.7,60Fig. 8 illustrates six representative examples of photocatalytic organic syntheses carried out using water as the sole reaction medium, which exemplify how employing traditional transition-metal catalysts under unconventional conditions can deliver remarkable outcomes.61–66
 | ||
| Fig. 8 Six representative examples of light-induced photocatalytic organic reactions allowed in water: (a) arylation of N-heteroarenes with aryldiazonium salts, first reported in 2014 by Xue et al.61 (b) Visible-light-promoted oxidative radical cyclization of N-N-biarylglycines, reported by Natarajan et al. in 2019.62 (c) Visible light-induced acylative epoxidation of vinylarenes in water, reported by Salles et al. in 2018.63 (d) Aromative dehydrogenation of cyclic amines, first reported by Balaraman's group in 2019.64 e) Visible-light induced hydrogenation of aromatic alkenes in water by Wenger et al., 2019.65 (f) Synthesis of 1,2-aminoalcohols by decarboxylative coupling of aminoacids, first reported by Zhong et al. in 2020.66 | ||
For instance, the arylation of N-heteroarenes with aryldiazonium salts (Fig. 8a) proceeds smoothly at room temperature, yielding monosubstituted products under the stated conditions.61 Interestingly, changing the reaction solvent (e.g., using formic acid) alters regioselectivity. Although water proved to be the ideal solvent, the substrate scope was limited to hydrophilic compounds, as solubility of both catalyst and substrates strongly influenced yields (e.g., 2,4-dimethylpyridine gave only 55% yield under these conditions).
As in the preceding example, water served as the preferred solvent for the photocatalytic dehydrogenation of cyclic amines with concurrent hydrogen evolution in water (Fig. 8d). The reaction proceeds efficiently at room temperature and tolerates a wide range of substrates,64 with yields dropping markedly when methanol (MeOH) or dimethylformamide (DMF) were employed. Importantly, when both the photocatalyst and substrates are water-soluble, mechanistic investigations become feasible through redox potential measurements and cyclic voltammetry, as demonstrated in this study.
The synthesis of 1,2-amino alcohols in water via decarboxylative coupling of amino acids (Fig. 8f), reported by Zhong et al.,66 provides straightforward access to diverse 1,2-amino alcohols from readily available starting materials without additives, achieving good yields even under sunlight and mild conditions. This reaction, too, was highly solvent-sensitive, with yields dropping in low-dielectric organic solvents (e.g., 55% in 1,4-dioxane).
Reaction e in Fig. 8 further illustrates how water as a reaction medium can enable novel and sometimes unexpected reactivities. Using the water-soluble photocatalyst trisodium fac-tris[2-(5′-sulfonatophenyl)pyridine]iridate(III) (Irsppy, Scheme 2), a variant of Ir(ppy)3, the authors observed light-intensity-dependent reactivity of trans-3-fluorocinnamic acid in water: focalized blue light yielded the saturated product, whereas non-focalized irradiation of equal power and wavelength produced the isomerized alkene.65 The authors hypothesized that modulating light intensity per unit area using a collimating optical element (such as a standard lens) enables switching between one-photon activation pathways (e.g., triplet–triplet sensitization or single-electron transfer) and two-photon processes involving the formation of solvated electrons. This phenomenon has also been observed with other water-soluble analogues of Irsppy.67 Interestingly, the same photocatalyst was later employed by this group to achieve efficient reduction of aliphatic imines to amines through a synergistic combination of photoredox catalysis and enzymatic processes in water, as reported in 2018.67
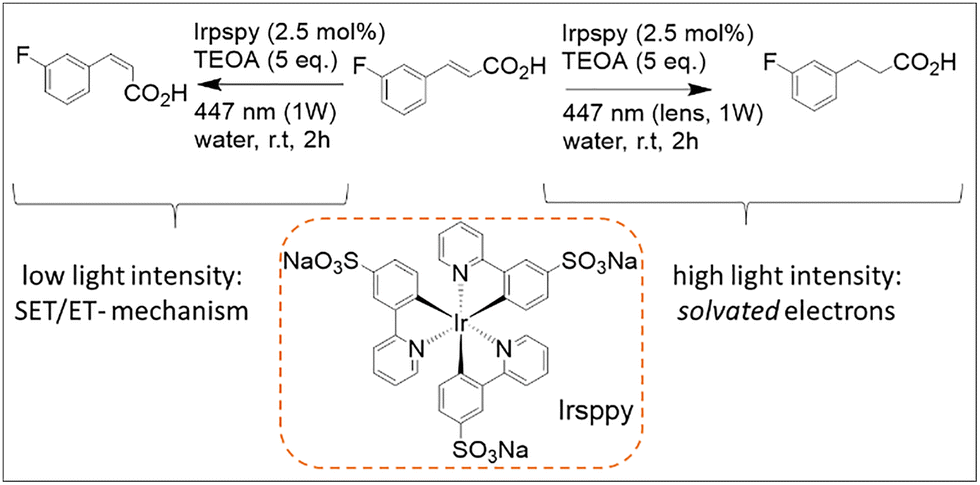 | ||
| Scheme 2 Reaction scheme of the light intensity dependent reaction modality allowed by the use of the photocatalyst Irsppy in water. Namely, the trans–cis isomerization (left, one-photon mechanism) and hydrogenation (right, two-photon pathway) with the substrates trans-3-fluorocinnamic acid using the blue-light driven photocatalytic system in water.65 | ||
