
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Chemoselective nickel-catalyzed difluoromethylation of aryl halides with (SIPr)AgCF2H as transmetalation reagent
Torben Rogge
Institut für Chemie, Technische Universität Berlin, Straße des 17. Juni 115, 10623 Berlin, Germany. E-mail: torben.rogge@chem.tu-berlin.de
First published on 8th December 2025
Abstract
A user-friendly method for the difluoromethylation of aryl iodides under nickel catalysis has been developed, employing readily accessible, light- and temperature-insensitive (SIPr)AgCF2H as CF2H source. A variety of electronically diverse arenes was smoothly transformed and the reaction displayed excellent levels of chemoselectivity, tolerating inter alia synthetically valuable triflates.
Difluoromethylated molecules have attracted considerable attention due to their unique properties, such as an increased lipophilicity, strong hydrogen bonding and high metabolic stability. Furthermore, difluoromethyl substituents are considered bioisosteres of hydroxy, thio and amino groups and have consequently found widespread application in agrochemicals1 and medicinal chemistry.2 Over the past decade, a number of difluoromethylation strategies has been developed under thermal as well as photochemical conditions.3 In particular, transition metal-mediated or -catalyzed approaches have proven highly efficient, utilizing inter alia4 copper5 and palladium.6 In a seminal report, Vicic reported the direct difluoromethylation of aryl (pseudo-)halides under nickel catalysis (Scheme 1, top).7 Key to success was the use of a novel difluoromethyl source, (DMPU)2Zn(CF2H)2, which was accessed from ZnEt2 and HCF2I.8 In addition, Shen demonstrated a palladium/silver co-catalyzed difluoromethylation of aryl iodides and bromides, proceeding via in situ-generation of the (NHC)silver(I)difluoromethyl complex (SIPr)AgCF2H.9 The same group later extended their strategy towards the transformation of (hetero)aryl (pseudo-)halides, albeit requiring the use of stoichiometric amounts of pre-formed (SIPr)AgCF2H.10
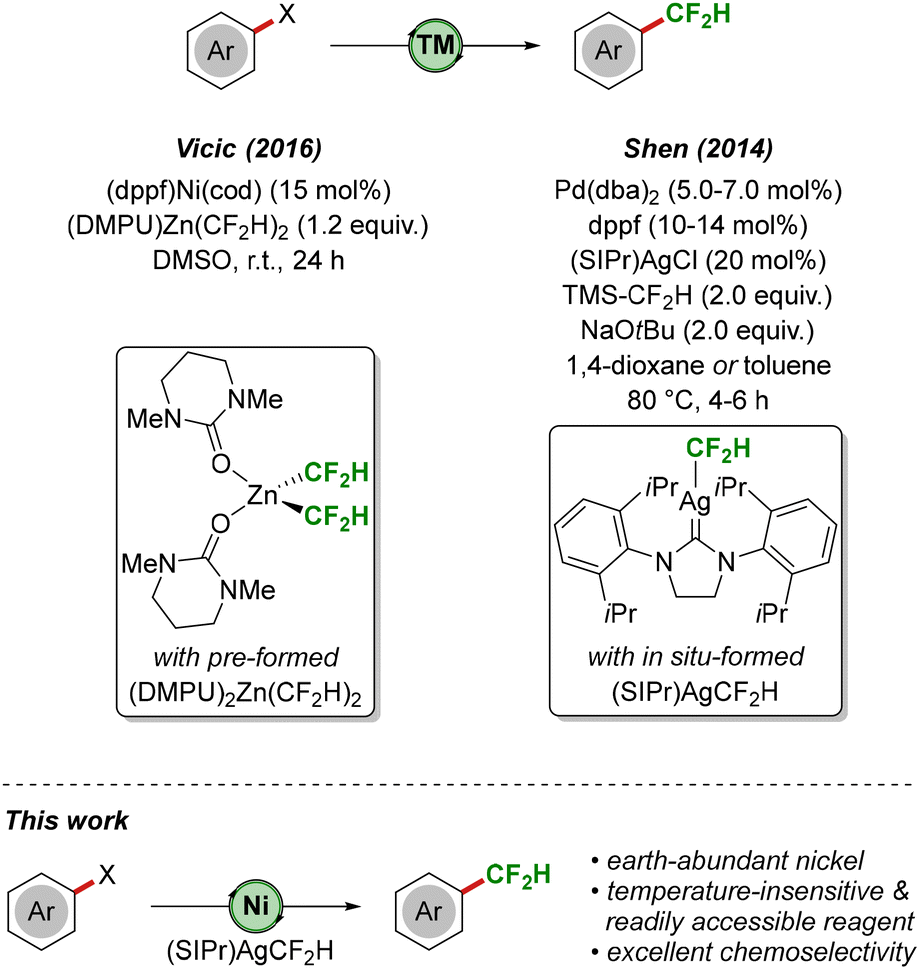 | ||
