
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Controlled gold-catalyzed 5-exo-dig cyclization of 3-sulfur-substituted 1,3-dien-5-ynes for synthesizing fulvenes and the in situ reaction with indoles
Clara
Martínez-Núñez
 ,
Carlos
Arribas-Torrecilla
,
Roberto
Sanz
* and
Samuel
Suárez-Pantiga
*
,
Carlos
Arribas-Torrecilla
,
Roberto
Sanz
* and
Samuel
Suárez-Pantiga
*
Departamento de Química, Facultad de Ciencias, Universidad de Burgos, Pza. Misael Bañuelos s/n, 09001-Burgos, Spain. E-mail: rsd@ubu.es; svsuarez@ubu.es
First published on 22nd December 2025
Abstract
Densely substituted fulvenes have been synthesized from 1,3-dien-5-ynes by gold-catalyzed 5-exo-dig cyclization, determined by the S-substituent. In addition, these fulvenes interact with the gold catalyst, forming fulvenium intermediates that react with indoles in a one-pot two-step protocol to synthesize 3-(alkylidene-cyclopent-2-en-1-yl)-1H-indoles, by the cooperative action of gold and Brønsted acid catalysts.
1,3-Dien-5-ynes exhibit diverse and rich reactivity, allowing the construction of cyclic compounds.1 Among the strategies used to activate these species, gold catalysis is a selective and efficient strategy for forming densely functionalized carbocyclic cores.2 After alkyne activation by gold, 1,5-enynes almost entirely undergo endo cyclizations (Scheme 1A),3 typically involving the attack of nucleophiles. Similarly, 1,3-dien-5-ynes I experience analogous reactions (Scheme 1A) via 5-endo-dig,4 6-endo-dig (Scheme 1B),5 or even 7-endo-dig cyclizations when using 1,3,5-trien-7-ynes.6 By contrast, the 5-exo-dig pathway in 1,5-enynes is significantly more challenging, as evidenced by its limited observation. This pathway has been reported as a side reaction in two pioneering works where endo-cyclization processes were the primary reaction.7 Only recently, we successfully directed the reactivity exclusively toward the 5-exo-dig pathway in the alkoxycyclization reactions of (E)-1-monosubstituted 1,3-dien-5-ynes (Scheme 1C).8 The complications in achieving the 5-exo cyclization have been attributed to the nature of the intermediate cyclopropyl gold carbene.9 The favorable formation of bicyclo[3.1.0]hexane-type intermediates II leads the cyclization to endo-type processes (III–V), as opposed to the elusive alternative bicyclo[2.1.0]pentane-type intermediates VI required for exo processes. DFT calculations10 have suggested that these gold intermediates display both notable cyclopropyl (VI) character and a marked cationic behavior (VII), which is particularly pronounced in the case of 1,3-dien-5-ynes, allowing the irreversible trapping of these intermediates by primary alcohols.8 In the absence of the external nucleophile, 1,3-dien-5-ynes exclusively evolve through endo pathways. In this context, dienynes adequately substituted with electron-rich groups could stabilize key intermediates. We hypothesized that a thioether at the C3 position of the 1,3-dien-5-ynes, which are easily accessible from the corresponding 1-halo-2-thio-1,3-dienes, could provide exclusively 5-exo-dig cyclization modes through a thio-Nazarov11 pathway (Scheme 1D).
 | ||
| Scheme 1 (A) Reaction patterns of 1,3-dien-ynes under Au(I) catalysis. (B) 6-Endo-dig cyclization of 1,3-dien-5-ynes. (C) Au(I)-catalyzed 5-exo-dig alkoxycyclization of 1,3-dien-5-ynes. (D) This work. | ||