It is worth noting that the functionalization of amino acids and peptides represents one of the most extensively explored applications of aqueous-phase organic photocatalysis. Several bioorthogonal modifications of both biotic and abiotic peptide systems have been successfully demonstrated. Examples include benzylation of dehydroalanine residues in peptides and proteins,68 trifluoromethylation of aromatic moieties in biologically active peptides,69 selective arylation of cysteine,70 benzylic alkylation of tryptophan residues in peptides,71 and decarboxylative alkylation of C-terminal sites in naturally occurring proteins,72 to name a few.
For a comprehensive overview of these transformations and their mechanistic implications, readers are referred to recently published reviews on the topic.7,73,74
4.2. From neat water to soft-matter nanoreactors
Having established the feasibility and limitations of naked photocatalysts operating in neat water, we now trace a progressive shift toward soft-matter nanoreactors that engineer microenvironments at increasing levels of organisation and specificity. This transition—from micellar assemblies to supramolecular nanocapsules and finally to single-chain polymeric nanoparticles (SCNPs) —reflects a deliberate response to the hydrophilic milieu, substrate solubility, excited-state stability, and reaction-selectivity challenges inherent to aqueous photocatalysis. The overarching premise is that controlled confinement, polarity tuning, and unimolecular precision can transform water from a quenching medium into an enabling solvent for visible-light photocatalysis, culminating in enzyme-mimetic platforms (i.e., artificial photo-synthases, APS) with manifold reactivity (vide infra). In brief, these assemblies address three core gaps in water: the hydrophobic gap (solubilizing/reactant partitioning), confinement/effective molarity (raising local concentration), and polarity control (tuning microenvironments around excited states/intermediates).Building on this principle, micellar photocatalysis allows both photocatalytic units and lipophilic reactants to be solubilized in water, positioning the reaction partners within the confined, nonpolar environment of the nanoaggregate. This unique microenvironment not only enhances substrate compatibility but also offers opportunities for improved selectivity and efficiency in photocatalytic transformations.
A recent study by Lipshutz et al.77 on the aqueous photocatalyzed β-sulfonylation of aromatic olefins exemplifies the potential of this approach for expanding photocatalytic protocols into water-based media. The group designed a ubiquinol-inspired amphiphilic photocatalyst (PQS-Ir, Fig. 9) featuring a hydrophilic poly(ethylene glycol) methyl ether (MPEG) segment, a 50-carbon lipophilic side chain, and a cyclometalated iridium(III) tripyridyl complex linked via a phenoxy carboxylate group. This architecture enabled efficient hydroxylsulfonylation of aromatic alkenes and enol acetates in excellent yields across a broad substrate scope, without any organic solvents or additives, using only blue LED light as the energy source.
 | ||
| Fig. 9 Structure of the ubiquinol-based, amphoteric photocatalyst PQS-Ir and the schematic representation of the self-assembled micelles in water with reaction schemes of the β-hydroxysulfonylation of aromatic alkenes and the di-functionalization of aromatic enol acetates, enabled in water via micellar photocatalysis.77 | ||
Notably, PQS-Ir demonstrated remarkable recyclability, maintaining catalytic performance over multiple cycles before any significant loss of activity was observed. This work underscores how rational design of amphiphilic photocatalysts can overcome solubility challenges and unlock sustainable photocatalytic transformations in purely aqueous environments.
