
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Fluorenyl-tethered N-heterocyclic carbene complexes of the heavy alkali metals
Stuart
Burnett
 a,
Alan R.
Kennedy
a,
Stephen M.
Mansell
*b and
Catherine E.
Weetman
*a
a,
Alan R.
Kennedy
a,
Stephen M.
Mansell
*b and
Catherine E.
Weetman
*a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, G1 1XL, UK. E-mail: catherine.weetman@strath.ac.uk
bInstitute of Chemical Sciences, School of Engineering and Physical Sciences, Heriot-Watt University, Edinburgh EH14 4AS, UK. E-mail: s.mansell@hw.ac.uk
First published on 8th December 2025
Abstract
The coordination chemistry of N-heterocyclic carbenes (NHCs) with Group 1 metals remains comparatively underdeveloped relative to their extensive application across the periodic table. Herein, we describe the synthesis of rubidium and caesium complexes incorporating a fluorenyl-tethered NHC ligand. Additionally, the first heterobimetallic Cs/Li NHC complex was obtained via treatment with a combination of alkali metal amide bases.
Since the first discovery of a stable N-heterocyclic carbene (NHC), this ligand class has become vital to modern synthetic chemistry.1,2 Their use as strong σ-donor ligands spans not only the transition metals but also increasingly into main group coordination chemistry, particularly in the stabilisation of reactive low oxidation state species that had previously been inaccessible via conventional routes.3 While NHCs have become one of the most commonly encountered ligand systems, their application in Group 1 metal complexes has remained largely confined to lithium,4–8 with only a select few examples of sodium and potassium NHC complexes reported in the literature.9–13 Notably, the only structurally characterised NHC complexes of Rb (Ic) and Cs (Id) were very recently detailed by Tamm and co-workers, who utilised a trimethylsilyl-protected anionic NHC to afford the complete series of alkali metal salt complexes via reaction with the corresponding alkali metal tert-butoxides (Fig. 1a).14 This scarcity of heavier alkali metal NHC complexes is unsurprising based on the trend of decreasing metal–carbene bond strength upon descending the group.15
 | ||
| Fig. 1 Reported examples of NHC Rb and Cs complexes (a) and tethered NHC alkali metal complexes (b) and (c). Dip = 2,6-diisopropylphenyl, Mes = 2,4,6-trimethylphenyl. | ||
Tethered NHC ligands – a typical carbene moiety bound to an ancillary donor group such as neutral N or P based groups,16–18 anionic groups such as aryl/alkoxide19,20 or polycyclic aromatic hydrocarbons e.g., fluorenyl, indenyl21 – have emerged as a versatile ligand subclass.22 Early notable examples using these anionic tethered NHC ligands in Group 1 metal complexes include a report from Arnold and co-workers in 2003 of the first structurally characterised, amido functionalised, NHC–Li complex.23 This was followed in 2005 by the same group where they describe the synthesis of the first potassium NHC complexes featuring an alkoxy-tethered NHC ligand (Fig. 1b).24 In the solid state, these form tetrameric [K-NHC] aggregates, where each potassium cation coordinates to three alkoxy-groups and one carbene centre. The Danopoulos group subsequently reported in 2007 a series of NHC–potassium complexes using fluorenyl- and indenyl-tethers (Fig. 1c), which served as effective ligand transfer reagents in the synthesis of various transition metal complexes.25–27 More recently, Mansell and co-workers were able to extend the use of fluorenyl-tethered saturated NHCs to access homobimetallic Li, Na and K complexes, employing a combination of metal hydrocarbyl/amide bases to initiate deprotonation and ring opening of the spirocyclic precursor.28,29 These complexes exhibit variable π-interactions between the alkali metal cation and the fluorenyl-tether with a variety of coordination modes observed (η2, η4, η5 and η6). Such π-stabilisation, which has been previously shown to play an important role in the isolation of heavy alkali metal complexes,30 suggested a promising potential route to the isolation of new Rb and Cs NHC complexes. Whilst the use of lighter alkali metals is commonplace within the synthetic chemist's toolkit,31,32 the heavier analogues are often overlooked. However, emerging trends suggest they shouldn’t be, with well-defined organocaesium complexes surpassing the catalytic activity of the lighter congeners.33–35 In this work, we describe the successful synthesis and isolation of homometallic Rb and Cs and heterobimetallic Cs/Li NHC complexes featuring fluorenyl-tethered NHC ligands. These findings illustrate the crucial role of the fluorenyl moiety in stabilising elusive heavy alkali metal complexes of this type.