Densely substituted pentafulvenes have exceptional electronic properties and are valuable precursors in diverse transformations.12 A fine-tuning of their electronic properties and reactivity is easily accomplished by the nature of the substituents of the fulvene. The synthesis of polysubstituted benzofulvenes13,14 and non-fused pentafulvenes15 has attracted continuous interest. Metal-catalyzed protocols are practical in several cyclization reactions.14,15 Gold complexes by alkyne activation16 have enabled the design of elegant fulvene synthesis. In most cases, benzofulvenes were formed, and only the gold-catalyzed cycloisomerization of furan/ynes with a two-carbon tether in between the furan and the alkyne afforded enal or enone-decorated non-fused pentafulvenes via a cascade reaction involving 6-endo-dig cyclization followed by a furan ring-opening reaction.17 As well, 1,5-diynes have been used for preparing benzofulvenes,18 also pentalenes19 and azulenes.20 Also, benzofulvenes have been obtained from 1,6-diynes as starting materials,21 by the 5-endo-dig cyclization of o-alkynyl styrenes,4e from benzophenones by addition of propargyl silanes,22 from o-alkynyl benzaldehydes and diazo compounds,23 by rearrangement of alkynyl α,β-epoxy ketones,24 and substituted 3-propargyl indoles.25
To test our hypothesis, we prepared 1,3-dien-5-yne 1a bearing at position C3 a p-tolylthio substituent through the coupling of an alkyne with a sulfur-decorated halodiene,26 available from propargyl sulfides.27 After some optimization, evaluating different catalysts and solvents (see the SI), 1a was efficiently converted into fulvene 2a in high yields, using IPrAuNTf2 as a catalyst in MeOH under open air (Scheme 2), achieving similar yields under an inert atmosphere. Interestingly, using toluene as the solvent also yielded fulvene 2a to a similar extent to that of MeOH. Then, we studied the reaction scope by varying the aryl substituent at the R1 position (Scheme 2). Various para-substituted arenes were successfully converted into the corresponding fulvenes with arenes substituted with an alkyl or an aryl group (2b and c), a strong electron-donating group (OMe) (2d), or halides (2e). Notably, strong electron-withdrawing groups (EWG) afforded the fulvenes 2f and g almost as a single diastereoisomer. Both meta (2h–j) and ortho-substituted arenes (2k) were well-tolerated, although in this last case, the catalytic system (tBu)3PAuCl/AgNTf2 was used. Although this phosphine and the IPr ligand bear similar buried volumes,28 the shorter Au–C bond length (1.99 Å) relative to the Au–P bond length in phosphines (typically 2.23 Å) could cause sterically unfavorable interactions with bulkier substrates, as shown using the topographic steric maps calculated with both ligands using SambVca2 (see the SI).29 A heteroaryl group at R1, such as 3-thienyl, also yielded fulvene 2l. In addition, dienynes bearing TMS (1m) or (cyclo)alkyl (1n and o) groups at R1 provided the fulvenes in high yields with excellent diastereoselectivities. Interestingly, with dienyne 1m, the C–Si bond cleavage afforded fulvene 2m, unsubstituted at the exocyclic position. Alkyl groups at R2, R3, or R4 positions afforded products 2p–r. The bulky tert-butyl group provided lower diastereoselectivity. Various aryl or alkyl groups at the S-atom were well tolerated, obtaining the fulvenes in high yields and comparable diastereoselectivity (2s–v).
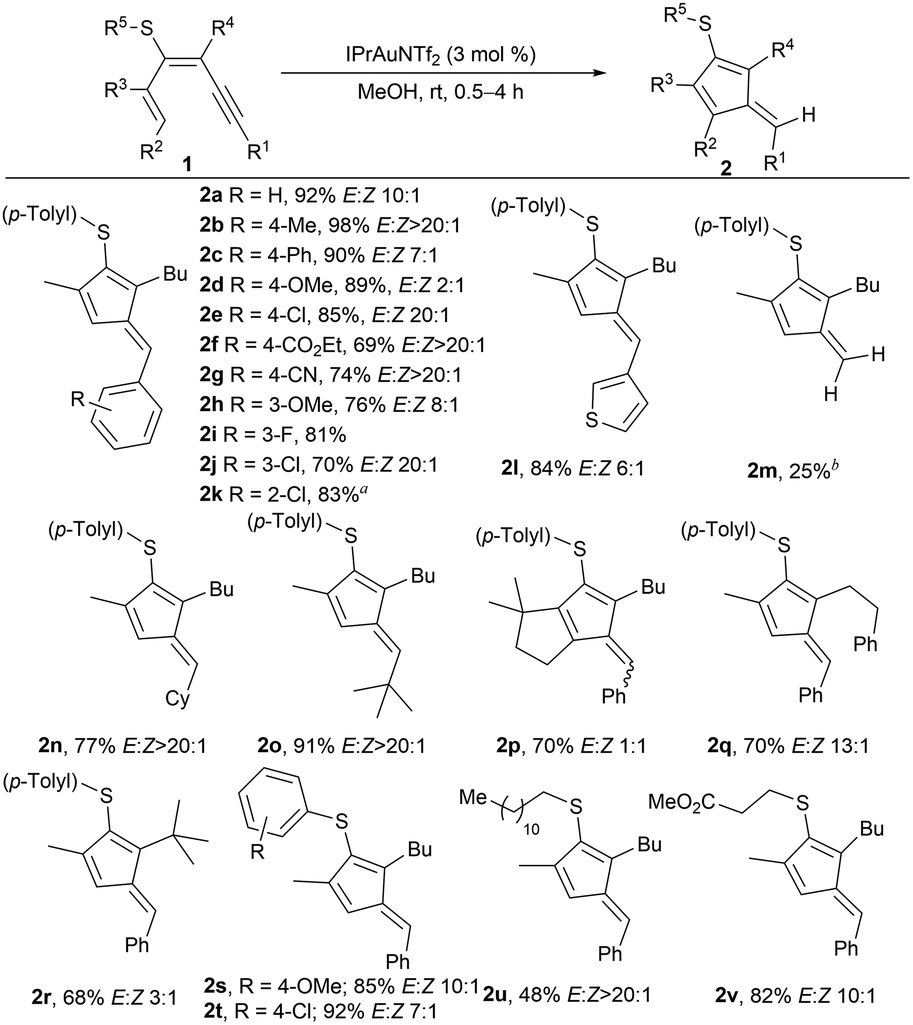 | ||
| Scheme 2 Scope of fulvenes 2 from 1,3-dien-5-ynes 1. Yields from isolated compounds after chromatography. Reaction conditions: 1 (0.3 mmol), IPrAuNTf2 (3 mol%), MeOH (3 mL) 30 min. (a) (tBu)3PAuCl/AgNTf2 (3 mol%) was used as the catalyst. (b) Dienyne 1m, where R1 = TMS, was used as the substrate. | ||
