
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Turn left and turn right: recent advances in selectivity controlled carbonylation
Yang
Yuan
 a,
Chang-Sheng
Kuai
a and
Xiao-Feng
Wu
*ab
a,
Chang-Sheng
Kuai
a and
Xiao-Feng
Wu
*ab
aDalian National Laboratory for Clean Energy, Dalian Institute of Chemical Physics, Chinese Academy of Sciences, Dalian 116023, China. E-mail: xwu2020@dicp.ac.cn
bLeibniz-Institut für Katalyse e.V, Rostock 18059, Germany
First published on 8th December 2025
Abstract
The quest for selectivity is a central theme in modern organic synthesis, aiming to construct complex molecules with precision and efficiency. Transition-metal-catalyzed carbonylation, which utilizes carbon monoxide as an ideal C1 building block, offers a powerful platform for this endeavor. However, the presence of multiple reactive sites or selectivities in a substrate often leads to a mixture of products, posing a significant challenge to synthetic utility. A paramount goal is to develop divergent synthetic methods where a single set of starting materials can be selectively converted into multiple, distinct products by subtle adjustments to the reaction conditions. This review summarizes key advances from 2018 to 2025 that demonstrate how selectivity in carbonylation can be effectively controlled not by the inherent reactivity of the substrate, but by the catalytic system itself. Strategic manipulation of key variables, especially ligand engineering, but also the choice of base, additives, and other conditions, allows precise steering of reaction pathways to turn “left” or “right”. These principles have been successfully applied across different substrate classes, including alkenes, alkynes, 1,3-enynes, alcohols, imines, oxime esters, and organohalides, enabling access to structurally diverse products from common starting materials which illustrates a general and tunable approach to divergent synthesis.
 Yang Yuan | Yang Yuan was born in Anhui, China, in 1991. He received his MS degree in 2018 from Hangzhou Normal University and earned his PhD in 2021 from the Leibniz Institute for Catalysis (LIKAT), Germany. From 2022 to 2024, he conducted postdoctoral research at the Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences. Following his postdoc, he continued his research at DICP as a staff member in Prof. Xiao-Feng Wu's group. His current research is focused on carbonylation chemistry and asymmetric catalysis. |
 Chang-Sheng Kuai | Chang-Sheng Kuai was born in Liaoning, China. He received his MS degree in 2016 from Huaqiao University. Currently, he is a PhD candidate at the Dalian Institute of Chemical Physics (DICP), Chinese Academy of Sciences, supervised by Prof. Xiao-Feng Wu. His research focuses on the selective carbonylation transformations of unsaturated bonds, with particular emphasis on constructing highly efficient catalytic systems with tunable selectivity. |
 Xiao-Feng Wu | Xiao-Feng Wu was born and raised in China. After being educated and trained in China (Zhejiang Sci-Tech University), France (Rennes 1 University) and Germany (Leibniz-Institute for Catalysis), he started his independent research at LIKAT and ZSTU where he was promoted to professor in 2013 and afterwards defended his Habilitation from Rennes 1 University (2017). In 2020, he joined in Dalian Institute of Chemical Physics (DICP) and established a group on light carbons transformation and practical synthesis. Xiao-Feng has authored more than 670 publications, edited more than 10 books and filed many patents. |
1. Introduction
Selectivity is a foundational concept in modern organic synthesis, representing the capacity to steer chemical reactions toward specific products when multiple outcomes are possible. Achieving high selectivity is essential for the efficient and sustainable construction of complex molecules, including pharmaceuticals, agrochemicals, and advanced materials.1 By suppressing the formation of undesired byproducts, selective transformations improve reaction yields, streamline purification, enhance atom economy, and reduce chemical waste. Control over selectivity, whether it is chemoselectivity between functional groups, regioselectivity at the site of reaction, or stereoselectivity in the spatial arrangement of atoms, is typically achieved by strategically designing catalysts and fine-tuning reaction parameters.2Transition-metal-catalyzed carbonylation reactions have emerged as a powerful and atom-economical platform for the synthesis of carbonyl-containing compounds, employing carbon monoxide (CO) as an inexpensive and abundant C1 building block.3 The general mechanism often involves the formation of a metal–carbon bond, followed by the insertion of carbon monoxide (CO) to create an acyl–metal intermediate, which is then intercepted by a nucleophile to form the final product.4 Each of these steps presents opportunities for precise control and potential divergence.
The inherent complexity of many substrates, which may contain multiple reactive sites such as different C–X bonds, competing C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C and C
C and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds, or competing nucleophilic groups like –OH versus –NH2, makes achieving selectivity in carbonylation challenging.5 Furthermore, the possibility of mono- versus double-carbonylation,6 alongside the formation of regioisomers (e.g., linear vs. branched products from alkenes),7 further expands the spectrum of accessible structures (Fig. 1(a)). Consequently, the development of catalytic systems capable of exquisitely controlling reaction pathways to furnish a desired product represents a major achievement in modern synthetic chemistry. A significant goal in this field is to develop divergent syntheses, where distinct products can be selectively accessed from the same starting materials by simply modifying the catalyst or reaction conditions.8 Controlling the chemo-, regio-, and stereoselectivity of carbonylation reactions thus presents a formidable challenge but also provides a compelling opportunity to rapidly build molecular complexity from simple feedstocks. A ligand bound to a transition metal is arguably the most powerful tool for controlling selectivity.9 Its electronic properties10 and steric profile11 directly modulate the metal center's reactivity, dictating which mechanistic pathway is favored. While ligands are a primary tool, selectivity can also be finely tuned through other parameters. The choice of base, the addition of specific additives, or adjustments made to reaction conditions, such as temperature and pressure, can profoundly influence the catalytic cycle, enabling access to divergent products.12