While the reported synthesis of imidazolium salt 1 requires heating dioxane solutions of mesitylimidazole and 9-(2-bromoethyl)-9H-fluorene to 100 °C for 5 days (79%),26,36 improved yields (85%) and similar purity (as judged by 1H NMR spectroscopy) could be obtained by simply heating the aforementioned starting materials to 100 °C overnight in the absence of solvent. In line with previous reports, addition of one equivalent of nBuLi to −80 °C toluene solutions of 1 affords the corresponding free carbene in situ, which upon subsequent addition to toluene solutions of RbN(SiMe3)2 or CsN(SiMe3)2, respectively, yields the Rb and Cs complexes 2 and 3 (Scheme 1). Complexes 2 and 3 were isolated as air- and moisture-sensitive red powders in yields of 25% and 34%, respectively. Both are essentially insoluble in typical non-donor hydrocarbon solvents (hexane, benzene, toluene) but dissolve readily in THF. NMR spectroscopic analysis of both suggested structures consistent with those proposed in Scheme 1, with only minor shifts observed in the resonances between the two spectra (e.g. Mes-o-CH3 appears at 1.63 ppm for 2 and 1.67 ppm for 3). The carbenic carbon gives rise to a very weak resonance at 210.0 ppm in the 13C{1H} NMR spectrum of 2, while no chemical shifts attributable to the carbenic carbon were observed for 3. However, coupling was observed between the ethyl-bridge CH2 and a weak resonance at 212.7 ppm in its 1H–13C HMBC NMR spectrum (Fig. S10), allowing for assignment of the carbene centre in 3. These are noticeably at higher chemical shift when compared with similar carbene chemical shift ranges in related fluorenyl-tethered alkali metal complexes (cf.IIa at 198.7 ppm and IIIa at 206.9 ppm)24,25 but are slightly upfield relative to Tamm's anionic NHC complexes (216.9 ppm Rb (Ic) and 216.2 ppm Cs (Id), respectively).14 Complex 2 shows good stability in solution – heating THF-d8 solutions to 80 °C for three days results in only minor decomposition. In contrast, complex 3 exhibits more notable decomposition under the same conditions (approximately 60% of 3 remaining), with formation of at least two new mesityl-containing fragments observed in the 1H NMR spectrum (Fig. S11). No resonances were observed in the 133Cs NMR spectrum of 3 between the range of −200 ppm and +200 ppm, likely due to issues arising from the quadrupolar nature of 133Cs.
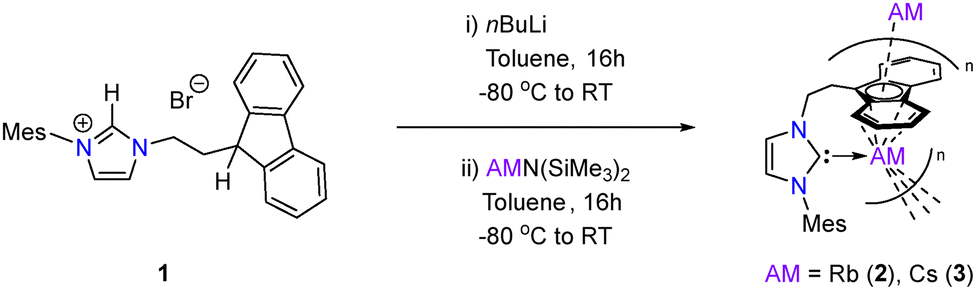 | ||
| Scheme 1 Synthesis of fluorenyl-tethered NHC Rb (2) and Cs (3) complexes. | ||
Recrystallisation of 2 from a layered THF/pentane solution, and 3 from a layered THF/benzene solution, afforded single crystals suitable for analysis by X-ray diffraction. The molecular structures of both are shown in Fig. 2 and are consistent with their spectroscopic data. Both complexes 2 and 3 crystallise in the monoclinic P21/c space group as a polymeric species, due to the cation–π interaction between the central Rb/Cs cation and the fluorenyl-tether, with the hapticity of the metal–aryl interactions ranging from η3 to η6 depending on the aryl substituent. No interaction between the metal centre and the mesityl substituent was observed, similar to the potassium derivatives reported by Danopoulos.25 The Rb–Ccarbene (3.067(3) Å and 3.088(3) Å) and Cs–Ccarbene (3.220(2) Å and 3.183(2) Å) bond lengths are among the longest reported for