Giustiniano et al.8 recently reported a photocatalytic micellar reaction between N-methyl-N-alkyl aromatic amines and both aliphatic and aromatic isocyanides for the synthesis of functionalized amides. In this approach, the authors employed a readily prepared photoactive micellar system formed simply by mixing the photocatalyst with selected surfactants in water (structures shown in Fig. 10). Remarkably, replacing a blue LED light source with natural sunlight did not affect reaction yields under optimized conditions. Supported by extensive NMR studies confirming interactions between the photocatalyst, surfactant, and substrates, the method demonstrated broad substrate scope, good functional group tolerance, and recyclability of the photocatalyst. This micellar photo-induced amide synthesis represents a straightforward yet innovative strategy for achieving more sustainable conditions in well-established organic transformations.
 | ||
| Fig. 10 (a) Chemical structures of the selected surfactants and photocatalyst for micellar photocatalysis by Giustiniano et. al.8 (b) Pictorial representation of the general approach to photo-micellar catalysis followed for the light-driven preparation of amides,8 and the in water photoactivation of carbon-halide bonds.78 (c) Reaction scheme of the photocatalysed synthesis of amides from N-methyl-N-alkyl aromatic amines and both aliphatic and aromatic isocyanides.8 | ||
Using a similar approach, Giedyk et al.78 developed a controllable micellar photocatalytic strategy that enables activation of carbon–halogen bonds in water under mild conditions. The system employs commercially available methylene blue as the photocatalyst, blue LED light as the energy source, and inexpensive, well-characterized surfactants such as sodium dodecyl sulfate (SDS), Triton X, and cetyltrimethylammonium bromide (CTAB). Under these conditions, o-chlorobenzamides—such as 2-chloro-N,N-diisopropylbenzamide (2-CNIPB)—can undergo either reductive N-dealkylation or intramolecular C–H arylation, depending on the selected reaction parameters (Fig. 11).
 | ||
| Fig. 11 (a) Reaction scheme of the micellar photocatalysed activation of carbon-halide bonds, enabling controllable mechanistic switch between aromatic alkylation and dealkylative hydrogenation. (b) Reaction scheme of the Minisci-like halogenation of inactivated heteroarenes, enabled by photo-micellar catalysis. Both works were reported by Giedyk et al.78,79 | ||
The crucial role of pre-aggregation in micellar photocatalysis was further emphasized by the same group, who reported the photocatalytic aromatic halogenation of unactivated heteroarenes (Fig. 11).79 Notably, this transformation was completely suppressed when performed in organic solvents instead of aqueous SDS, highlighting the importance of micellar organization. These recent findings demonstrate that leveraging micellar effects—whether using standard or designer surfactants—offers virtually limitless possibilities in photocatalysis, enabling broad reaction scope, regioselectivity control, and straightforward preparation of reaction mixtures.
 | ||
| Fig. 12 Pictorial representation of the in water self-assembly of the nanocavities prepared by Akita et al.10 The V-shaped organic amphiphile spontaneously aggregate in aqueous environment, generating hydrophobic nanocavities in which lipophilic photocatalyst as PC can be hosted. | ||
Building on this concept, the same group employed this amphiphile to encapsulate three different photocatalysts (Scheme 3), enabling demethoxylative reductive cleavage of N–O bonds in Weinreb amides under mild aqueous conditions. This approach delivered good yields (62–88%) across a reasonably broad substrate scope, demonstrating the versatility of supramolecular assemblies for promoting photocatalytic transformations in water.80

Recently, a new class of polymeric scaffolds has been developed to mimic the folding behaviour of proteins into their native state.82,83 These structures -known as single-chain nanoparticles (SCNPs)- consist of individual synthetic polymer chains that collapse into compact conformations through intrachain interactions.84 SCNPs with ultra-small dimensions (2–20 nm) are emerging as ideal candidates for the development of advanced, next-generation light-activated catalysts operating in water.82–85 Despite their extensive use as nanoreactors for a wide range of organic transformations, only a handful of studies have explored their application in aqueous photocatalysis.