The influence of the organosulfur substituent was evaluated by subjecting 1,3-dien-5-ynes bearing sulfoxide (3a) or sulfone (4a) groups to the reaction (Scheme 3). Using MeOH as a solvent, no fulvenes were observed, and only products derived from the competitive addition of MeOH to the triple bond were detected. To avoid this path, the reaction was performed in toluene. In this case, sulfoxide 3a gave rise to fulvene 5a derived from the 5-exo-dig pathway, and a benzene derivative 6a arising from a 6-endo-dig cyclization in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, under heating at 60 °C, to achieve full conversion of 3a. The sulfone 4a at rt also delivered both cyclization products, fulvene 7a and diaryl sulfone 8a, although in this case the ratio was more favorable to the 6-endo-dig product 8a. Under these reaction conditions, no full conversion of 4a was observed, whereas heating produced similar results. These observations support a higher stabilization of cationic intermediates by the electron-donating nature of the thioether substituent and thus favor 5-exo-dig cyclization. Additionally, we evaluated whether MeOH plays a more active role in the process, facilitating protodemetallation. When dienyne 1a was subjected to the reaction in MeOD, double deuteration was observed at the exocyclic alkene position and at C4 of the cycle, affording di-deuterated 2a-D2 (Scheme 4a). A plausible mechanism involves alkyne activation by the gold complex (A), followed by 5-exo-dig cyclization, affording intermediate B that is stabilized by the heteroatomic substituent (Scheme 4b). The loss of a proton generates an organogold intermediate C, which, after solvent-facilitated deutero-demetallation, releases fulvene 2a-D and regenerates the gold catalyst. Product 2a-D could be activated by the cationic gold complex through the more nucleophilic C1 (C4) position of the fulvene, affording a fulvenium intermediate D that evolves upon elimination into organogold fulvene intermediate E. Finally, deutero-demetallation enabled by the deuterated solvent would give rise to the doubly deuterated fulvene 2a-D2. Based on these findings, we envisioned that fulvenes 2, upon activation by a gold catalyst, could evolve through nucleophilic addition reactions. Notably, this unconventional reactivity pattern involving the activation of fulvenes by electrophilic species has been scarcely reported, partly due to the tendency of fulvenes to experience oligomerization or polymerization processes after activation with a Brønsted acid.30 NMR studies at low temperature under superacid conditions revealed the formation of fulvenium cations.31 However, the reaction with an external nucleophile is challenging due to competitive oligomerization.30 In this context, Cu(OTf)2 has been reported to activate symmetric 6-mono- or 6,6-disubstituted fulvenes, followed by a reaction with indoles.32 Inspired by these results, we envisaged that fulvenes 2 could undergo analogous reactions upon activation with the gold catalyst present in the reaction media. Moreover, the different substitution patterns at positions C2 and C3 of the fulvene could play a crucial role in achieving regiospecific nucleophilic addition to one of these positions. Based on the formation of di-deuterated fulvene 2a-D2, we hypothesized that the addition of indole 9a (1 equiv.) to the reaction media could trigger a nucleophilic addition to the activated fulvene, achieving a one-pot two-step procedure to synthesize the 3-(alkylidene-cyclopent-2-en-1-yl)-1H-indole derivative 10aa (Scheme 5). In a preliminary experiment, the expected indole 10aa was obtained in a 54% yield directly from 1,3-dien-5-yne 1a. Interestingly, a Brønsted acid co-catalyst such as p-TsOH enhanced fulvene activation, affording compound 10aa in 88% yield. Under these conditions, regioisomer 11aa was also isolated in trace amounts. This by-product from a nucleophilic attack on the position C6 of the fulvene supported the participation of fulvenium ions (Scheme 5). Control experiments with isolated fulvene 2a, catalytic amounts of p-TsOH, and indole 9a (1 equiv.) in MeOH in the absence of the gold complex showed that compound 10aa was also formed, albeit in a lower yield, suggesting that the cooperative effect between the gold and the acid enables the fulvene heterohydroarylation.