NHC–alkali metal bonds, as expected with the large ionic radii of both alkali metals, and represent some of the longest reported of the fluorenyl-tethered NHC alkali metal complexes.37 These are also in line with those reported for Tamm's NHC Rb and Cs complexes (Rb (Ic): 3.066(5) Å and 3.078(2) Å; Cs (Id): 3.3815(16) Å). As would be expected, the deviation from linearity of the M–NHC bond (pitch angle)38 increases from 17.8° in 2 to 21.6° in 3. A similar trend is noted across the series of alkali metal NHC complexes reported by Tamm.14
 | ||
| Fig. 2 Molecular structures (50% thermal ellipsoids) of 2 (polymer expansion of Rb2 network (A) and monomeric unit of Rb1 network (B)) and 3 (monomeric unit (C)). The fluorenyl ring system of the connecting unit in B and C is displayed in wireframe. Hydrogen atoms have been omitted from both for clarity. Selected bond lengths (Å) and angles (°): Complex 2 – Rb1–C1: 3.067(3), Rb1′–C6: 3.277(3), Rb1′–C7: 3.088(3), Rb1–C7: 3.284(3), Rb1–C8: 3.402(4), Rb1–C9: 3.421(4), Rb1–C10: 3.342(4), Rb1–C11: 3.251(3), Rb1′–C12: 3.238(3), Rb1–C12: 3.209(3), Rb1′–C13: 3.528(3), Rb1–C18: 3.543(3), Rb2–C28: 3.088(3), Rb2′–C34: 3.157(3), Rb2–C34: 3.327(4), Rb2′–C35: 3.363(4), Rb2–C35: 3.430(4), Rb2–C36: 3.472(4), Rb2–C37: 3.414(4), Rb2–C38: 3.310(4), Rb2–C39: 3.241(3), Rb2′–C39: 3.438(3); Rb1–C1–N1: 136.4(2), Rb1–C1–N2: 116.8(2). Complex 3 – Cs1–C1: 3.220(2), Cs1–C9: 3.361(2), Cs1′–C9: 3.523(2), Cs1–C10: 3.439(2), Cs1′–C10: 3.293(2), Cs1–C11: 3.562(2), Cs1′–C11: 3.405(2), Cs1–C12: 3.625(3), Cs1′–C12: 3.687(3), Cs1–C13: 3.589(3), Cs11–C14: 3.454(3), Cs2–C28: 3.183(2), Cs2′–C33: 3.524(2), Cs2–C40: 3.410(2), Cs2′–C40: 3.352(2), Cs2–C41: 3.410(3), Cs2–C42: 3.568(3), Cs2–C43: 3.651(3), Cs2–C44: 3.594(3), Cs2′–C44: 3.521(2), Cs2–C45: 3.426(2), Cs2′–C45: 3.186(2); Cs1–C1–N1: 110.6(15), Cs1–C1–N2: 140.2(16). | ||
From one such attempted synthesis of 3 a red crystalline solid was obtained, which was revealed to be the heterobimetallic Cs/Li complex 4 by single crystal X-ray diffraction analysis (Fig. 3). The formation of 4 likely arises from contamination of the CsN(SiMe3)2 used in this synthesis. A previous report from O’Hara and co-workers showed that altering the ratio of LiN(SiMe3)2 to CsF in the synthesis of CsN(SiMe3)2 affords a mixed metal [Cs/Li{N(SiMe3)}2]∞ species.39 The aforementioned molecular structure of 4 showed poor quality and thus is only included here for completeness, although the connectivity is consistent with solution state spectroscopic data (vide infra).
 | ||
| Fig. 3 Molecular structure of 4. Central Cs, Li, Si and N atoms shown with 50% thermal ellipsoids, carbon in capped sticks, methyl groups and connecting fluorenyl unit displayed in wireframe. Connectivity data only. | ||
Complex 4 could be independently synthesised using a mixture of pure CsN(SiMe3)2 and LiN(SiMe3)2 in a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio (Scheme 2). Similarly, addition of preformed [CsLi{N(SiMe3)2}2] to benzene-d6 suspensions of 3 resulted in the formation of 4 as judged by 1H NMR spectroscopy, albeit at a slower rate than the aforementioned method due to the poor solubility of 3 in benzene-d6. 4 was isolated as a red crystalline solid and showed good solubility in toluene and benzene. No decomposition of 4 was observed after heating benzene-d6 solutions to 80 °C for 3 days.