Palmans et al. reported two seminal studies demonstrating that water-soluble photoactive SCNPs can efficiently catalyse metal-free reductions and aromatic C–C cross-coupling reactions in water without any cosolvent.12,13 In these works, the authors introduced a systematic methodology for preparing stable, self-assembled photocatalytic SCNPs with defined structure and chemical composition.
In their first study, high-molecular-weight poly(pentafluorophenyl) acrylate precursors were post-functionalized with four distinct groups, including phenothiazine pendants, to ensure uniform molecular weight and impart photoredox activity (Fig. 13). The resulting amphiphilic copolymers spontaneously self-assembled in water to form well-defined noncovalent SCNPs, which were successfully applied to UV-light-induced reduction of haloarenes and cross-coupling of 2-cyanoiodobenzene with N-methylpyrrole in water in the presence of triethylamine (Scheme 4).12
 | ||
| Fig. 13 On top, the formation of non-covalent, photocatalytic SCNP upon spontaneous folding/collapse in water is depicted. The unimolecular polymeric aggregate enables the photocatalytic process to occur in its lipophilic, confined core. On the bottom, the synthetic route explored by Palmans et al. for the preparation of the amphiphilic photoredox SCNP polymeric precursor is shown.12 | ||
 | ||
| Scheme 4 Reaction schemes of the “in water”, light-driven transformation explored by Palmans’ group in their works on photocatalytic SCNPs (PC-SCNPs).12,13 On top, the reduction of C-halogen bonds of aromatic substrates. On the bottom, the cross-coupling of N-methyl pyrrolidone and iodoarenes. | ||
More recently, Palmans et al.86 reported direct C–H trifluoromethylation of (hetero)arenes in water using photoredox-active amphiphilic SCNPs, achieving high conversions, excellent recyclability, and operation under low-intensity LED or sunlight, thereby highlighting the utility of SCNPs for late-stage -CF3 introduction in aqueous media.
Barner-Kowollik et al.14 recently reported a SCNP-based nanoreactor capable of performing photooxidation of fatty acids in water under visible-light irradiation (Fig. 14). Through precise control of the polymer chain folding/collapse process using a rose bengal-derived crosslinker, the authors were able to tune the hydrophobicity of lipophilic pockets within the nanoreactor core. Remarkably, the photocatalytic SCNP exhibited up to a threefold increase in efficiency compared to the free photosensitizer. Molecular dynamics simulations supported this enhancement, attributing it to the increased polarity and high confinement within the active pockets provided by the SCNP architecture.
 | ||
| Fig. 14 Schematic illustration of SCNPs developed by Barner-Kowollik et al. capable of performing photooxidation of fatty acids in water under visible-light irradiation.14Fig. 14 reproduced from ref. 14, licensed under a Creative Commons Attribution-Noncommercial License (CC BY-NC). | ||
More recently, Pomposo et al.15 introduced the concept of artificial photo-syntases (APS), i.e. SCNPs with manifold visible-light photocatalytic activity for challenging “in water” organic reactions. In a pioneering work, a first generation of APS was reported enabling four distinct organic transformations in aqueous solution at room temperature under LED illumination (λmax = 450 nm): two reactions unprecedented in water—namely, [2 + 2] photocycloaddition of vinyl arenes and α-arylation of N-arylamines (a transformation first reported by MacMillan, 2021 Nobel Laureate in Chemistry)—alongside aerobic oxidation of 9-substituted anthracenes and β-sulfonylation of α-methylstyrene (Fig. 15).
 | ||
| Fig. 15 Schematic illustration of artificial photo-synthases (APS) developed by Pomposo et al.15 as enzyme-mimetic SCNPs endowed with manifold visible-light photocatalytic activity toward “in water” reactions. Fig. 15 reproduced from ref. 15, licensed under a Creative Commons Attribution 4.0 International License (CC BY 4.0). | ||
Due to their enzyme-like characteristics, APS-mediated [2 + 2] photocycloaddition kinetics were analyzed using the classical Michaelis–Menten model, yielding apparent kcat and KM values of 2.6 s−1 and 4.6 × 10−2 M, respectively.15 For comparison to kcat and KM values of enzymes, chymotrypsin exhibits kcat = 0.14 s−1 and KM = 1.5 × 10−2 M, pepsin kcat = 0.50 s−1 and KM = 3.0 × 10−4 M, and tRNA synthetase kcat = 7.6 s−1 and KM = 9.0 × 10−4 M.