:1 ratio, under heating at 60 °C, to achieve full conversion of 3a. The sulfone 4a at rt also delivered both cyclization products, fulvene 7a and diaryl sulfone 8a, although in this case the ratio was more favorable to the 6-endo-dig product 8a. Under these reaction conditions, no full conversion of 4a was observed, whereas heating produced similar results. These observations support a higher stabilization of cationic intermediates by the electron-donating nature of the thioether substituent and thus favor 5-exo-dig cyclization. Additionally, we evaluated whether MeOH plays a more active role in the process, facilitating protodemetallation. When dienyne 1a was subjected to the reaction in MeOD, double deuteration was observed at the exocyclic alkene position and at C4 of the cycle, affording di-deuterated 2a-D2 (Scheme 4a). A plausible mechanism involves alkyne activation by the gold complex (A), followed by 5-exo-dig cyclization, affording intermediate B that is stabilized by the heteroatomic substituent (Scheme 4b). The loss of a proton generates an organogold intermediate C, which, after solvent-facilitated deutero-demetallation, releases fulvene 2a-D and regenerates the gold catalyst. Product 2a-D could be activated by the cationic gold complex through the more nucleophilic C1 (C4) position of the fulvene, affording a fulvenium intermediate D that evolves upon elimination into organogold fulvene intermediate E. Finally, deutero-demetallation enabled by the deuterated solvent would give rise to the doubly deuterated fulvene 2a-D2. Based on these findings, we envisioned that fulvenes 2, upon activation by a gold catalyst, could evolve through nucleophilic addition reactions. Notably, this unconventional reactivity pattern involving the activation of fulvenes by electrophilic species has been scarcely reported, partly due to the tendency of fulvenes to experience oligomerization or polymerization processes after activation with a Brønsted acid.30 NMR studies at low temperature under superacid conditions revealed the formation of fulvenium cations.31 However, the reaction with an external nucleophile is challenging due to competitive oligomerization.30 In this context, Cu(OTf)2 has been reported to activate symmetric 6-mono- or 6,6-disubstituted fulvenes, followed by a reaction with indoles.32 Inspired by these results, we envisaged that fulvenes 2 could undergo analogous reactions upon activation with the gold catalyst present in the reaction media. Moreover, the different substitution patterns at positions C2 and C3 of the fulvene could play a crucial role in achieving regiospecific nucleophilic addition to one of these positions. Based on the formation of di-deuterated fulvene 2a-D2, we hypothesized that the addition of indole 9a (1 equiv.) to the reaction media could trigger a nucleophilic addition to the activated fulvene, achieving a one-pot two-step procedure to synthesize the 3-(alkylidene-cyclopent-2-en-1-yl)-1H-indole derivative 10aa (Scheme 5). In a preliminary experiment, the expected indole 10aa was obtained in a 54% yield directly from 1,3-dien-5-yne 1a. Interestingly, a Brønsted acid co-catalyst such as p-TsOH enhanced fulvene activation, affording compound 10aa in 88% yield. Under these conditions, regioisomer 11aa was also isolated in trace amounts. This by-product from a nucleophilic attack on the position C6 of the fulvene supported the participation of fulvenium ions (Scheme 5). Control experiments with isolated fulvene 2a, catalytic amounts of p-TsOH, and indole 9a (1 equiv.) in MeOH in the absence of the gold complex showed that compound 10aa was also formed, albeit in a lower yield, suggesting that the cooperative effect between the gold and the acid enables the fulvene heterohydroarylation.
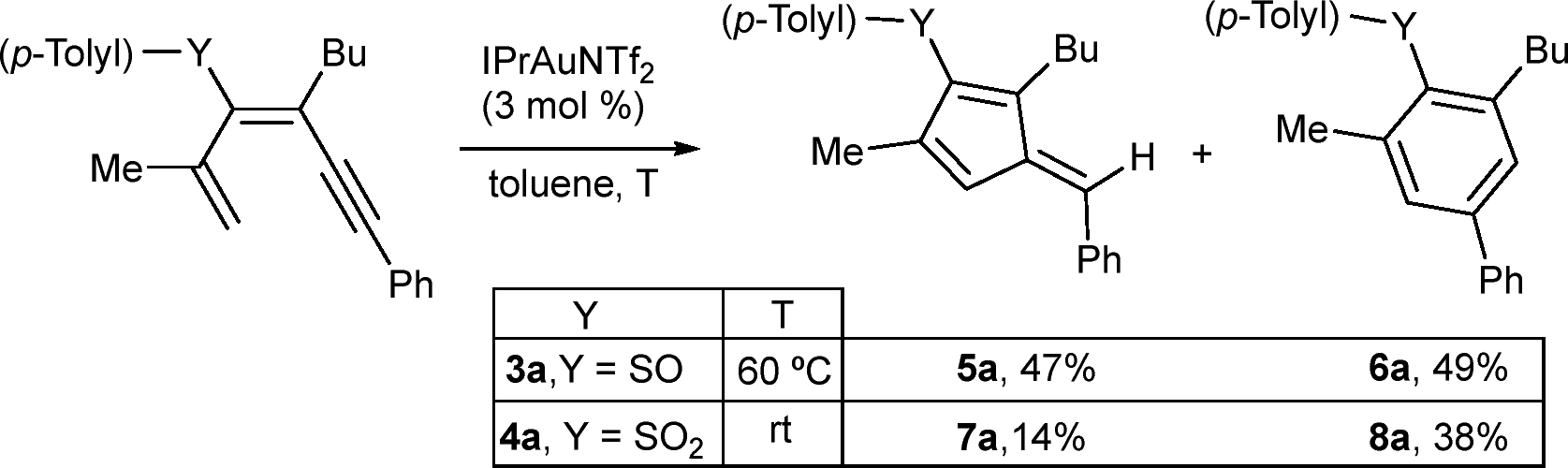 | ||