:1 ratio (Scheme 2). Similarly, addition of preformed [CsLi{N(SiMe3)2}2] to benzene-d6 suspensions of 3 resulted in the formation of 4 as judged by 1H NMR spectroscopy, albeit at a slower rate than the aforementioned method due to the poor solubility of 3 in benzene-d6. 4 was isolated as a red crystalline solid and showed good solubility in toluene and benzene. No decomposition of 4 was observed after heating benzene-d6 solutions to 80 °C for 3 days.
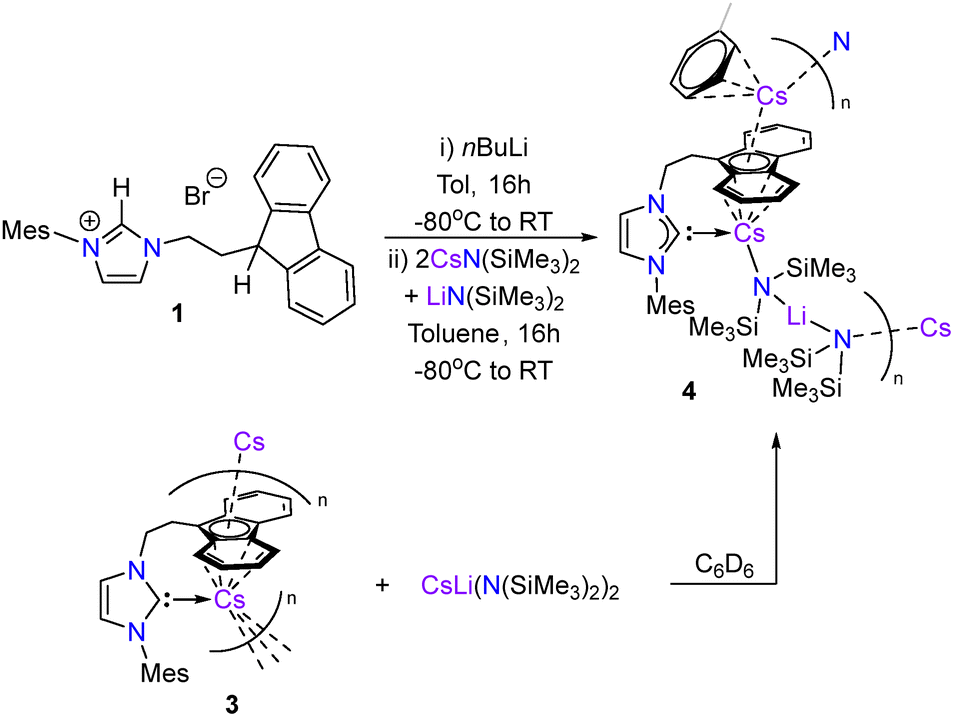 | ||
| Scheme 2 Synthesis of Cs/Li heterobimetallic NHC complex 4. | ||
All expected chemical shifts based on the solid-state structure were observed in the 1H NMR spectrum of 4, with similar environments observed for the mesityl- and fluorenyl-substituents as those observed in 3, while exhibiting a notable downfield shift at 0.33 ppm of the N(Si(CH)3)2 group in comparison to that previously reported for [CsLi{N(SiMe3)2}2] (0.27 ppm). Similar to 3, no resonance for the carbene centre was observed in the 13C{1H} NMR spectrum of 4, however, coupling was again seen in the 1H–13C HMBC spectrum between the ethyl-bridged CH2 and a weak resonance at 210.9 ppm. Only one broad resonance was observed in the 133Cs NMR spectrum (Fig. S18) as opposed to the expected two, again likely due to the quadrupolar nature of 133Cs, or the presence of a dynamic process in solution.
It is of note that the resonances attributable to the coordinated toluene molecule are observed at the same chemical shift as free toluene, suggesting dissociation in solution. Subsequent analysis by 1H-DOSY NMR spectroscopy confirms this (Fig. S19), whereby notable separation of the resonances attributable to monomeric complex 3, [CsLi{N(SiMe3)2}2] and toluene was observed. Multiple attempts to crystallise and analyse complex 4 by single crystal X-ray diffraction resulted in unit cell checks matching that found for 3, again affirming the ease with which 4 can dissociate in solution.