Hence, while seminal SCNPs studies have demonstrated the feasibility of metal-free photoredox catalysis in water and visible light photooxidations within polymer derived nanoreactors, APS move beyond the “nanoreactor” paradigm by (i) organizing catalytic elements and substrate handling features to approximate active site–like functions within a single polymer chain, (ii) delivering multi-reaction capability in a single platform, and (iii) being quantitatively benchmarked through enzyme style kinetic analysis (Michaelis–Menten), thereby aligning catalytic performance metrics with those used for natural biocatalysts (kcat = 2.6 s−1; KM = 4.6 × 10−2 M for APS-mediated [2 + 2] photocycloaddition).
5. Summary and future outlook
The integration of soft-matter assemblies into photocatalytic systems represents a paradigm shift toward sustainable, water-compatible photochemistry (see Table 1).| Platform | Size range | Catalyst confinement | Catalyst location | Typical loading | Recyclability | Scope |
|---|---|---|---|---|---|---|
| Micellar systems | 5–20 nm | Lipophilic core | Into the core | Catalyst: µM–mM | Good | Broad scope |
| Substrates: mM | ||||||
| Supramolecular nanocapsules | 2–10 nm | Host–guest binding | Within the cavity | Catalyst: µM–mM | Moderate | Enhanced selectivity |
| Substrates: mM | ||||||
| Single-chain nanoparticles | 2–20 nm | Pocket polarity | Ultra-small pockets or within the core | Catalyst: µM–mM | Moderate to good | Multifunctionality |
| Substrates: mM |
Future research on soft-matter nanoreactors should prioritize the development of programmable assemblies with tuneable polarity, charge distribution and dynamic responsiveness, enabling selective activation of complex substrates and multi-step cascade reactions. In particular, APS-mediated visible-light photocatalysis opens new directions for green chemistry and biomimetic catalyst design by enabling challenging “in-water” reactions. Next-generation of APS could incorporate cooperative catalytic sites and adaptive folding mechanisms, bridging the gap between synthetic photocatalysis and natural photosynthesis.
The rational design of these systems will be accelerated by combining molecular dynamics simulations with artificial intelligence-driven predictive modelling, allowing performance optimization prior to experimental validation. Regarding reaction scope, future efforts will extend beyond reductions and oxidations to encompass asymmetric photocatalysis, stereo-selective transformations, and late-stage functionalization of bioactive molecules. Additionally, the integration of visible-light and solar-driven platforms with continuous-flow reactors and recyclable catalysts will be critical for achieving industrial-scale applications aligned with modern circular economy principles.
In the near term, we anticipate asymmetric photocatalysis in water and programmable cascades/late-stage functionalization enabled by the tandem co-immobilization of next-generation soft-matter nanoreactors (APS) and natural enzymes in continuous-flow photoreactors.
Interdisciplinary collaboration across photochemistry, polymer science, supramolecular chemistry and computational modelling will be essential to advance this field. Furthermore, the application of advanced spectroscopic techniques and ultrafast photodynamics will provide mechanistic insights, guiding the design of catalysts that combine efficiency, selectivity and environmental compatibility. In summary, the convergence of molecular design, soft-matter science, and photochemistry promises a future where light-driven processes become central to future green chemical manufacturing.
Conflicts of interest
There are no conflicts to declare.Data availability
Data sharing is not applicable to this article as no new data were created in this study.Acknowledgements
This work was supported by Grant PID2024-157988NB-I00 funded by MICIU/AEI/10.13039/501100011033 and ERDF/EU, Grant IT-1820-26 by Eusko Jaurlaritza (Basque Government), and Grant RED2025-CIE4-000037-01 by Gipuzkoako Foru Aldundia (Provincial Council of Gipuzkoa).Notes and references
- J. Zhang, B. Tia, L. Wang, M. Xing and J. Lei, Photocatalysis: Fundamentals, Materials and Applications, Springer, 2018 Search PubMed.
- V. Balzani, P. Ceroni and A. Juris, Photochemistry and Photophysics: Concepts, Research, Applications, Wiley-VCH, 2nd edn, 2024 Search PubMed.
- C. K. Prier, D. A. Rankic and D. W. C. MacMillan, Chem. Rev., 2013, 113, 5322 CrossRef CAS PubMed.