| Scheme 3 The effect of the nature of the S-atom substituent on the cyclization. | ||
 | ||
| Scheme 4 (a) Control experiments in MeOD. (b) Mechanism proposal. | ||
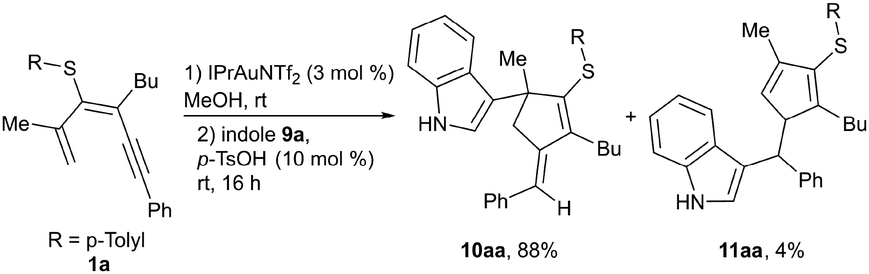 | ||
| Scheme 5 One-pot two-step synthesis of indole functionalized alkylidene cyclopentenes 10 from dienynes 1 and indoles 9. Reaction scale: 1 (0.3 mmol). | ||
Next, we evaluated the scope of the reaction with a selection of indoles 9 and 1,3-dien-5-ynes 1 in equimolar amounts, thereby accessing indole-decorated alkylidenecyclopentenes 10 (Scheme 6). Various dienynes 1 subjected to the one-pot two-step procedure afforded 3-(alkylidene-cyclopent-2-en-1-yl)-1H-indole products 10 (10ea, 10na, 10oa, 10qa, and 10ua). More nucleophilic N-methylindole 9b also led to the formation of compound 10ab, although other by-products were also detected in the crude. Substituents, such as methoxy (9c) or halogens (9d and e), at the benzenoid ring of the indole are well-tolerated, giving rise to the 3-functionalized indoles 10ac–ae. The structure of 10ae was further confirmed by single-crystal X-ray analysis (CCDC 2499730). Remarkably, in all cases, compounds 10 were obtained selectively as the E-isomer. A switch in the regioselectivity of the nucleophile addition was achieved with more nucleophilic 2-phenylindole (9f), leading to a product derived from the addition at the exocyclic C6 position of the fulvene. Purification attempts yielded only hydration and cleavage of the thioorganyl moiety, yielding the 2-cyclopentenone-decorated indole 12 as a mixture of diastereoisomers (Scheme 6). Its structure was also confirmed by single-crystal X-ray analysis (CCDC 2499841).
 | ||
| Scheme 6 Scope of indole functionalized alkylidene cyclopentenes 10 from dienynes 1. Reaction conditions: 1 (0.3 mmol), IPrAuNTf2 (3 mol%), MeOH (3 mL), 30 min; then indole 9 (0.3 mmol) and p-TsOH (10 mol%) 16 h. | ||
In summary, we have described a new gold-catalyzed approach for the synthesis of densely substituted fulvenes from readily available 3-thio-1,3-dien-5-ynes. This transformation is enabled through a scarcely reported gold-catalyzed 5-exo-dig cyclization mode of the activated 1,3-dien-5-yne. Control experiments have demonstrated that the nature of the S-atom substituent of the dienyne is crucial in favoring the selective 5-exo-dig cyclization over the more commonly described endo-dig cyclization pathways. Additional mechanistic studies have demonstrated that these fulvenes can be activated by the gold catalyst in the reaction media, leading to fulvenium intermediates. This interaction of the pentafulvene with electrophilic π-acid catalysts enables unusual reactivity patterns on the synthesized fulvenes that involve nucleophilic addition to the C3 (C2) position of the fulvene. To the best of our knowledge, this is the first report of a gold catalyst being practical in the electrophilic activation of a fulvene, enabling subsequent reaction with nucleophiles. Also, starting dienynes have been engaged in a one-pot two-step protocol to synthesize 3-(alkylidene-cyclopent-2-en-1-yl)-1H-indole derivatives. This process implies a gold-catalyzed 5-exo-dig cyclization of the 1,3-dien-5-yne affording a fulvene intermediate that, after addition of indole and p-TsOH as co-catalysts, evolves through functionalization of the indole at the C3 position by a nucleophilic attack on the fulvenium ion. The combination of the gold complex and the Brønsted acid facilitates this hydroheteroarylation step, achieving higher yields. Also, the gold complex plays a dual role in the one-pot process, acting as a catalyst in both steps. We expect that ongoing studies exploring the potential synthetic applications of these compounds and their optoelectronic properties will yield diverse applications for sulfur-decorated fulvenes.