| Scheme 1 Transition metal-catalyzed difluoromethylation of aryl (pseudo-)halides. | ||
In light of the continuing demand for new, user-friendly strategies for the selective installation of difluoromethyl groups in organic molecules, we set out to explore whether (SIPr)AgCF2H can serve as a light- and temperature-insensitive, easily accessible reagent11 in earth-abundant nickel-catalyzed difluoromethylations of aryl halides, on which we report herein (Scheme 1, bottom). Notable results of this study comprise (i) an efficient transformation of aryl iodides bearing electron-donating as well as electron-withdrawing functional groups, (ii) excellent levels of chemoselectivity (I > Br ≫ OTf, Cl), (iii) very good scalability of the reaction, and (iv) the use of apolar reaction media and mild reaction conditions.
We initiated our investigation by exploring different reaction conditions for the envisioned difluoromethylation with (SIPr)AgCF2H, employing methyl 4-iodobenzoate (1a) as substrate (Table 1 and Table S-1 in the SI). While the use of 4,4′-bis(tert-butyl)-2,2′-bipyridine (dtbbpy) or dppe as ligand resulted in an unsatisfactory reaction outcome, significant amounts of product 2a were formed with dppf and Xantphos, with Xantphos proving optimal (entries 1–4). Increasing the amount of Xantphos to 15 mol% and employing toluene as reaction medium led to a further improved outcome (entries 5 and 6). Surprisingly, lowering the temperature to 60 °C proved beneficial, which can likely be attributed to a higher stability of the employed (SIPr)AgCF2H reagent at milder reaction temperatures (entry 7). The reaction also proceeded smoothly at even lower temperatures of 40 °C and room temperature, albeit with a decreased yield (entries 8-9). Replacing aryl iodide 1a with less reactive methyl 4-bromobenzoate (1a′) led to formation of the desired product 2a in 63% (entry 10). In contrast, 2a was detected in only trace amounts, when the corresponding triflate (1a″) or nonaflate (1a‴) was employed as the substrate (entry 11). Furthermore, no desired product was formed in the absence of either Xantphos or Ni(cod)2 (entries 12-13).