Using a fluorenyl-tethered NHC system, we have synthesised and structurally characterised only the second examples of N-heterocyclic carbene-stabilised Rb and Cs complexes, and the first examples of such heavy alkali metals stabilised by a fluorenyl-tethered NHC donor. A related Cs/Li heterobimetallic species could also be synthesised using a combination of the respective metal amide bases. As expected, these species contain some of the longest reported alkali metal–carbene bond lengths due to the large ionic radii of the metals involved. These species also highlight the importance of π-interactions in the stabilisation of such heavy alkali metal complexes whereas conventional monodentate-NHC ligands have in the past fallen short. With the rise of well-defined organocaesium complexes in catalytic applications, these compounds represent a significant synthetic achievement and one for future reactivity studies.
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. SB: writing – original draft, formal analysis, data curation. ARK: writing – review & editing, validation, formal analysis. SMM: writing – review & editing, validation, formal analysis, conceptualization. CEW: writing – review & editing, validation, supervision, project administration, funding acquisition, formal analysis, conceptualization.
We thank Craig Irving for assistance with NMR spectroscopy and Janie-Anne Pickrell for technical support. This work was supported by the EPSRC New Investigator Award [EP/Y015754/1]. CEW would like to thank the University of Strathclyde for the award of a Chancellor's Fellowship.
Conflicts of interest
There are no conflicts to declare.Data availability
Data that supports the findings of this study are available from the University of Strathclyde Knowledge Base Pure data repository at https://doi.org/10.15129/c6ec8b54-de92-4b76-9eb2-bb54af4c2fb4.The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: general synthetic experimental details, NMR spectra, X-ray crystallography. See DOI: https://doi.org/10.1039/d5cc06477e.
CCDC 2501678–2501680 contain the supplementary crystallographic data for this paper.40a–c
References
- F. E. Hahn and M. C. Jahnke, Angew. Chem., Int. Ed., 2008, 47, 3122–3172 CrossRef CAS PubMed.
- M. N. Hopkinson, C. Richter, M. Schedler and F. Glorius, Nature, 2014, 510, 485–496 CrossRef CAS PubMed.
- V. Nesterov, D. Reiter, P. Bag, P. Frisch, R. Holzner, A. Porzelt and S. Inoue, Chem. Rev., 2018, 118, 9678–9842 CrossRef CAS PubMed.
- R. Fränkel, C. Birg, U. Kernbach, T. Habereder, H. Nöth and W. P. Fehlhammer, Angew. Chem., Int. Ed., 2001, 40, 1907–1910 CrossRef.
- A. J. Arduengo, M. Tamm, J. C. Calabrese, F. Davidson and W. J. Marshall, Chem. Lett., 1999, 1021–1022 CrossRef CAS.
- M. Brendel, J. Wenz, I. V. Shishkov, F. Rominger and P. Hofmann, Organometallics, 2015, 34, 669–672 CrossRef CAS.
- J. Obenlüneschloß, N. Boysen, K. Rönnby, A. Muriqi, V. Hoffmann, C. Abad, D. Rogalla, U. Brokmann, E. Rädlein, M. Nolan and A. Devi, Angew. Chem., Int. Ed., 2025, e202513066 Search PubMed.
- A. Stasch, S. P. Sarish, H. W. Roesky, K. Meindl, F. Dall’Antonia, T. Schulz and D. Stalke, Chem. – Asian J., 2009, 4, 1451–1457 CrossRef CAS.
- M. S. Hill, G. Kociok-Köhn and D. J. MacDougall, Inorg. Chem., 2011, 50, 5234–5241 CrossRef CAS.
- D. R. Armstrong, S. E. Baillie, V. L. Blair, N. G. Chabloz, J. Diez, J. Garcia-Alvarez, A. R. Kennedy, S. D. Robertson and E. Hevia, Chem. Sci., 2013, 4, 4259 RSC.
- R. W. Alder, M. E. Blake, C. Bortolotti, S. Bufali, C. P. Butts, E. Linehan, J. M. Oliva, A. Guy Orpen and M. J. Quayle, Chem. Commun., 1999, 241–242 RSC.
- S. Bellemin-Laponnaz and S. Dagorne, Chem. Rev., 2014, 114(18), 8747–8774 CrossRef CAS PubMed.
- M. Bhandari and S. Yruegas, Eur. J. Inorg. Chem., 2025, e202400560 CrossRef CAS.
- L. J. Groth, D. Bockfeld and M. Tamm, Chem. Commun., 2025, 61, 15806–15809 RSC.