The authors acknowledge the financial support from Ministerio de Ciencia, Innovación y Universidades (PID2023-148198NB-C21/AEI/10.1039/501100011033), and Junta de Castilla y León and FEDER (BU028P23). C.M.-N. thanks Consejería de Educación (Junta de Castilla y León) for a predoctoral contract. S.S.-P. thanks Ministerio de Ciencia, Innovación y Universidades and “NextGenerationEU”/PRTR EU for a Ramón y Cajal contract (RYC2021-031533-I).
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details and spectroscopic data. See DOI: https://doi.org/10.1039/d5cc06478c.Data for this article, including NMR raw data, are available at Zenodo at https://doi.org/10.5281/zenodo.17519909.
CCDC 2499730 (10ae) and 2499841 (12) contain the supplementary crystallographic data for this paper.33a,b
References
- (a) E. Aguilar, R. Sanz, M. A. Fernández-Rodríguez and P. García-García, Chem. Rev., 2016, 116, 8256–8311 CrossRef PubMed; (b) A. S. K. Hashmi, T. M. Frost and J. W. Bats, J. Am. Chem. Soc., 2000, 122, 11553–11554 CrossRef.
- (a) A. S. K. Hashmi, Chem. Rev., 2007, 107, 3180–3211 Search PubMed; (b) R. Dorel and A. M. Echavarren, Chem. Rev., 2015, 115, 9028–9072 CrossRef PubMed; (c) C. M. Hendrich, K. Sekine, T. Koshikawa, K. Tanaka and A. S. K. Hashmi, Chem. Rev., 2021, 121, 9113–9163 CrossRef PubMed; (d) A. S. K. Hashmi, Chem. Rev., 2021, 121, 8309–8310 CrossRef PubMed.
- (a) P. Y. Toullec, T. Blarre and V. Michelet, Org. Lett., 2009, 11, 2888–2891 CrossRef PubMed; (b) C. H. M. Amijs, V. Lopez-Carrillo, M. Raducan, P. Perez-Galan, C. Ferrer and A. M. Echavarren, J. Org. Chem., 2008, 73, 7721–7730 CrossRef PubMed; (c) A. K. Buzas, F. M. Istrate and F. Gagosz, Angew. Chem., Int. Ed., 2007, 46, 1141–1144 CrossRef PubMed; (d) L. Zhang, J. Sun and S. A. Kozmin, Adv. Synth. Catal., 2006, 348, 2271–2296 CrossRef; (e) L. Zhang and S. A. Kozmin, J. Am. Chem. Soc., 2005, 127, 6962–6963 CrossRef PubMed; (f) F. Gagosz, Org. Lett., 2005, 7, 4129–4132 CrossRef PubMed; (g) M. R. Luzung, J. P. Markham and F. D. Toste, J. Am. Chem. Soc., 2004, 126, 10858–10859 CrossRef PubMed.
- (a) A. Martínez, P. García-García, M. A. Fernández-Rodríguez, F. Rodríguez and R. Sanz, Angew. Chem., Int. Ed., 2010, 49, 4633–4637 CrossRef; (b) P. García-García, M. A. Rashid, A. M. Sanjuán, M. A. Fernández-Rodríguez and R. Sanz, Org. Lett., 2012, 14, 4778–4781 CrossRef; (c) A. M. Sanjuán, P. García-García, M. A. Fernández-Rodríguez and R. Sanz, Adv. Synth. Catal., 2013, 355, 1955–1962 CrossRef; (d) A. M. Sanjuán, M. A. Rashid, P. García-García, A. Martínez-Cuezva, M. A. Fernández-Rodríguez, F. Rodríguez and R. Sanz, Chem. Eur. J., 2015, 21, 3042–3052 CrossRef; (e) A. M. Sanjuán, C. Virumbrales, P. García-García, M. A. Fernández-Rodríguez and R. Sanz, Org. Lett., 2016, 18, 1072–1075 CrossRef PubMed; (f) C. Virumbrales, S. Suárez-Pantiga, M. Solas, M. A. Fernández-Rodríguez and R. Sanz, Org. Biomol. Chem., 2018, 16, 2623–2628 RSC; (g) C. Virumbrales, M. A. E. A. A. A. El-Remaily, S. Suárez-Pantiga, M. A. Fernández-Rodríguez, F. Rodríguez and R. Sanz, Org. Lett., 2022, 24, 8077–8082 CrossRef PubMed.