C bonds, or competing nucleophilic groups like –OH versus –NH2, makes achieving selectivity in carbonylation challenging.5 Furthermore, the possibility of mono- versus double-carbonylation,6 alongside the formation of regioisomers (e.g., linear vs. branched products from alkenes),7 further expands the spectrum of accessible structures (Fig. 1(a)). Consequently, the development of catalytic systems capable of exquisitely controlling reaction pathways to furnish a desired product represents a major achievement in modern synthetic chemistry. A significant goal in this field is to develop divergent syntheses, where distinct products can be selectively accessed from the same starting materials by simply modifying the catalyst or reaction conditions.8 Controlling the chemo-, regio-, and stereoselectivity of carbonylation reactions thus presents a formidable challenge but also provides a compelling opportunity to rapidly build molecular complexity from simple feedstocks. A ligand bound to a transition metal is arguably the most powerful tool for controlling selectivity.9 Its electronic properties10 and steric profile11 directly modulate the metal center's reactivity, dictating which mechanistic pathway is favored. While ligands are a primary tool, selectivity can also be finely tuned through other parameters. The choice of base, the addition of specific additives, or adjustments made to reaction conditions, such as temperature and pressure, can profoundly influence the catalytic cycle, enabling access to divergent products.12
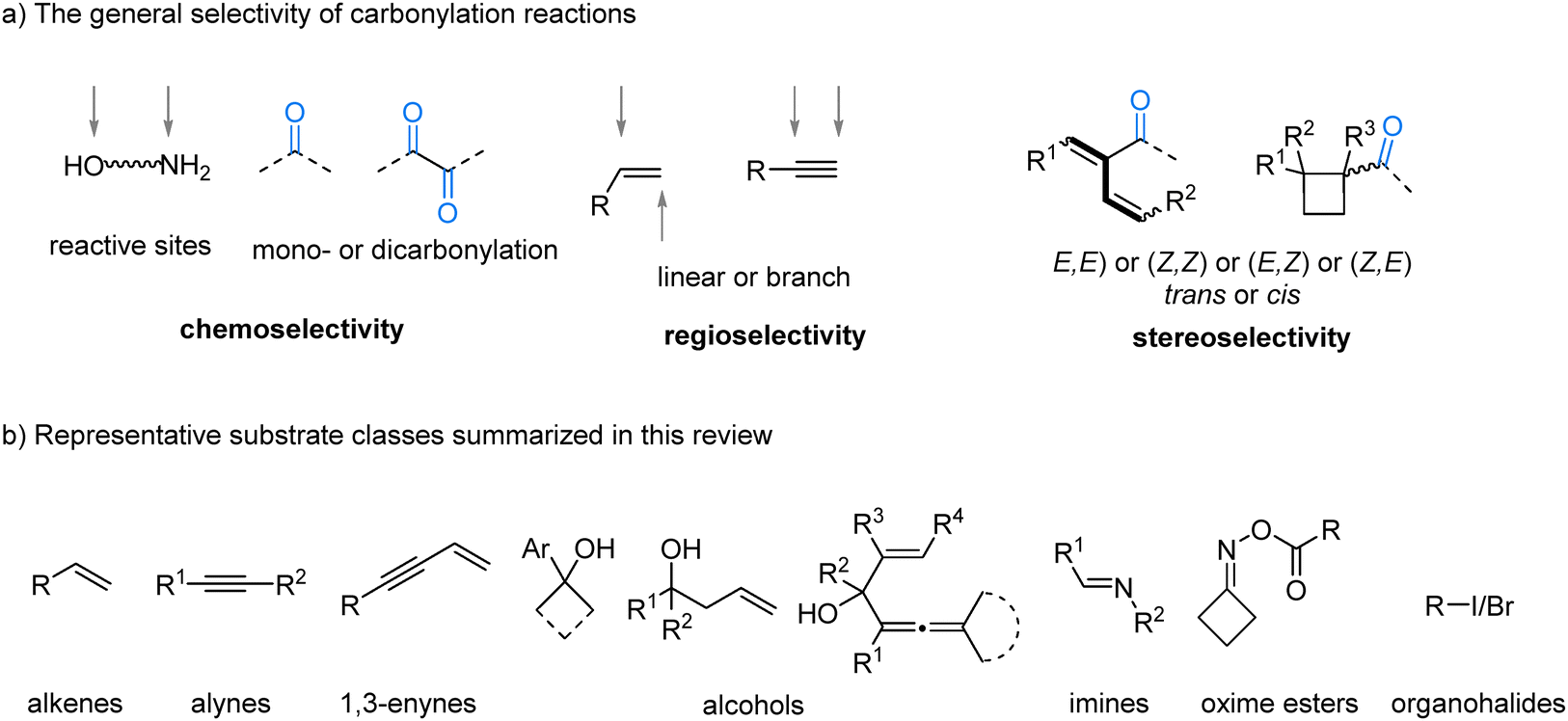 | ||
| Fig. 1 The general selectivity of carbonylation reactions and representative substrate classes. | ||
In this review, we summarize recent advances in controlling selectivity in carbonylative transformations using gaseous CO as the C1 source, through the systematic modulation of reaction parameters since 2018. By exploring diverse substrate classes, including alkenes, alkynes, 1,3-enynes, alcohols, imines, oxime esters, and organohalides, we illustrate how selective carbonylative reactions can be achieved (Fig. 1(b)). This strategy is grounded in the fundamental principles of selectivity control, with particular emphasis placed on tailored ligand design to govern regio- and chemoselectivity, and the ability to redirect reaction pathways through precise adjustments of reaction conditions.
2. Selectivity controlled carbonylation of alkenes
Alkenes serve as fundamental and highly versatile chemical feedstocks, amenable to transformation into a diverse array of functional compounds.13 Among these transformations, transition-metal-catalyzed carbonylation reactions provide an efficient pathway to valuable carboxylic acid derivatives. A key characteristic of alkene carbonylation is the potential formation of two distinct regioisomers: linear and branched products.7 As these isomers often possess different properties and applications, achieving control over the regioselectivity is of significant importance. Considerable efforts have been devoted to developing complementary catalyst systems for key carbonylation processes, including hydroformylation,14 alkoxycarbonylation,15 aminocarbonylation,16 and various hydrofunctional carbonylation.17 These systems enable selective formation of either linear or branched products, with regioselectivity primarily controlled by the choice of ligand. For instance, in palladium catalysis, monodentate phosphine ligands typically favor branched products, while bidentate chelating phosphine ligands tend to promote the formation of linear products.14–172.1. Cu-catalyzed regiodivergent borocarbonylation of unactivated alkenes
Copper-catalyzed carbonylative difunctionalization of alkenes18 has attracted considerable attention in the field of carbonylation chemistry, as it represents an effective strategy that allows two functional groups to be attached across a double bond in one step, thereby enabling the rapid assembly of molecular complexity. In 2020, Wu and co-workers established a copper-catalyzed borocarbonylative coupling of unactivated alkenes with primary alkyl iodides for synthesizing β-boryl ketones.19 This method provides a broad range of β-boryl ketone derivatives with complete regioselectivity and in moderate to excellent yields. The use of Xantphos as an additive ligand was found to be critical for facilitating alkyl halide activation by the alkylcopper intermediate. They propose that the LCuBpin complex undergoes insertion into the CC bond of the alkene to generate a borylcuprated intermediate A,20 which subsequently reacts with the alkyl iodide to form the Cu(III) complex Bvia a radical pathway. Migratory insertion of CO into complex B affords the acylcopper intermediates C or C′, which undergo reductive elimination to deliver the final products (Fig. 2, proposed mechanism right). To probe the mechanism, an alkyl–copper intermediate was prepared and reacted with 1-iodobutane. In the absence of Xantphos, no product was detected; however, adding 1.0 equivalent of Xantphos afforded the desired product in 32% yield. This reveals that Xantphos is essential for alkyl halide activation by the alkyl–copper intermediate.
 | ||
| Fig. 2 Cu-Catalyzed regioselective borocarbonylation of unactivated alkenes. | ||
Later, the same group discovered that a complete reversal of regioselectivity could be accomplished. By employing a CuOAc/BuPAd2 catalytic system with sterically hindered alkyl bromides, a series of β-boryl ketones with inverted regioselectivity were synthesized in moderate yields.21 In contrast to the prior study, they proposed a catalytic cycle for this regioselectivity-reversed borocarbonylation, proceeding via a radical-relay mechanism.22 The LCu(I)Bpin species undergoes single-electron transfer (SET) with the alkyl bromide to generate an LCu(II)Bpin intermediate D and an alkyl radical E. Under high CO pressure, radical E undergoes carbonylation to form an acyl radical F, which subsequently adds to the alkene to generate a new carbon-centered radical G. The radical then recombines with the LCu(II)Bpin D, followed by reductive elimination to afford the desired β-boryl ketone, while regenerating the LCu(I)X species. (Fig. 2, proposed mechanism left).