|
|||
|---|---|---|---|
| Entry | Ligand (x mol%) | T/°C | Yield/% |
| a Reaction conditions: 1a (0.10 mmol), Ni(cod)2 (10 mol%), L (10–15 mol%), (SIPr)AgCF2H (0.11 mmol), 1,4-dioxane (1.0 mL), 16 h. Yields were determined by GC analysis with n-decane as internal standard. Isolated yield (0.20 mmol scale) is shown in parenthesis.b In toluene (1.0 mL).c Aryl bromide (1a′) used instead of 1a.d Aryl triflate (1a″) or aryl nonaflate (1a‴) used instead of 1a.e Without Ni(cod)2. | |||
| 1 | dtbbpy (10) | 90 | 8 |
| 2 | dppe (10) | 90 | 9 |
| 3 | dppf (10) | 90 | 27 |
| 4 | Xantphos (10) | 90 | 46 |
| 5 | Xantphos (15) | 90 | 67 |
| 6b | Xantphos (15) | 90 | 80 |
| 7b | Xantphos (15) | 60 | 89 (70) |
| 8b | Xantphos (15) | 40 | 84 |
| 9b | Xantphos (15) | r.t. | 68 |
| 10bc | Xantphos (15) | 60 | 63 |
| 11bd | Xantphos (15) | 60 | Traces |
| 12b | — | 60 | n.d. |
| 13be | Xantphos (15) | 60 | n.d. |
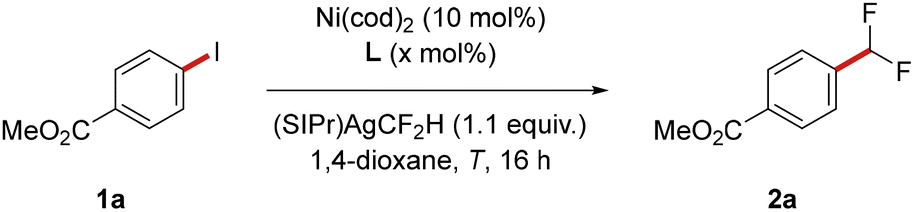
With the optimized reaction conditions identified, the scope of the nickel-catalyzed difluoromethylation was explored with diversely substituted aryl iodides and as well as some aryl bromides (Scheme 2). A broad range of electron-withdrawing substituents, such as ester (2a and 2b), acetyl (2m) and trifluoromethyl (2p), was well-tolerated in the para- and meta-position, even on 1.0 mmol scale. Substrates bearing sterically demanding substituents in ortho-position delivered the desired products 2c and 2i only in low yield and significant amounts of unreacted starting material were observed. Furthermore, substrates bearing electron-donating functional groups, e.g. ortho-, meta- and para-methylether (2d–2f), phenylether (2g) and thioether (2l), reacted smoothly. In contrast, alkyl substitution (2k and 2s) resulted in a significantly diminished conversion – an effect that was also previously noted by Vicic.7 In addition to phenyl substrates, the transformation was likewise applicable to naphthyl, phenanthryl and dibenzofuranyl halides, delivering the desired products 2h, 2i and 2j in excellent to low yield. Other heterocyclic substrates, such as benzofuran and benzothiophene, have thus far proven unsuccessful. Gratifyingly, and in contrast to previous reports,7 excellent levels of chemoselectivity were observed, when aryl di(pseudo-)halides were employed as substrates. When 4-iodophenyl triflate (1v) or 4-iodophenyl bromide (1w) were subjected to the reaction conditions, the reaction took place at the iodide, delivering products 2v and 2w in 52% and 64% yield, respectively, and preserving the (pseudo-)halide motif as a convenient synthetic handle for further diversifications. Surprisingly, when 4-iodobenzonitrile (1q), 4-iodo-nitrobenzene (1r) or 4-bromostyrene (1t) were employed as substrates, the desired products were only obtained in low yields, which can likely be attributed to an unfavorable coordination of the cyano, nitro and vinyl substituent to either the nickel catalyst or the silver reagent. Indeed, when the transformation of 1a was performed in the presence of 0.5 equiv. of styrene or benzonitrile, the yield dropped from 89% (Table 1, entry 7) to 77% and 54%, respectively (for details, see the SI). A carbaldehyde substituent was not tolerated under the reaction conditions and decomposition of the substrate was observed.