- (a) P. García-García, A. Martínez, A. M. Sanjuán, M. A. Fernández-Rodríguez and R. Sanz, Org. Lett., 2011, 13, 4970–4973 CrossRef; (b) J. Aziz, G. Frison, P. Le Menez, J.-D. Brion, A. Hamze and M. Alami, Adv. Synth. Catal., 2013, 355, 3425–3436 CrossRef.
- R. Eshagh Saatlo, J. Oczlon, J. F. Wunsch, M. Rudolph, F. Rominger, T. Oeser, F. Shiri, A. Ariafard and A. S. K. Hashmi, Angew. Chem., Int. Ed., 2024, 63, e202402481 CrossRef PubMed.
- (a) T. Shibata, Y. Ueno and K. Kanda, Synlett, 2006, 0411–0414 Search PubMed; (b) C. H. M. Amijs, V. López-Carrillo, M. Raducan, P. Pérez-Galán, C. Ferrer and A. M. Echavarren, J. Org. Chem., 2008, 73, 7721–7730 CrossRef PubMed.
- C. Virumbrales, S. Suárez-Pantiga, M. Marín-Luna, C. Silva López and R. Sanz, Chem. – Eur. J., 2020, 26, 8443–8451 CrossRef PubMed.
- (a) A. S. K. Hashmi, Angew. Chem., Int. Ed., 2008, 47, 6754–6756 CrossRef PubMed; (b) V. Lopez-Carrillo, N. Huguet, A. Mosquera and A. M. Echavarren, Chem. Eur. J., 2011, 17, 10972 CrossRef; (c) Y. Wang, M. E. Muratore and A. M. Echavarren, Chem. Eur. J., 2015, 21, 7332–7339 CrossRef PubMed; (d) R. Dorel and A. M. Echavarren, J. Org. Chem., 2015, 80, 7321–7332 CrossRef PubMed.
- E. García-Padilla, I. Escofet, F. Maseras and A. M. Echavarren, ChemPlusChem, 2024, 89, e202300502 CrossRef PubMed.
- N. Velasco, C. Martínez-Núñez, M. A. Fernández-Rodríguez, R. Sanz and S. Suárez-Pantiga, Adv. Synth. Catal., 2022, 364, 2932–2938 CrossRef.
- P. Preethalayam, K. S. Krishnan, S. Thulasi, S. S. Chand, J. Joseph, V. Nair, F. Jaroschik and K. V. Radhakrishnan, Chem. Rev., 2017, 117, 3930–3989 Search PubMed.
- (a) Muskan and A. K. Verma, Org. Lett., 2025, 27, 2328–2333 CrossRef PubMed; (b) J. Kikuchi, R. Nakajima, E. Kwon and N. Yoshikai, Org. Lett., 2025, 27, 9559–9564 CrossRef PubMed; (c) T. Okitsu, T. Yoshikawa, M. Morohashi, K. Aoki, T. Yakura, K. Sakata and M. Hatano, Org. Lett., 2024, 26, 1652–1656 CrossRef PubMed; (d) M. Humanes, E. Sans-Panadés, C. Virumbrales, A. Milián, R. Sanz, P. García-García and M. A. Fernández-Rodríguez, Org. Lett., 2024, 26, 6568–6573 CrossRef PubMed.
- (a) C. Li, Z. Wang and Z. Song, Adv. Synth. Catal., 2024, 366, 4503–4508 Search PubMed; (b) H. Goto, R. Shiomi, T. Shimizu, T. Kochi and F. Kakiuchi, Org. Lett., 2024, 26, 10152–10157 CrossRef PubMed; (c) G. Sreenivasulu, B. Sridhar and G. V. Karunakar, Org. Biomol. Chem., 2023, 21, 7799–7807 RSC.
- (a) H. Ohki, H. Kinoshita and K. Miura, Org. Lett., 2023, 25, 1331–1335 Search PubMed; (b) L.-J. Li, X. Wang, H. Xu and H.-X. Dai, Chem. Commun., 2023, 59, 3269–3272 RSC; (c) K. Goyal, G. A. Kukier, X. Chen, A. Turlik, K. N. Houk and R. Sarpong, Chem. Sci., 2023, 14, 11809–11817 RSC; (d) B. Español-Sánchez, J. Moradell, M. Galiana-Cameo, E. Barrenas, J. J. Pérez-Torrente, V. Passarelli and R. Castarlenas, Angew. Chem., Int. Ed., 2025, 64, e202507424 CrossRef.
- (a) Y. Fukuda and K. Utimoto, Synthesis, 1991, 975–978 CrossRef; (b) Y. Fukuda and K. Utimoto, J. Org. Chem., 1991, 56, 3729–3731 CrossRef; (c) Y. Fukuda and K. Utimoto, Bull. Chem. Soc. Jpn, 1991, 64, 2013–2015 CrossRef; (d) A. S. K. Hashmi, L. Schwarz, J.-H. Choi and T. M. Frost, Angew. Chem., Int. Ed., 2000, 39, 2285–2288 CrossRef; (e) M. Pernpointner and A. S. K. Hashmi, J. Chem. Theory Comput., 2009, 5, 2717–2725 CrossRef PubMed.