2.2. Cu/Pd-Catalyzed divergent borocarbonylation of aryl alkenes
While the aforementioned catalytic systems exhibited good reactivity with alkyl halides, they proved ineffective for aryl halides. To address this limitation, Wu and co-workers strategically introduced a palladium co-catalyst, thereby establishing a Cu/Pd dual catalytic system. In 2020, they developed a cooperative Cu/Pd-catalyzed four-component borocarbonylative reaction for the selective synthesis of either β-boryl ketones or β-boryl vinyl esters from vinylarenes, aryl iodides or aryl triflates, B2Pin2, and CO.23 When aryl iodides are employed as substrates, the reaction affords synthetically useful β-boryl ketones in good to high yields. A broad range of aryl iodides with electron-donating or electron-withdrawing substituents, as well as disubstituted aryl iodides and iodoheterocycles, are well-tolerated. Likewise, various substituted styrenes undergo the reaction efficiently. The resulting β-boryl ketones can be further transformed via oxidation to alcohols or via Suzuki–Miyaura cross-coupling. Notably, when aryl triflates are used in place of aryl iodides as the coupling partners, the reaction delivers β-boryl vinyl esters with similarly broad functional group tolerance. For the synthesis of β-boryl vinyl esters, MeIPrCuCl was identified as the optimal copper catalyst (Fig. 3). | ||
| Fig. 3 Cu/Pd-catalyzed borocarbonylation of aryl alkenes. | ||
Mechanistic experiments excluded a possible pathway involving two interconnected catalytic cycles for copper24 and palladium. For the formation of β-boryl ketones, the copper cycle begins with the borylcupration of the vinylarene, generating alkyl copper species I. Meanwhile, the aryl iodide undergoes oxidative addition to a LnPd(0) species, followed by CO insertion to form an acyl–palladium intermediate. Subsequent transmetalation between the alkyl copper and acyl palladium species furnishes the final β-boryl ketone product via reductive elimination. In the case of β-boryl vinyl ester formation, the alkyl copper species I undergoes CO insertion to form an acyl copper intermediate II, which is proposed to tautomerize into a vinyl alkoxide copper species III. This species then engages in transmetalation with an acyl–palladium species A (formed from the aryl triflate and CO in the palladium cycle), followed by reductive elimination to afford the final product.
The divergent products obtained from aryl triflates and aryl iodides are attributed to kinetic differences in the generation rates of the acyl–palladium species and the vinyl alkoxide copper intermediate. Due to the higher reactivity of the C–I bond in aryl iodides compared to the C–OTf bond in aryl triflates, oxidative addition occurs more readily, resulting in a rate of acyl–palladium formation that matches well with that of the alkyl copper species. In contrast, during the borocarbonylation of aryl triflates, the rate of acyl–palladium intermediate formation is more closely aligned with that of the vinyl alkoxide copper species III, thereby influencing the reaction pathway and determining the product selectivity.
2.3. Cu/Pd-Catalyzed regiodivergent borocarbonylation of MCPs
Methylenecyclopropanes (MCPs), a class of strained carbocycles, are widely recognized as versatile and readily accessible synthons in organic synthesis. Their inherent ring strain enables a wide range of transformations, including ring-opening and ring-retaining reactions, which can be initiated by transition metals, Lewis acids, Brønsted acids, or radical species.24 Among these, transition metals, particularly copper, have demonstrated high catalytic efficiency and enable switchable reaction pathways of MCPs under mild conditions (Fig. 4(a)).25 | ||
| Fig. 4 Regiodivergent borocarbonylation of MCPs & divergent borocarbonylation of α-substituted styrenes. | ||
In 2022, the Wu group reported the Cu/Pd-catalyzed regioselective borofunctionalization of MCPs where the reaction pathway could be directed to furnish either cycloaddition or ring-opening products. Specifically, when IPrCuCl and Pd(dppp)Cl2 were used as co-catalysts, γ-vinylboryl ketones were obtained via a ring-opening pathway.26 The use of the strong electron-donating and sterically bulky IPr ligand is crucial for this selectivity. Conversely, the combination of Cu(dppp)Cl, [Pd(η3-cinnamyl)Cl]2/Xantphos led to the formation of β-cyclopropylboryl ketones. The borocarbonylation of methylenecyclopropanes (MCPs) with aryl iodides yielded a range of γ-vinylboryl and β-cyclopropylboryl ketones in good yields and with excellent regioselectivity. These products could be further derivatized into other value-added compounds (Fig. 4(b)).
2.4. Cu-Catalyzed chemodivergent borocarbonylation of α-substituted styrenes
Building on the continuous interest in copper-catalyzed carbonylative borofunctionalization of alkenes,27 Wu and co-workers recently disclosed a Cu-catalyzed, ligand-controlled selective borocarbonylation of α-substituted styrenes. This protocol enables the synthesis of either β-boryl aldehydes or cyclopropyl boronate esters from the same starting materials.28 The reaction outcome is controlled by the choice of ligand. The N-heterocyclic carbene (IMes) ligand promotes O–H bond insertion of a proposed carbene intermediate with an alcohol, affording β-boryl quaternary aldehydes. In contrast, the phosphine ligand DPPE directs the carbene intermediate towards intramolecular C–H bond insertion, furnishing cyclopropyl boronate esters with a quaternary carbon center (Fig. 4(c)). This ligand-controlled reactivity of the key carbene intermediate offers a versatile strategy for accessing structurally distinct and synthetically valuable organoboron compounds.The reaction begins with borocupration of α-substituted styrenes to generate a β-boroalkylcopper intermediate. Subsequent carbonylation via CO insertion into the C–Cu bond affords an acyl–copper intermediate, which rapidly undergoes isomerization to form the key carbene intermediate A. When IMes is employed as the ligand, intermediate A undergoes O–H insertion with tBuOH to afford intermediate B, which then undergoes β-elimination to furnish β-boryl aldehydes. Alternatively, using DPPE as the ligand promotes C–H insertion of carbene intermediate A to yield intermediate C, which subsequently transforms into the final cyclopropyl boronate esters. However, the detailed mechanism requires further investigation.