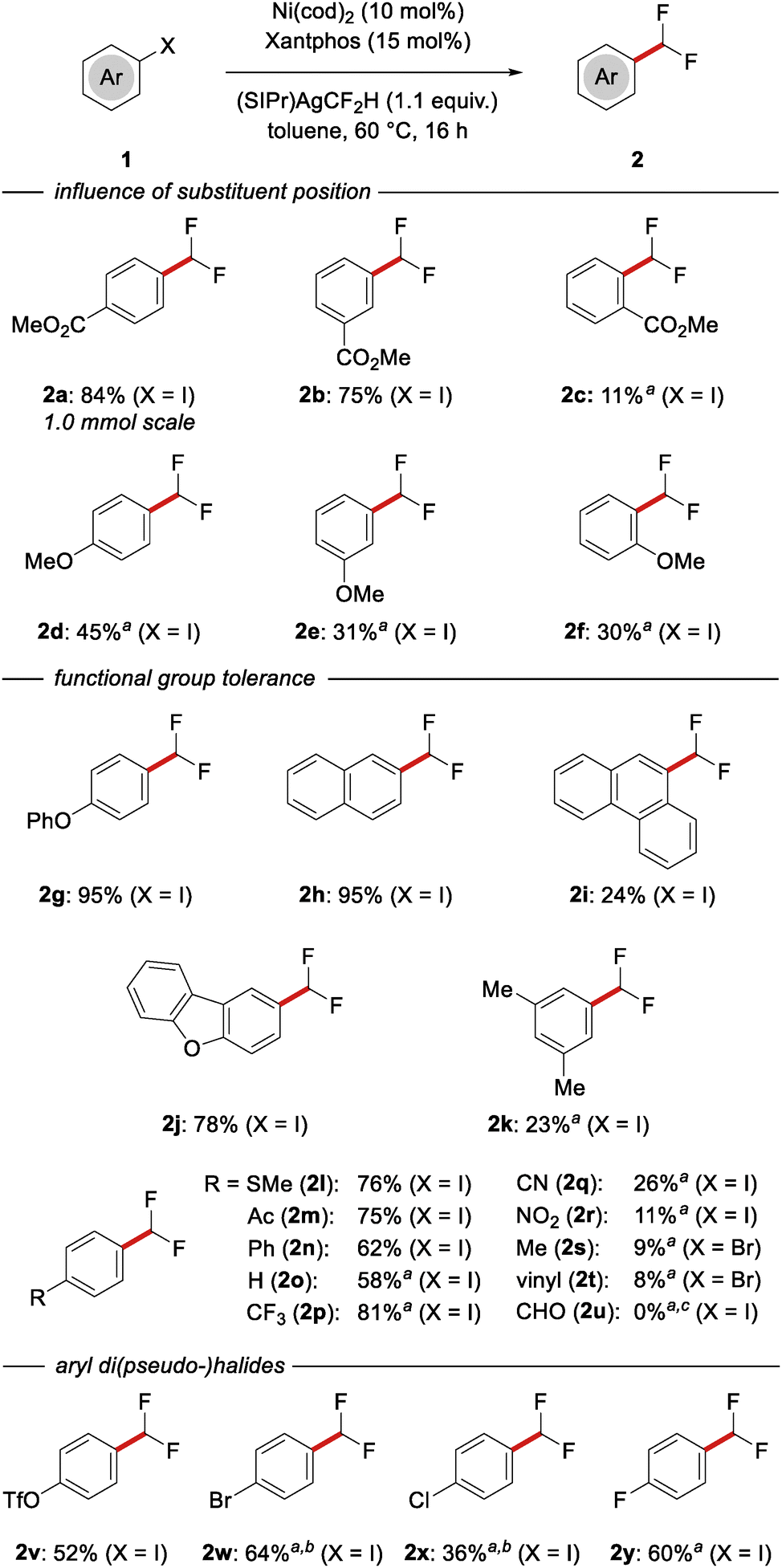 | ||
Scheme 2 Scope of the nickel-catalyzed difluoromethylation. a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Determined by 19F NMR spectroscopy with 4-fluorotoluene as internal standard. bTraces (< 2%) of disubstituted product were detected. cDecomposition of starting material was observed. Determined by 19F NMR spectroscopy with 4-fluorotoluene as internal standard. bTraces (< 2%) of disubstituted product were detected. cDecomposition of starting material was observed. | ||
In a next step, we investigated whether our approach is amendable to the in situ-formation of (SIPr)AgCF2H from bench-stable (SIPr)AgCl, (difluoromethyl)trimethylsilane and sodium tert-butoxide. When catalytic amounts of (SIPr)AgCl were employed, negligible amounts of desired product were detected (Scheme 3, top). However, in the presence of stoichiometric amounts of (SIPr)AgCl, (difluoromethyl)naphthalene (2h) was obtained in 34% isolated yield. A one-pot/two-step approach led to an improved efficacy of the transformation, delivering 2h in 63% yield and thus confirming that the use of isolated (SIPr)AgCF2H is not strictly required although being highly beneficial for an optimal reaction outcome (Scheme 3, bottom).