- K. C. Edwards, M. Vasiliu, J. W. Maxwell, C. E. Castillo, D. M. Marion, R. Craciun, J. F. Hall, D. Tapu and D. A. Dixon, J. Phys. Chem. A, 2023, 127, 10838–10850 CrossRef CAS PubMed.
- D. Pugh and A. A. Danopoulos, Coord. Chem. Rev., 2007, 251, 610–641 CrossRef CAS.
- O. Kühl, Chem. Soc. Rev., 2006, 36, 592–607 RSC.
- T. Simler, L. Karmazin, C. Bailly, P. Braunstein and A. A. Danopoulos, Organometallics, 2016, 35, 903–912 CrossRef CAS.
- S. Hameury, P. De Frémont and P. Braunstein, Chem. Soc. Rev., 2017, 46, 632–733 RSC.
- D. Zhang and G. Zi, Chem. Soc. Rev., 2015, 44, 1898–1921 RSC.
- K. J. Evans and S. M. Mansell, Chem. – Eur. J., 2020, 26, 5927–5941 CrossRef CAS PubMed.
- B. Royo and E. Peris, Eur. J. Inorg. Chem., 2012, 1309–1318 CrossRef CAS.
- P. L. Arnold, S. A. Mungur, A. J. Blake and C. Wilson, Angew. Chem., Int. Ed., 2003, 42, 5981–5984 CrossRef CAS PubMed.
- P. L. Arnold, M. Rodden and C. Wilson, Chem. Commun., 2005, 1743 RSC.
- S. P. Downing and A. A. Danopoulos, Organometallics, 2006, 25, 1337–1340 CrossRef CAS.
- S. P. Downing, S. C. Guadaño, D. Pugh, A. A. Danopoulos, R. M. Bellabarba, M. Hanton, D. Smith and R. P. Tooze, Organometallics, 2007, 26, 3762–3770 CrossRef CAS.
- S. P. Downing, P. J. Pogorzelec, A. A. Danopoulos and D. J. Cole-Hamilton, Eur. J. Inorg. Chem., 2009, 1816–1824 CrossRef CAS.
- K. J. Evans and S. M. Mansell, Chem. – Eur. J., 2019, 25, 3766–3769 CrossRef CAS PubMed.
- K. J. Evans, C. L. Campbell, M. F. Haddow, C. Luz, P. A. Morton and S. M. Mansell, Eur. J. Inorg. Chem., 2019, 4894–4901 CrossRef CAS.
- M. G. Davidson, D. Garcia-Vivo, A. R. Kennedy, R. E. Mulvey and S. D. Robertson, Chem. – Eur. J., 2011, 17, 3364–3369 CrossRef CAS PubMed.
- T. X. Gentner and R. E. Mulvey, Angew. Chem., Int. Ed., 2021, 60, 9247–9262 CrossRef CAS PubMed.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef CAS.
- P. A. Macdonald, S. Banerjee, A. R. Kennedy, A. van Teijlingen, S. D. Robertson, T. Tuttle and R. E. Mulvey, Angew. Chem., Int. Ed., 2023, 62, e202304966 CrossRef CAS.
- H.-Z. Du, J.-Z. Fan, Z.-Z. Wang, N. A. Strotman, H. Yang and B.-T. Guan, Angew. Chem., Int. Ed., 2023, 62, e202214461 CrossRef CAS PubMed.
- F. Krämer, M. H. Crabbe, I. Fernández and R. E. Mulvey, Angew. Chem., Int. Ed., 2025, 64, e202516376 CrossRef.
- J. Kukral, P. Lehmus, M. Klinga, M. Leskelä and B. Rieger, Eur. J. Inorg. Chem., 2002, 1349–1356 CrossRef CAS.
- Based on a search of the Cambridge Structural Database (CSD). Accessed 10th November 2025.
- N. Schwarz, X. Sun, R. Yadav, R. Köppe, T. Simler and P. W. Roesky, Chem. – Eur. J., 2021, 27, 12857–12865 CrossRef CAS.
- A. I. Ojeda-Amador, A. J. Martínez-Martínez, A. R. Kennedy and C. T. O’Hara, Inorg. Chem., 2016, 55, 5719–5728 CrossRef CAS.
- (a) CCDC 2501678: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pz696; (b) CCDC 2501679: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pz6b7; (c) CCDC 2501680: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pz6c8.
| This journal is © The Royal Society of Chemistry 2026 |