3. Selectivity controlled carbonylation of alkynes
The catalytic carbonylation of alkynes is a powerful and atom-economical transformation for accessing valuable carbonyl compounds. The mechanism typically involves alkyne coordination, migratory CO insertion, and nucleophilic attack. By enabling precise control over regioselectivity, this method serves as an indispensable tool in modern synthesis for constructing pharmaceutical and fine chemical intermediates.3.1. Ligand-controlled Pd-catalyzed regioselective alkoxycarbonylation of alkynes
In 2024, Dong and co-workers reported a palladium-catalyzed, ligand-controlled regiodivergent alkoxycarbonylation of trifluoromethylthiolated (SCF3) internal alkynes using gaseous CO as the carbonyl source (Fig. 5).29 A key feature of this method is its ability to access either α- or β-SCF3-substituted acrylate esters selectively from the same alkyne precursor simply by switching the ligand class: phosphine-oxazoline ligand (tBuPhox) favors the α-isomer with high regioselectivity (up to 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), while bis-phosphine ligand (DPEPhos) promotes formation of the β-isomer (up to 99:1). Mechanistic insights suggest that ligand bite angle and steric environment critically influence regiocontrol, and solvent effects further modulate selectivity, likely by accelerating the alcoholysis step. The methodology demonstrates broad substrate scope, accommodating diverse aryl, heteroaryl, and alkyl substituents, as well as various alcohols under mild conditions.
:4), while bis-phosphine ligand (DPEPhos) promotes formation of the β-isomer (up to 99:1). Mechanistic insights suggest that ligand bite angle and steric environment critically influence regiocontrol, and solvent effects further modulate selectivity, likely by accelerating the alcoholysis step. The methodology demonstrates broad substrate scope, accommodating diverse aryl, heteroaryl, and alkyl substituents, as well as various alcohols under mild conditions.
 | ||
| Fig. 5 Pd-catalyzed regioselective alkoxycarbonylation of alkynes. | ||
3.2. Pd-catalyzed regioselective thiocarbonylation of alkynes
While regioselective hydroformylation, alkoxycarbonylation, and aminocarbonylation of alkynes have seen success, the development of regioselective thiocarbonylation has been limited.30 This difficulty might be partly due to the strong binding affinity of sulfur compounds to late-transition metals, which can affect ligand coordination. Previous reports on alkyne thiocarbonylation have also been limited by poor selectivity and narrow functional group compatibility.To address these limitations, Wu's group established a palladium-based catalytic system capable of achieving high regioselectivity and excellent functional group compatibility. The key to the regioselectivity control lies in the choice of ligand on the palladium catalyst. By using the bidentate ligand DPEphos, along with B(OH)3 and 5-chlorosalicylic acid (5-Cl-SA) forming 5-chloroborosalicylic acid (5-Cl-BSA) in situ as an additive, which preferentially yields linear α,β-unsaturated thioesters. However, switching to the monodentate ligand tri(2-furyl)phosphine steers the reaction to selectively form branched α,β-unsaturated thioesters.31 This ligand-controlled approach was successfully applied to a wide variety of terminal aryl and alkyl alkynes, demonstrating broad substrate scope and functional group compatibility for both linear and branched products (Fig. 6(a)).
 | ||
| Fig. 6 Pd-catalyzed regioselective thiocarbonylation of alkynes. | ||
In 2023, Luo, Zhu, and colleagues reported a palladium-catalyzed regioselective thiocarbonylation of terminal alkynes under exceptionally mild conditions, using atmospheric pressure of CO and triethylsilane (Et3SiH) as a hydride source instead of traditional strong acids (Fig. 6(b)).32 In palladium-catalyzed hydrocarbonylation, Pd–H species are usually generated from Pd(0) and an acid additive; here the use of silane as a hydride source can successfully avoid the need for acid. A key finding is the unusual counter anion-controlled regioselectivity for terminal alkynes: with PdCl2, the reaction favors the branched thioester, whereas switching to Pd(CH3CN)4(BF4)2 switches selectivity to the linear isomer with high regiocontrol (L/B >20:1). In their study, Pd(TFA)2 and PdI2 were also tested and linear α,β-unsaturated thioester was formed preferentially. This contrasts with conventional ligand-dominated selectivity and highlights the critical role of the palladium counter anion in directing the hydropalladation step. This method tolerates a broad scope of thiols, including primary, secondary, aromatic, and even acid-sensitive glucosinolate derivatives, delivering α,β-unsaturated thioesters in moderate to excellent yields.
3.3. Ligand-controlled Pd-catalyzed chemo- and regioselective carbonylative cyclization
Indolo[3,2-c]coumarins and benzofuro[3,2-c]quinolinones are important polybenzoheterocyclic scaffolds in numerous bioactive molecules, representing key structural motifs for pharmaceuticals with diverse biological activities (Fig. 7(a)).33 In 2018, Jiang and co-workers reported a palladium-catalyzed regiodivergent carbonylative cyclization of 2-hydroxy-2′-amino-diphenylethyne derivatives to selectively synthesize either benzofuro[3,2-c]quinolinones or indolo[3,2-c]coumarins.34 The chemo- and regioselectivity are controlled by the ligand choice. Employing a rigid, electron-deficient nitrogen ligand, 1,10-phenanthroline-5,6-dione (L1) promotes an O-attack/N-carbonylation sequence to selectively afford benzofuro[3,2-c]quinolinones. Switching to a sterically bulky and electron-rich phosphine ligand (DPPM) redirects the reaction towards an N-attack/O-carbonylation pathway, furnishing indolo[3,2-c]coumarins (Fig. 7). On the basis of the experimental results, they proposed a plausible mechanism to rationalize this ligand-controlled divergence. The reaction begins with the coordination of the Pd(II) catalyst to the substrate, with the ligand's properties determining the initial coordination site. When the electron-deficient ligand 1,10-phenanthroline-5,6-dione is used, the resulting palladium center becomes more electrophilic and preferentially coordinates to the amino group. This activates the alkyne for an intramolecular O-attack by the hydroxyl group, followed by CO insertion and reductive elimination to yield the benzofuro[3,2-c]quinolinone. Conversely, the sterically bulky and electron-rich DPPM ligand is proposed to favor coordination to the hydroxyl group. This initial O-coordination facilitates a subsequent N-attack from the amino group, followed by CO insertion and reductive elimination to deliver the indolo[3,2-c]coumarin product (Fig. 7). | ||
| Fig. 7 Pd-Catalyzed chemo- and regioselective carbonylative cyclization. | ||
3.4. Ligand-controlled Pd-catalyzed chemoselective carbonylation of 1,3-diynes
Palladium-catalyzed carbonylation reactions offer powerful tools for constructing complex organic molecules. The carbonylation of readily available 1,3-diynes, in particular, presents an atom-economical route to valuable conjugated systems, such as substituted dienes and enynes. However, controlling the selectivity of this transformation poses significant challenges due to the presence of multiple reactive sites and the potential for various reaction pathways, including mono- versus dicarbonylation, regioisomer formation, and stereoisomer control. Recent studies from the Beller group have highlighted the importance of ligand design in controlling the selectivity of these reactions.They demonstrated how ligand design critically controls selectivity in palladium-catalyzed carbonylation of 1,3-diynes. For stereoselective dicarbonylation, the bidentate phosphine ligand L5, featuring a ferrocene backbone with a pyridyl “built-in-base” moiety, enabled the double alkoxycarbonylation of 1,3-diynes with high (E,E)-selectivity (up to 97:3) under mild conditions.35 On the other hand, switching to the BINAP-derived ligand L6 (Neolephos) shifted selectivity dramatically toward chemoselective monoalkoxycarbonylation, with exclusive E-stereocontrol (Fig. 8).36 Kinetic studies showed that Neolephos allows the first carbonylation to proceed to completion with almost no formation of the double carbonylation product, suggesting a large difference in the reaction rates for the first and second carbonylation steps. The different selectivities highlight how subtle ligand modifications, particularly in the backbone structure (ferrocene versus binaphthyl), critically modulate reactivity: L5 promotes sequential carbonylation, while L6 kinetically suppresses the second step, enabling precise chemoselectivity.