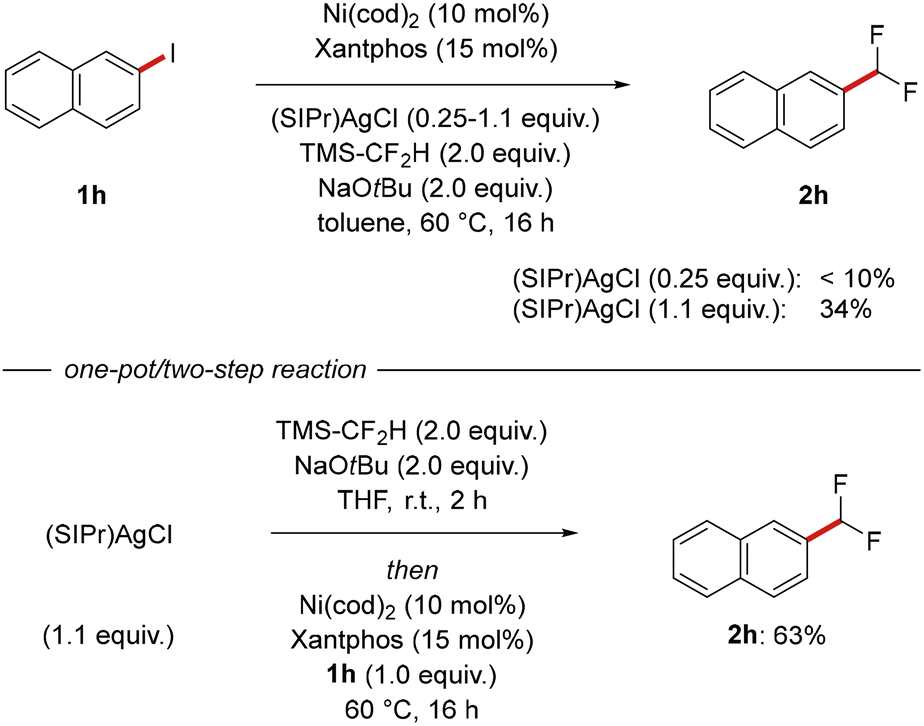 | ||
| Scheme 3 Difluoromethylation with in situ-generated (SIPr)AgCF2H. | ||
A plausible reaction mechanism proceeds with formation of a [(Xantphos)Ni]0 species, which can likely be stabilized through reversible coordination of a second Xantphos ligand. Thereafter, facile oxidative addition of aryl halide 1 takes place, followed by transmetalation with (SIPr)AgCF2H. Finally, reductive elimination results in the formation of difluoromethylated arene 2 and regenerates the catalytically active [(Xantphos)Ni]0 complex (Scheme 4).