- Y. Chen and Y. Liu, J. Org. Chem., 2011, 76, 5274–5282 Search PubMed.
- (a) A. S. K. Hashmi, M. Wieteck, I. Braun, P. Nösel, L. Jongbloed, M. Rudolph and F. Rominger, Adv. Synth. Catal., 2012, 354, 555–562 CrossRef CAS; (b) P. Nösel, T. Lauterbach, M. Rudolph, F. Rominger and A. S. K. Hashmi, Chem. Eur. J., 2013, 19, 8634–8641 CrossRef PubMed; (c) A. Ahrens, J. Schwarz, D. M. Lustosa, R. Pourkaveh, M. Hoffmann, F. Rominger, M. Rudolph, A. Dreuw and A. S. K. Hashmi, Chem. Eur. J., 2020, 26, 5280–5287 CrossRef PubMed; (d) S. T. Fard, K. Sekine, K. Farshadfar, F. Rominger, M. Rudolph, A. Ariafard and A. S. K. Hashmi, Chem. Eur. J., 2021, 27, 3552–3559 CrossRef PubMed; (e) K. Sekine, K. Fuji, K. Kawashima, T. Mori and Y. Kuninobu, Chem. Eur. J., 2024, 30, e202403163 CrossRef PubMed.
- (a) T. Wurm, E. C. Ruediger, J. Schulmeister, S. Koser, M. Rudolph, F. Rominger, U. H. F. Bunz and A. S. K. Hashmi, Chem. – Eur. J., 2018, 24, 2735–2740 CrossRef PubMed; (b) K. Sekine, F. Stuck, J. Schulmeister, T. Wurm, D. Zetschok, F. Rominger, M. Rudolph and A. S. K. Hashmi, Chem. Eur. J., 2018, 24, 12515–12518 CrossRef PubMed.
- A. V. Mackenroth, A. Ahrens, J. F. Wunsch, R. Berger, F. Rominger, M. Rudolph and A. S. K. Hashmi, Adv. Synth. Catal., 2024, 366, 1331–1340 CrossRef.
- J.-J. Lian, P.-C. Chen, Y.-P. Lin, H.-C. Ting and R.-S. Liu, J. Am. Chem. Soc., 2006, 128, 11372–11373 CrossRef PubMed.
- S. Fernández, J. Santamaría and A. Ballesteros, Adv. Synth. Catal., 2022, 364, 1286–1294 CrossRef.
- A. S. Narode, Y.-S. Ho, M.-J. Cheng and R.-S. Liu, Org. Lett., 2023, 25, 1589–1594 CrossRef PubMed.
- L.-Z. Dai and M. Shi, Chem. – Eur. J., 2010, 16, 2496 CrossRef PubMed.
- E. Álvarez, D. Miguel, P. García-García, M. A. Fernández-Rodríguez, F. Rodríguez and R. Sanz, Synthesis, 2012, 1874–1884 Search PubMed.
- C. Martínez-Núñez, N. Velasco, R. Sanz and S. Suárez-Pantiga, Chem. Commun., 2024, 60, 1794–1797 RSC.
- N. Velasco, A. Suárez, F. Martínez-Lara, M. Á. Fernández-Rodríguez, R. Sanz and S. Suárez-Pantiga, J. Org. Chem., 2021, 86, 7078–7091 CrossRef PubMed.
- (a) H. Clavier and S. P. Nolan, Chem. Commun., 2010, 46, 841–861 RSC; (b) A. Gómez-Suárez, D. J. Nelson and S. P. Nolan, Chem. Commun., 2017, 53, 2650–2660 RSC.
- (a) L. Falivene, Z. Cao, A. Petta, L. Serra, A. Poater, R. Oliva, V. Scarano and L. Cavallo, Nat. Chem., 2019, 11, 872–879 CrossRef PubMed; (b) L. Falivene, R. Credendino, A. Poater, A. Petta, L. Serra, R. Oliva, V. Scarano and L. Cavallo, Organometallics, 2016, 35, 2286–2293 Search PubMed.
- C. Rentsch, M. Slongo, S. Schönholzer and M. Neuenschwander, Makromol. Chem., 1980, 181, 19–29 Search PubMed.
- (a) G. A. Olah, G. K. S. Prakash and G. Liang, J. Org. Chem., 1977, 42, 661–666 CrossRef; (b) G. K. S. Prakash, J. Org. Chem., 2006, 71, 3661–3676 Search PubMed.
- S. S. Chand, G. Gopalan, P. V. Santhini, P. Preethanuj, J. John, D. Harakat, F. Jaroschik and K. V. Radhakrishnan, Org. Lett., 2016, 18, 964–967 CrossRef PubMed.
- (a) CCDC 2499730: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2px5g8; (b) CCDC 2499841: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2px91z.
| This journal is © The Royal Society of Chemistry 2026 |