 | ||
| Fig. 8 Palladium-catalyzed selective carbonylation of 1,3-diynes. | ||
4. Pd-Catalyzed multiselective carbonylation of 1,3-enynes
The functionalization of 1,3-enynes is highly important in organic synthesis because they are a versatile class of readily available multisite substrates containing both conjugated double and triple bonds. This distinctive structure enables their use as strategic building blocks for constructing complex molecular frameworks through chemo-, regio-, and stereoselective transformations, including direct functionalizations and efficient tandem cyclizations.37 Developing methods to precisely control the reactivity of these multiple sites represents a significant challenge, holding potential for advancements in drug discovery and materials science.Recently, a multimodal palladium-catalyzed strategy for the selective carbonylation of 1,3-enynes has been developed in the Wu group, enabling access to five distinct classes of products from the same starting materials through the precise control of ligands and additives.38 This system facilitates two direct hydroaminocarbonylation reactions and three different tandem cyclization pathways. The selective transformations include (1) 1,2-hydroaminocarbonylation to yield linear α,β-unsaturated amides, which is achieved using the BINAP ligand with PTSA in DMF; (2) 2,1-hydroaminocarbonylation to form branched α,β-unsaturated amides, which is accomplished with the bulky monophosphine ligand PAd3 and NaH2PO4; (3) 2,4-tandem cyclization to produce 5-endo-trig lactams, which is performed with the Xantphos ligand and PTSA under 1 bar CO pressure; (4) 2,3-tandem dicarbonylation to give E-configured succinimides, which requires the TFP ligand and aniline hydrochloride under high (50 bar) CO pressure; and (5) 1,3-tandem cyclization to yield 5-exo-trig lactams, which uses the PAd3 ligand with PTSA. The substrate scope for each transformation is broad, tolerating a variety of anilines with both electron-donating (e.g., Me, OMe, and OPh) and electron-withdrawing groups (e.g., CF3, Cl, and Br), as well as a wide range of aryl- and alkyl-substituted 1,3-enynes (Fig. 9).
 | ||
| Fig. 9 Multimodal Pd-catalyzed carbonylation of 1,3-enynes. | ||
The multimodal selectivity is rationalized by distinct mechanistic pathways originating from two key hydroaminocarbonylation intermediates, the linear amide (3a) and the branched amide (4a), whose roles were confirmed by kinetic and control experiments. The initial regioselectivity is dictated by the active palladium species; a Pd–H species, formed with the ligand BINAP, favors 1,2-insertion to give 3a (Fig. 9, catalytic cycle A, upper part). The acyl–palladium complex C, generated in situ from PdCl2/PAd3 in the presence of amine and CO, undergoes coordination and insertion with an enyne substrate to form palladium intermediate Int G. Subsequent isomerization yields the more stable allylic palladium species Int G′, which upon protonation with HCl affords product 4a while regenerating the active palladium catalyst (catalytic cycle C, left part). The amide 3a can undergo 2,4-cyclization via a Pd–H catalyzed hydroamination–transamination sequence (promoted by the ligand Xantphos) leading to 5a (Fig. 9, catalytic cycle A), or a 2,3-dicarbonylation (promoted by the ligand TFP) to afford 6a (catalytic cycle B). This divergence is attributed to the ligand's coordination properties; the monophosphine TFP provides an open coordination site for the second CO insertion, while the bidentate Xantphos does not. Amide 4a serves as the key intermediate in the 1,3-tandem cyclization (catalytic cycle C). The amino–palladium complex undergoes selective olefin insertion into 4a, generating hydroamination intermediate Int I. Subsequently, under palladium catalysis with CO participation, intramolecular transamination occurs to furnish product 7a while regenerating the active acyl–palladium catalyst C. Notably, CO is essential for this cyclization step.
5. Selective carbonylation of alcohols
5.1. Ligand-controlled Pd-catalyzed regioselective carbonylation of alcohols
Alcohols are attractive, abundant feedstocks for carbonylation, yet their direct use is challenging due to the hydroxyl's poor leaving group ability and strong nucleophilicity.39 Traditionally, alcohols require pre-functionalization to halides or sulfonates to facilitate carbonylation, a process that adds synthetic steps and generates stoichiometric waste.40 This has motivated the development of a more atom-economical route involving in situ olefin generation from alcohols via dehydration, followed by regioselective hydroaminocarbonylation. In 2024, Wu's group realized this concept by developing a ligand-controlled, regiodivergent aminocarbonylation of cyclobutanols to selectively afford either 1,1- or 1,2-disubstituted cyclobutanecarboxamides (Fig. 10(a)).41 Later, alcohols were also demonstrated as the nucleophiles to be compatible with this process, enabling the formation of 1,1- or 1,2-disubstituted cyclobutanecarboxylates.42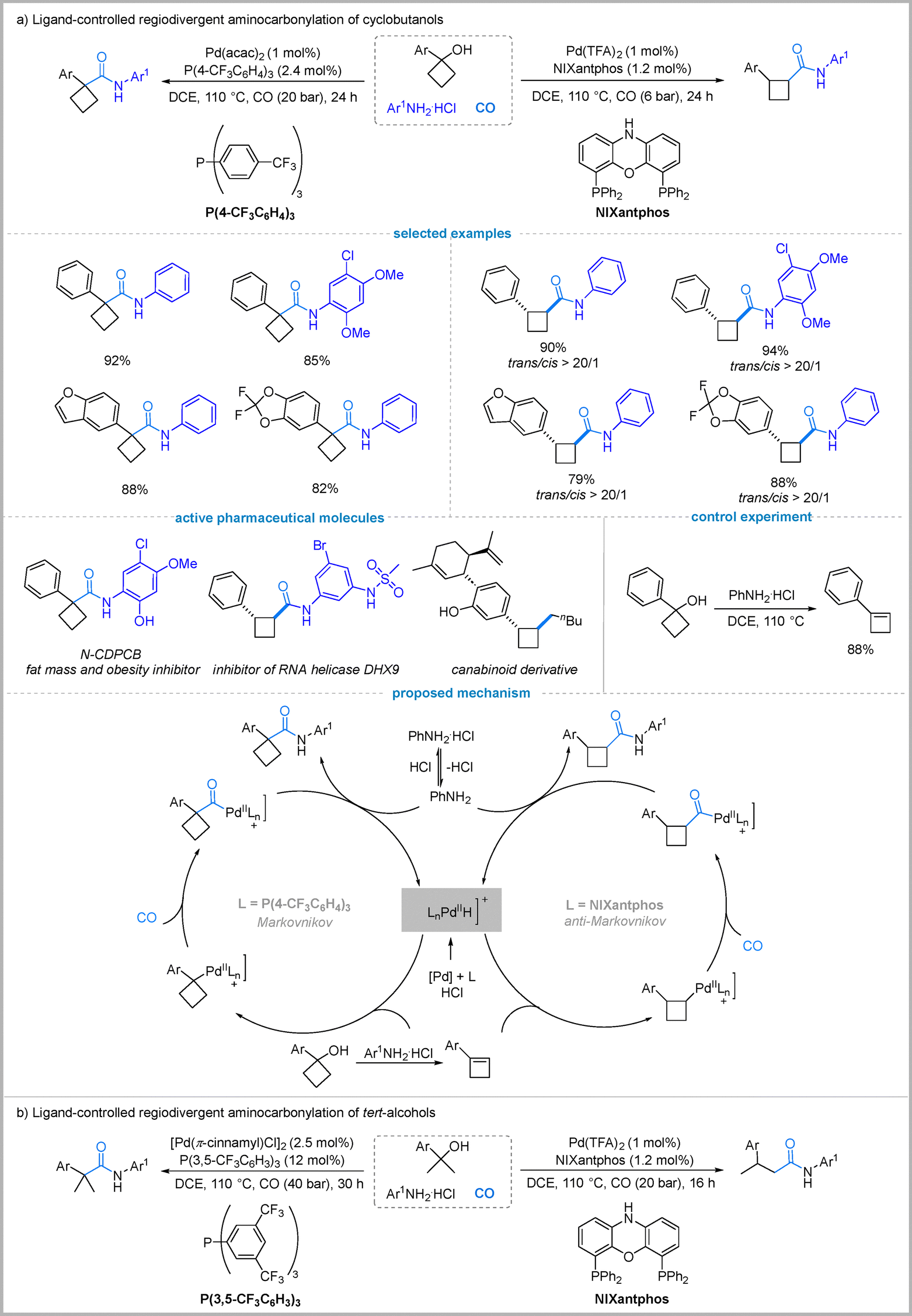 | ||
| Fig. 10 Pd-catalyzed regioselective carbonylation of alcohols. | ||