- J. W. Tucker and C. R. J. Stephenson, J. Org. Chem., 2012, 77, 1617 CrossRef CAS PubMed.
- J. M. R. Narayanam and C. R. J. Stephenson, Chem. Soc. Rev., 2011, 40, 102 RSC.
- P. Li, J. A. Terret and J. R. Zbieg, ACS Med. Chem. Lett., 2020, 11, 2120 CrossRef CAS PubMed.
- C. Russo, F. Brunelli, G. C. Tron and M. Giustiniano, J. Org. Chem., 2023, 88, 6284 CrossRef CAS.
- R. Cannalire, F. Santoro, C. Russo, G. Graziani, G. C. Tron, A. Carotenuto, D. Brancaccio and M. Giustiniano, ACS Org. Inorg. Au, 2022, 2, 66 CrossRef CAS PubMed.
- M. Cybularezyk-Cecotka, J. Predygier, S. Crespi, J. Szczepanik and M. Giedik, ACS Catal., 2022, 12, 3543 CrossRef.
- N. Noto, Y. Hyodo, M. Yoshizawa, T. Koike and M. Akita, ACS Catal., 2020, 10, 14283 CrossRef CAS.
- J. F. Thümmler and W. H. Binder, Chem. Commun., 2024, 60, 14332 RSC.
- F. Eisenreich, E. W. Meijer and A. R. A. Palmans, Chem. – Eur. J., 2020, 26, 10355 CrossRef CAS PubMed.
- F. Eisenreich, T. H. R. Kuster, D. van Krimpen and A. R. A. Palmans, Molecules, 2021, 26, 5882 CrossRef CAS PubMed.
- K. Mundsinger, B. T. Tuten, L. Wang, K. Neubauer, C. Kropf, M. L. O’Mara and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2023, 62, e202302995 CrossRef CAS PubMed.
- D. Arena, E. Verde-Sesto, I. Rivilla and J. A. Pomposo, J. Am. Chem. Soc., 2024, 146, 14397 CrossRef CAS PubMed.
- https://www.collinsdictionary.com/dictionary/english/photocatalysis (11 November 2025).
- https://www.oed.com/search/dictionary/?scope=Entries&q=photocatalysis (11 November 2025).
- https://www.merriam-webster.com/medical/photocatalysis (11 November 2025).
- https://goldbook.iupac.org/terms/view/P04580 (11 November 2025).
- H. Davy, Philos. Trans. R. Soc. London, 1814, 104, 557 CrossRef.
- T. Martin, J. Sci. Instrum., 1931, 8, 379 CrossRef CAS.
- https://catalogue.museogalileo.it/object/Lens.html (11 November 2025).
- https://goldbook.iupac.org/terms/view/P04588 (11 November 2025).
- H. Trommsdorff, Ann. Pharm., 1834, 11, 190 CrossRef.
- E. Becquerel, C. R. Acad. Sci. Paris, 1839, 9, 561 Search PubMed.
- A. Eibner, Chem.-Ztg., 1911, 35, 753 CAS.
- G. Ciamician, Science, 1912, 36, 385 CrossRef CAS PubMed.
- A. J. Allmand, Annu. Rep. Prog. Chem., 1925, 22, 333 RSC.
- H. Megan, J. Twilton and D. W. C. MacMillan, J. Org. Chem., 2016, 81, 6898 CrossRef PubMed.
- S. Dutta, J. E. Erchinger, F. Strieth-Kalthoff, R. Kleinmans and F. Glorius, Chem. Soc. Rev., 2024, 53, 1068 RSC.
- F. Strieth-Kalthoff, M. J. James, M. Teders, L. Pitzer and F. Glorius, Chem. Soc. Rev., 2018, 47, 7190 RSC.
- E. P. Farney and T. Yoon, Angew. Chem., Int. Ed., 2014, 53, 793 CrossRef CAS PubMed.
- C. Zhou, Z. Liu, Y.-Q. Liang, T. Lei, B. Chen, R.-Z. Liao, C.-H. Tung and L.-Z. Wu, Org. Lett., 2024, 26, 1116 CrossRef CAS.
- M. Jung, J. Ham and J. Song, Org. Lett., 2002, 4, 2763 CrossRef CAS PubMed.