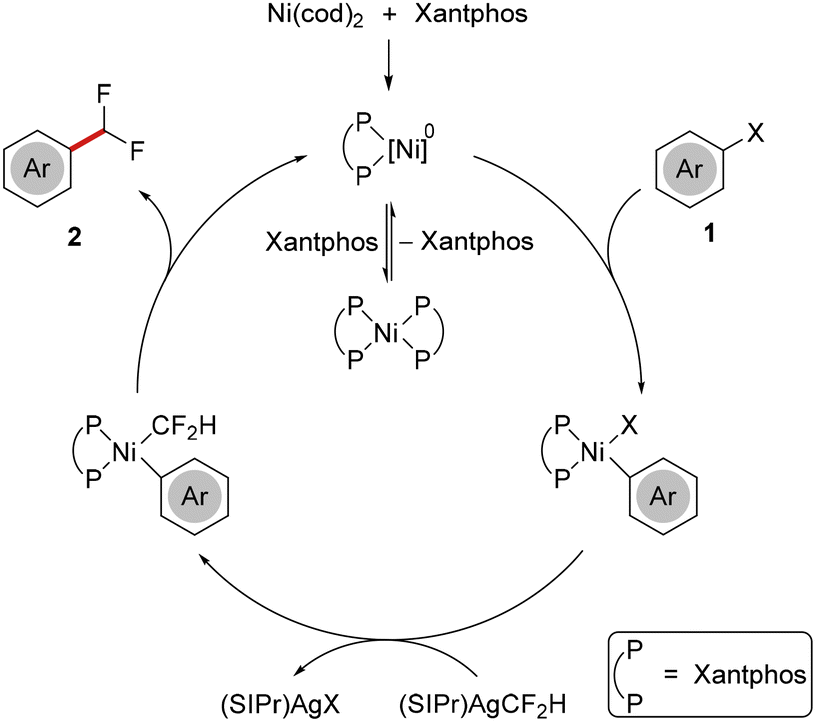 | ||
| Scheme 4 Plausible catalytic cycle (X = I, Br). | ||
In conclusion, a new strategy for difluoromethylations of aryl iodides and bromides via earth-abundant nickel catalysis with readily accessible and easy-to-handle (SIPr)AgCF2H as difluoromethyl source was developed. The transformation is applicable to a broad range of diversely substituted aryl iodides, tolerating inter alia synthetically useful triflates and bromides, and delivering the desired products in up to 95% isolated yield. Furthermore, in situ-formed (SIPr)AgCF2H could also be employed in our strategy, thus allowing for the installation of difluoromethyl motifs in a convenient manner.
Generous support by the Fonds der Chemischen Industrie (Liebig fellowship to T. R.) is gratefully acknowledged. The author thanks Prof. Dr Martin Oestreich and Dr Hendrik F. T. Klare (both TU Berlin) for helpful discussions and suggestions.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc06479a.References
- W. Zhang, P. Guo, Y. Zhang, Q. Zhou, Y. Sun and H. Xu, J. Agric. Food Chem., 2024, 72, 21344–21363 Search PubMed.
- (a) J. Ford, S. Ortalli and V. Gouverneur, Angew. Chem., Int. Ed., 2024, 63, e202404957 Search PubMed; (b) J. B. I. Sap, C. F. Meyer, N. J. W. Straathof, N. Iwumene, C. W. Am Ende, A. A. Trabanco and V. Gouverneur, Chem. Soc. Rev., 2021, 50, 8214–8247 Search PubMed.
- For reviews, see: (a) Z. Sun, X. S. Zhang, S. W. Bian, C. Zhang, Y. P. Han and Y. M. Liang, Org. Biomol. Chem., 2025, 23, 3029–3075 Search PubMed; (b) L.-J. Tang, Y. Wen, H.-X. Jin, W. Luo, L. Long, J. Shen, H. Luo and D. Yu, Org. Chem. Front., 2025, 12, 2052–2075 Search PubMed; (c) D. E. Yerien, S. Barata-Vallejo and A. Postigo, Chem. – Eur. J., 2017, 23, 14676–14701 Search PubMed; (d) Y. Lu, C. Liu and Q.-Y. Chen, Curr. Org. Chem., 2015, 19, 1638–1650 Search PubMed ; for selected examples, see: ; (e) R. Giri, E. Zhilin and D. Katayev, Chem. Sci., 2024, 15, 10659–10667 Search PubMed; (f) H. Zhao, X. B. Leng, W. Zhang and Q. Shen, Angew. Chem., Int. Ed., 2022, 61, e202210151 CrossRef CAS PubMed; (g) H.-S. Shi, S.-H. Li, F.-G. Zhang and J.-A. Ma, Chem. Commun., 2021, 57, 13744–13747 Search PubMed; (h) Q. Xiao, M. Lu, Y. Deng, J.-X. Jian, Q.-X. Tong and J.-J. Zhong, Org. Lett., 2021, 23, 9303–9308 CrossRef CAS PubMed; (i) V. Bacauanu, S. Cardinal, M. Yamauchi, M. Kondo, D. F. Fernández, R. Remy and D. W. C. MacMillan, Angew. Chem., Int. Ed., 2018, 57, 12543–12548 Search PubMed; (j) S. Liu, K. Kang, S. Liu, D. Wang, P. Wei, Y. Lan and Q. Shen, Organometallics, 2018, 37, 3901–3908 CrossRef CAS.