Ligand screening in this work revealed that using the bidentate phosphine ligand NIXantphos selectively afforded trans-1,2-disubstituted cyclobutanecarboxamides in high yields (up to 90%) with excellent regio- and diastereoselectivity under optimized conditions. This outcome is consistent with an anti-Markovnikov Pd–H insertion into the cyclobutene intermediate. In contrast, switching to the electron-deficient monophosphine ligand (4-CF3C6H4)3P under slightly modified conditions favored 1,1-disubstituted cyclobutanecarboxamides in high yields (up to 92%), suggesting a Markovnikov-type Pd–H insertion. Both protocols showed a broad substrate scope and utility in late-stage functionalization, enabling the synthesis of bioactive analogues. Mechanistic study confirmed the involvement of a cyclobutene intermediate (Fig. 10(a)).
Next, they extended this ligand-controlled strategy to tertiary alcohols, demonstrating that the electron-deficient monophosphine ligand ((3,5-(CF3)2C6H3)3P) with [Pd(π-cinnamyl)Cl]2 selectively afforded α-quaternary amides via Markovnikov hydroaminocarbonylation of in situ-formed alkenes, while switching to NIXantphos/Pd(TFA)2 reversed regioselectivity to deliver β-substituted linear amides through anti-Markovnikov Pd–H insertion, with both systems tolerating diverse anilines and tertiary alcohols (Fig. 10(b)).43
5.2. EDA-complex-controlled chemoselective carbonylation of homoallylic alcohols
Recently, a significant strategy for controlling selectivity in carbonylative transformations has been developed by Wu and Huang, enabling the divergent synthesis of γ-lactones and 1,4-diones from the same homoallylic alcohol starting materials (Fig. 11).44 This process operates through a transition-metal-free, radical tandem carbonylation, where the reaction's chemoselectivity is dictated by the choice of tertiary amine used as an electron donor. The amine serves a dual function: it not only mediates the initial electron transfer to form an electron donor–acceptor (EDA) complex but also acts as a “tuner” that governs the subsequent reaction pathway by controlling the nature of the key acyl intermediate. | ||
| Fig. 11 Chemoselective carbonylation of homoallylic alcohols. | ||
The synthesis of γ-lactones is achieved using the sterically unhindered amine quinuclidine as the electron donor. Mechanistic studies reveal that after the initial formation of an acyl radical, the quinuclidine radical cation, which has a high oxidation potential, oxidizes this intermediate into a more electrophilic acyl cation. This electrophilic species then readily undergoes intramolecular lactonization with the pendant hydroxyl group to yield the cyclized product. Density functional theory (DFT) calculations support this pathway, indicating that the energy barrier for this oxidation process (outer-sphere single-electron transfer, OSET) is lower than that of the competing migration pathway, making lactonization the kinetically favored outcome with quinuclidine.
On the other hand, when a sterically hindered amine N,N-diisopropylethylamine (DIPEA) is used, the reaction selectively yields 1,4-diones through a remote aryl group migration. In this scenario, the bulkier DIPEA radical cation is a weaker oxidant, and the OSET pathway to form an acyl cation is energetically less favorable, with a higher calculated energy barrier than the migration step. As a result, the initial nucleophilic acyl radical intermediate persists long enough to undergo a radical-mediated, long-distance migration of an aryl group to the acyl carbon, ultimately affording the 1,4-dione product.
5.3. Pd-Catalyzed chemo- and diastereoselective carbonylation of enallenol
In 2018, Qiu, Bäckvall, and colleagues reported a palladium-catalyzed oxidative tandem carbonylation of enallenols that enables chemodivergent and highly diastereoselective synthesis of either cyclopentenone-fused γ-lactones or simple γ-lactones (Fig. 12).45 The selectivity is controlled not by ligands or additives, but by the nature of the catalyst: a heterogeneous palladium catalyst (PdII-AmP-MCF, palladium immobilized on amino-functionalized mesocellular foam) selectively delivers the tandem carbonylation–carbocyclization–carbonylation–lactonization product in up to 80% yield, whereas a homogeneous Pd(II) salt (Pd(TFA)2) favors early lactonization to give the simpler γ-lactone in up to 86% yield. The switch in chemoselectivity is attributed to the porous support's ability to concentrate CO and stabilize Pd(0)/Pd(II) species, promoting sequential CO insertions only in the heterogeneous system. | ||
| Fig. 12 Pd-Catalyzed chemo- and diastereoselective carbonylation of enallenol. | ||
The methodology exhibits broad substrate scope, accommodating various alkyl, aryl, and functionalized substituents on the enallenol framework, and consistently delivers products as single diastereomers, often with multiple stereocenters, including quaternary carbon centers. The heterogeneous catalyst is recyclable without loss of activity (up to 9 times), and no Pd leaching is detected (<0.1 ppm), confirming true heterogeneous catalysis. Furthermore, enantiopure γ-lactones are accessible via kinetic resolution of the starting enallenols followed by the tandem process. This work represents a rare example of catalyst-phase-controlled chemodivergence in carbonylative catalysis, offering a step- and atom-economical route to valuable heterocyclic scaffolds.