- J. B. Metternich, D. G. Artiukhin, M. C. Holland, M. von Bremen-Kuhne, J. Neugebauer and R. Gilmour, J. Org. Chem., 2017, 82, 9955 CrossRef CAS PubMed.
- K. Teegardin, J. I. Day, J. Chan and J. Weaver, Org. Process Res. Dev., 2016, 20, 1156 CrossRef CAS PubMed.
- T. J. van Bergen, D. M. Hedstrand, W. H. Kruizinga and R. M. Kellogg, J. Org. Chem., 1979, 44, 4953 CrossRef CAS.
- K. Hironaka, S. Fukuzumi and T. Tanaka, J. Chem. Soc., Perkin Trans. 2, 1984, 1705 RSC.
- H. Cano-Yelo and A. Deronzier, Tetrahedron Lett., 1984, 25, 5517 CrossRef CAS.
- L. Buglioni, F. Raymenants, A. Slattery, S. D. A. Zondag and T. Noël, Chem. Rev., 2022, 122, 2752 CrossRef CAS PubMed.
- N. Zheludev, Nat. Photonics, 2007, 1, 189 CrossRef CAS.
- A. Bhattacharyya, S. De Sarkar and A. Das, ACS Catal., 2021, 11, 710 CrossRef CAS.
- N. Vallavoju and L. Sivaguru, Chem. Soc. Rev., 2014, 43, 4084 RSC.
- M. Raynal, P. Ballester, A. Vidal-Ferran and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2014, 43, 1660 RSC.
- M. J. Wiester, P. A. Ulmann and C. A. Mirkin, Angew. Chem., Int. Ed., 2011, 50, 114 CrossRef CAS PubMed.
- T. S. Koblenz, J. Wassenaar and J. N. H. Reek, Chem. Soc. Rev., 2008, 37, 247 RSC.
- C. J. Brown, F. D. Toste, R. G. Bergman and K. N. Raymond, Chem. Rev., 2015, 115, 3012 CrossRef CAS PubMed.
- J. Meeuwissen and J. N. H. Reek, Nat. Chem., 2010, 2, 615 CrossRef CAS PubMed.
- M. Yoshizawa, J. K. Klosterman and M. Fujita, Angew. Chem., Int. Ed., 2009, 48, 3418 CrossRef CAS PubMed.
- M. Morimoto, S. M. Bierschenk, K. T. Xia, R. G. Bergman, K. N. Raymond and F. D. Toste, Nat. Catal., 2020, 3, 969 CrossRef CAS.
- M. Yoshizawa, M. Tamura and M. Fujita, Science, 2006, 312, 251 CrossRef CAS PubMed.
- Q. Zhang, L. Catti, J. Pleiss and K. Tiefenbacher, J. Am. Chem. Soc., 2017, 139, 11482 CrossRef CAS PubMed.
- J. T. B. Govindjee, H. Gest and J. F. Allen, Discoveries in Photosynthesis, Advances in Photosynthesis and Respiration, Springer, 2005, vol. 20 Search PubMed.
- E. C. C. Baly, I. M. Heilbron and W. F. Barker, J. Chem. Soc., Trans., 1921, 119, 1025 RSC.
- F. Wöhler, Ann. Phys., 1828, 88, 253 CrossRef.
- B. H. Lipshutz, S. Ghorai and M. Cortes-Clerget, Chem. – Eur. J., 2018, 24, 6672 CrossRef CAS PubMed.
- H. Gröger, F. Gallou and B. H. Lipshutz, Chem. Rev., 2023, 123, 5262 CrossRef PubMed.
- J. K. Virdi, A. Dusunge and S. Handa, JACS Au, 2024, 4, 301 CrossRef CAS PubMed.
- G. La Sorella, G. Strukul and A. Scarso, Green Chem., 2015, 17, 644 RSC.
- Y. Hao, Y.-L. Lu, Z. Jiao and C.-Y. Su, Angew. Chem., Int. Ed., 2024, 63, e202317808 CrossRef CAS PubMed.
- D. Xue, Z.-H. Jia, C.-J. Zhao, Y.-Y. Zhang, C. Wang and J. Xiao, Chem. – Eur. J., 2014, 20, 2960 CrossRef CAS PubMed.
- P. Natarajan, D. Chuskit and S. Priya, Green Chem., 2019, 21, 4406 RSC.