- (a) W. Du, Q. Luo, Z. Wei, X. Wang, C. Ni and J. Hu, Sci. China: Chem., 2023, 66, 2785–2790 Search PubMed; (b) P. García-Domínguez, Organometallics, 2021, 40, 2923–2928 Search PubMed; (c) C. Xu, W. H. Guo, X. He, Y. L. Guo, X. Y. Zhang and X. Zhang, Nat. Commun., 2018, 9, 1170 Search PubMed; (d) H. Motohashi and K. Mikami, Org. Lett., 2018, 20, 5340–5343 Search PubMed.
- (a) Y. Gu, C. Lu, Y. Gu and Q. Shen, Chin. J. Chem., 2018, 36, 55–58 CrossRef CAS; (b) J. R. Bour, S. K. Kariofillis and M. S. Sanford, Organometallics, 2017, 36, 1220–1223 CrossRef CAS; (c) H. Serizawa, K. Ishii, K. Aikawa and K. Mikami, Org. Lett., 2016, 18, 3686–3689 Search PubMed; (d) C. Matheis, K. Jouvin and L. J. Goossen, Org. Lett., 2014, 16, 5984–5987 Search PubMed; (e) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 5524–5527 CrossRef CAS PubMed.
- (a) X. Jiang, W. Gong, X. Li, S. Wang, Z. Gu, Y. Yang and X. Zeng, ACS Catal., 2024, 14, 13557–13566 Search PubMed; (b) D. M. Ferguson, C. A. Malapit, J. R. Bour and M. S. Sanford, J. Org. Chem., 2019, 84, 3735–3740 Search PubMed; (c) F. Pan, G. B. Boursalian and T. Ritter, Angew. Chem., Int. Ed., 2018, 57, 16871–16876 Search PubMed; (d) Z. Feng, Q. Q. Min, X. P. Fu, L. An and X. Zhang, Nat. Chem., 2017, 9, 918–923 CrossRef CAS PubMed; (e) K. Aikawa, H. Serizawa, K. Ishii and K. Mikami, Org. Lett., 2016, 18, 3690–3693 Search PubMed; (f) Z. Feng, Q.-Q. Min and X. Zhang, Org. Lett., 2016, 18, 44–47 Search PubMed; (g) X.-Y. Deng, J.-H. Lin and J.-C. Xiao, Org. Lett., 2016, 18, 4384–4387 Search PubMed.
- L. Xu and D. A. Vicic, J. Am. Chem. Soc., 2016, 138, 2536–2539 Search PubMed.
- S. Monfette, Y.-Q. Fang, M. M. Bio, A. R. Brown, I. T. Crouch, J.-N. Desrosiers, S. Duan, J. M. Hawkins, C. M. Hayward, N. Peperni and J. P. Rainville, Org. Process Res. Dev., 2020, 24, 1077–1083 CrossRef CAS.
- Y. Gu, X. Leng and Q. Shen, Nat. Commun., 2014, 5, 5405 CrossRef CAS PubMed.
- (a) C. Lu, H. Lu, J. Wu, H. C. Shen, T. Hu, Y. Gu and Q. Shen, J. Org. Chem., 2018, 83, 1077–1083 Search PubMed; (b) C. Lu, Y. Gu, J. Wu, Y. Gu and Q. Shen, Chem. Sci., 2017, 8, 4848–4852 Search PubMed.
- (a) H. Zhao, Y. Gu and Q. Shen, Chem. Rec., 2023, 23, e202300124 Search PubMed; (b) Y. Gu, D. Chang, X. Leng, Y. Gu and Q. Shen, Organometallics, 2015, 34, 3065–3071 Search PubMed; (c) D. Chang, Y. Gu and Q. Shen, Chem. – Eur. J., 2015, 21, 6074–6078 Search PubMed.
| This journal is © The Royal Society of Chemistry 2026 |