6. Ligand-controlled Cu-catalyzed regioselective carbonylation of imines
While significant progress has been made with CC bonds, the regiodivergent carbonylative functionalization of imines remains a challenge. This difficulty stems from the inherent polarity of the CN bond, where the electrophilic carbon atom favors attack by nucleophiles, making it difficult to control the regioselectivity.46 Consequently, inverting this innate electronic preference is key to achieving a divergent transformation.47 Addressing this, Wu and co-workers have developed a regiodivergent protocol for the carbonylative functionalization of imines with B2pin2 and alkyl iodides that selectively affords either α-amino ketones or α-boryl amides. The key to this selectivity is the choice of ligand: the electron-withdrawing phosphine ligand (p-CF3C6H4)3P promotes the formation of α-amino ketones in good yields, while the N-heterocyclic carbene (NHC) ligand MeIMes directs the reaction to furnish the corresponding α-boryl amides (Fig. 13).48
 | ||
| Fig. 13 Cu-Catalyzed regioselective carbonylation of imines. | ||
Based on the control experiments and literature precedents,49 a plausible mechanism was proposed. The reaction begins with the insertion of a LCu-Bpin species into the CN bond of an imine to generate a key α-boryl amido–copper intermediate A. The electronic properties of the ligand dictate the subsequent reaction pathway from intermediate A. When an electron-withdrawing phosphine ligand is employed, the resulting N–Cu bond is weaker, facilitating an intramolecular 1,2-rearrangement to an α-amino alkyl–copper complex B; this intermediate then undergoes carbonylation and reductive elimination to yield the α-amino ketone after work up with MeOH. In contrast, under the assistance of the MeIMes ligand, the more electron-rich amido–copper intermediate undergoes a direct cross-coupling reaction with the alkyl iodide to intermediate C, followed by carbonylation and reductive elimination to produce the α-boryl amide.
7. Philicity-controlled carbonylation of oxime esters
In 2022, Xiao, and Chen's group presented a photoredox-catalyzed strategy for switchable radical carbonylation of alkyl radicals derived from oxime esters, enabling selective access to either single-carbonylated amides or double-carbonylated α-ketoamides from identical starting substrates—oxime esters, amines, and carbon monoxide (Fig. 14).50 The key innovation lies in philicity regulation: by modulating the electronic character of reaction partners through visible-light photoredox catalysis, the reaction pathway can be precisely controlled. Under standard conditions using fac-Ir(ppy)3 as the photocatalyst in DMF, amines are oxidized to aminium radical cations, which add to CO to form carbamoyl radicals; these then cross-couple with cyanoalkyl acyl radicals (generated from oxime esters + CO) to afford α-ketoamides selectively.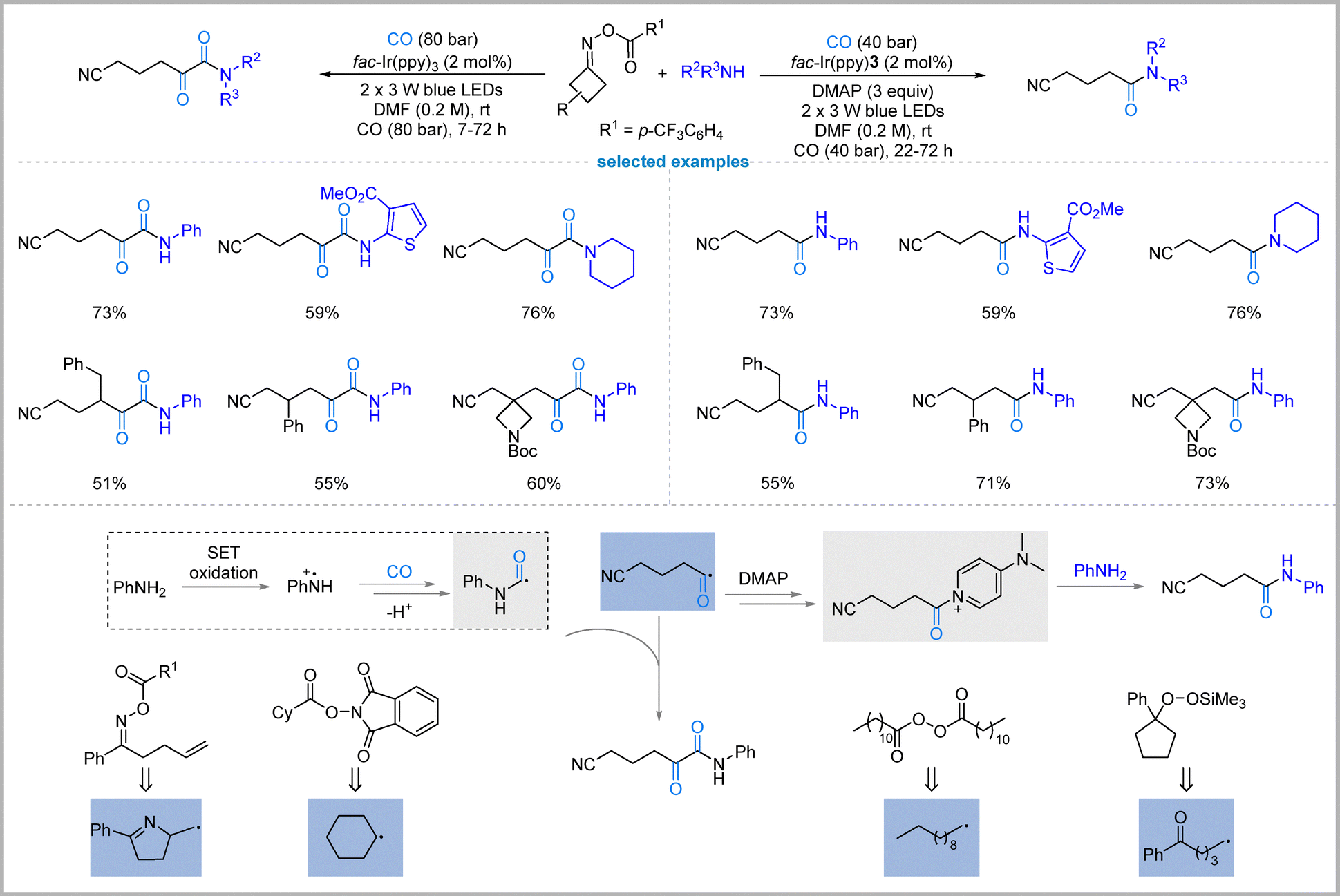 | ||
| Fig. 14 Philicity-controlled carbonylation of oxime esters. | ||
In contrast, single carbonylation is switched on by adding stoichiometric DMAP (4-dimethylaminopyridine). DMAP intercepts the cyanoalkyl acyl radical to form a stabilized acyl-DMAP salt, which then undergoes nucleophilic substitution with the amine to yield amides with high chemoselectivity at lower CO pressure (40 bar). Mechanistic and DFT studies support that DMAP acts not as a base but as a π-nucleophile that traps the acyl radical, diverting the pathway away from double carbonylation. This approach overcomes a long-standing challenge in radical carbonylation: the difficulty of achieving a second CO insertion into acyl radicals, which is thermodynamically and kinetically disfavored under conventional conditions.