- G. F. P. De Souza, J. A. Bonacin and A. G. Salles Jr, J. Org. Chem., 2018, 83, 8331 CrossRef CAS PubMed.
- M. K. Sahoo and E. Balaraman, Green Chem., 2019, 21, 2119 RSC.
- C. Kerzig and O. S. Wenger, Chem. Sci., 2019, 10, 11023 RSC.
- S. Pan, M. Jiang, J. Hu, R. Xu, X. Zeng and G. Zhong, Green Chem., 2020, 22, 336 RSC.
- X. Guo, Y. Okamoto, M. R. Schreier, T. R. Ward and O. S. Wenger, Chem. Sci., 2018, 9, 5052 RSC.
- R. C. W. von Lier, A. D. de Brujin and G. Roelfes, Chem. – Eur. J., 2021, 27, 1430 CrossRef PubMed.
- T.-T. H. Nguyen, C. J. O’Brien, L. N. T. Minh, S. H. Olson, N. S. Settineri, S. B. Prusiner, N. A. Paras and J. Conrad, Org. Lett., 2021, 23, 3823 CrossRef CAS PubMed.
- C. Bottecchia, M. Rubens, S. B. Gunnoo, V. Hessel, A. Madder and T. Noël, Angew. Chem., Int. Ed., 2017, 56, 12702 CrossRef CAS PubMed.
- Y. Yu, L.-K. Zhong, A. V. Buevich, G. Li, H. Tang, P. Vachal, S. L. Colletti and Z. C. Shi, J. Am. Chem. Soc., 2018, 140, 6797 CrossRef CAS PubMed.
- S. Bloom, C. Liu, D. K. Kölmel, J. X. Quiao, Y. Zhang, M. A. Poss, W. R. Ewing and D. W. C. MacMillan, Nat. Chem., 2018, 10, 205 CrossRef CAS PubMed.
- K. Sun, Q.-Y. Lv, X.-L. Chen, L.-B. Qua and B. Yu, Green Chem., 2021, 23, 232 RSC.
- Q. Dou and H. Zeng, Curr. Opin. Green Sustainable Chem., 2023, 40, 100766 CrossRef CAS.
- J. H. Fendler and E. J. Fendler, Adv. Phys. Org. Chem., 1970, 8, 271 CrossRef.
- L. Brüss, R. Jeyaseelan, J. C. G. Kürschner, M. Utikal and L. Naesborg, ChemCatChem, 2023, 15, e2022011 CrossRef.
- M.-J. Bu, C. Cai, F. Gallou and B. H. Lipshutz, Green Chem., 2018, 20, 1233 RSC.
- M. Cybularezyk-Cecotka, J. Predygier, S. Crespi, J. Szczepanik and M. Giedik, ACS Catal., 2022, 12, 3543 CrossRef.
- M. S. Santos, M. Cybularczyk-Cecotka, B. König and M. Giedyk, Chem. – Eur. J., 2020, 26, 15323 CrossRef CAS PubMed.
- Y. Hyodo, K. Takashi, Y. Chitose, M. Abe, M. Yoshizawa, T. Koike and M. Akita, Synlett, 2022, 1184 CAS.
- Y. Kita and Y. Amao, Sustainable Energy Fuels, 2021, 5, 6004 RSC.
- T. Terashima, T. Mes, T. F. A. De Greef, M. A. J. Gillissen, P. Besenius, A. R. A. Palmans and E. W. Meijer, J. Am. Chem. Soc., 2011, 133, 4742 CrossRef CAS PubMed.
- J. A. Pomposo, Polym. Int., 2014, 63, 589 CrossRef CAS.
- Single-Chain Polymer Nanoparticles: Synthesis, Characterization, Simulations, and Applications, ed. J. A. Pomposo, Wiley-VCH, 2017 Search PubMed.
- K. Mundsinger, A. Izuagbe, B. T. Tuten, P. W. Roesky and C. Barner-Kowollik, Angew. Chem., Int. Ed., 2024, 63, e202311734 CrossRef CAS PubMed.
- F. Eisenreich and A. R. A. Palmans, Chem. – Eur. J., 2022, 28, e202201322 CrossRef CAS PubMed.
Footnote |
| † Current address: Institute for Bioengineering of Catalonia (IBEC), 08028 Barcelona, Spain. |
| This journal is © The Royal Society of Chemistry 2026 |