The method exhibits broad scope, tolerating diverse primary/secondary alkyl and aryl amines, including complex drug molecules, and various functionalized oxime esters, all at room temperature. In addition, other radical precursors, such as γ-alkenyl O-acyl oxime, aliphatic carboxylic acid-derived N-(acyloxy)phthalimide, alkyl diacyl peroxide, and alkylsilyl peroxide, can also undergo these switchable radical carbonylation reactions. Both amides and α-ketoamides are valuable motifs in medicinal chemistry, and this strategy provides a rare, general, and tunable platform to access either class selectively from identical feedstocks. By exploiting photoredox-mediated polarity switching, the work establishes a new paradigm for controlling chemoselectivity in radical carbonylative transformations.
8. Divergent carbonylation of organohalides
8.1. Palladium-catalyzed carbonylative synthesis of isoquinoline-1,3(2H,4H)-diones and indanones
Since the pioneering work of Heck and co-workers in 1974, palladium-catalyzed carbonylation of aryl halides has undergone remarkable development, enabling a broad array of carbonylation with diverse nucleophiles.51 This methodology provides efficient access to carboxylic acid derivatives, and also serves as a powerful approach for constructing heterocyclic compounds, which represent key structural motifs in pharmaceuticals, biologically active molecules, and agrochemicals.Recently, Wu reported a palladium-catalyzed carbonylation strategy for the selective synthesis of isoquinoline-1,3(2H,4H)-dione and indanone derivatives from 1-bromo-2-vinylbenzenes and amines. The chemoselectivity is controlled by tuning the reaction conditions to generate distinct active palladium species that initiate divergent reaction pathways. The synthesis of isoquinoline-1,3(2H,4H)-diones is achieved using the monodentate ligand PCy3 with NaH2PO4 as the base and TsOH·H2O as an additive in CH3CN. Switching to the bidentate ligand DPPB with Et3N as the base in 1,4-dioxane selectively furnishes the indanone derivatives. The catalytic system exhibited excellent tolerance toward a wide range of amines and o-bromostyrenes for both products (Fig. 15).52
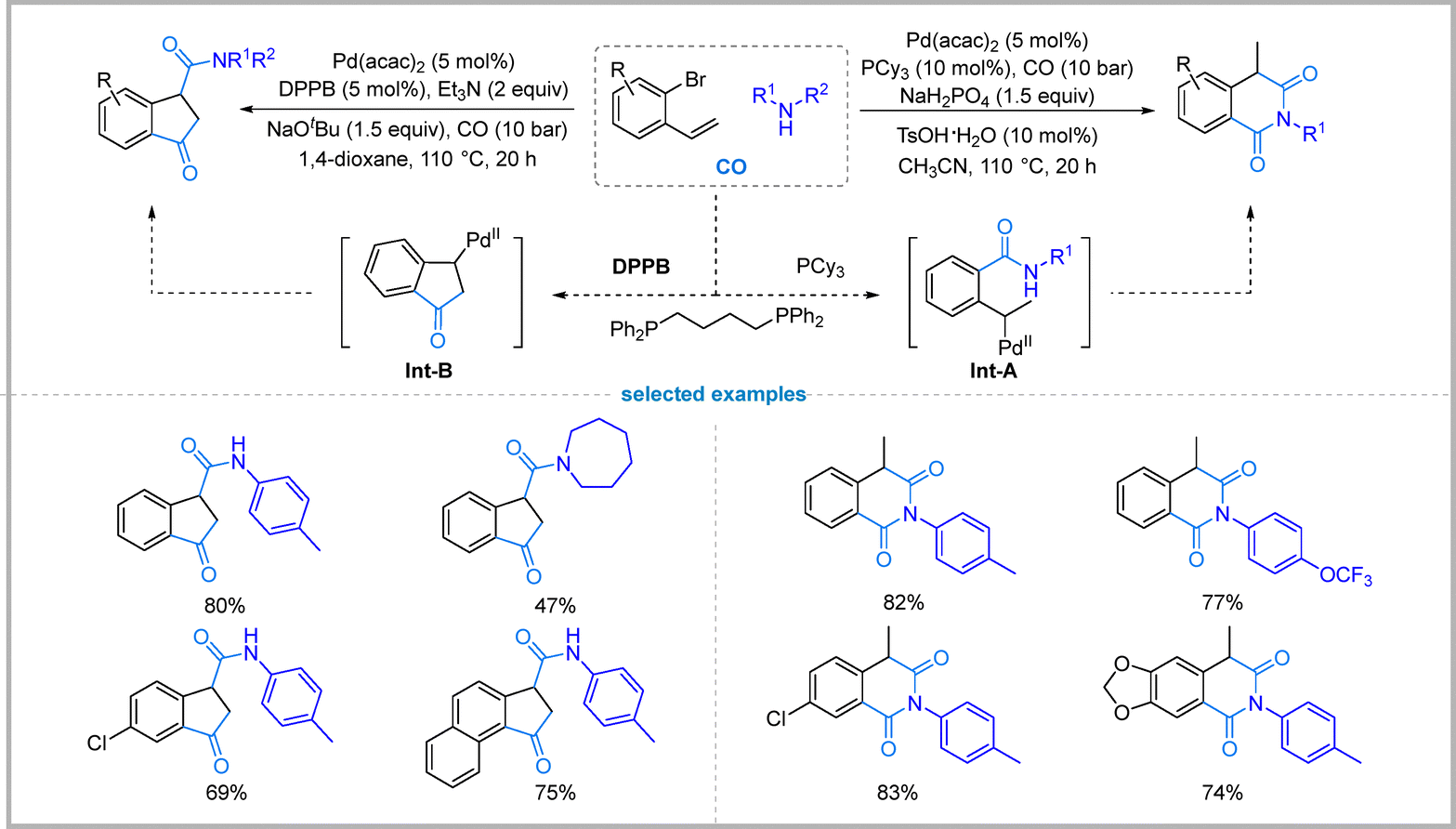 | ||
| Fig. 15 Carbonylative synthesis of isoquinoline-1,3(2H,4H)-diones. | ||
The formation of isoquinoline-1,3(2H,4H)-diones is suggested to proceed through a pathway involving a palladium hydride species. An amide intermediate is first formed, which then undergoes hydro-palladation across its vinyl group by the Pd–H species to Int-A, followed by a second CO insertion and intramolecular cyclization. The synthesis of indanones is proposed to follow a different pathway involving an initial oxidative addition of the aryl bromide to the palladium catalyst, followed by CO insertion. Subsequent intramolecular migratory insertion into the vinyl group forms the five-membered ring Int-B. A second CO insertion, followed by reaction with the amine, leads to the final indanone product.
8.2. Copper-catalyzed borylative methylation of alkyl iodides
Transition metal-catalyzed cross-couplings are indispensable for constructing C–C and C-heteroatom bonds. Recent efforts have focused on alkyl halides, which offer milder conditions and fewer side reactions than classical substitution methods.53 However, coupling unactivated alkyl halides bearing β-hydrogens remains challenging due to sluggish oxidative addition of C(sp3)–X bonds and competing β-hydride elimination of the resulting alkylmetal intermediates.54 In light of the interest in CuBpin and CuH chemistry in carbonylation,55 Wu and colleagues reported a CuH- and CuBpin-catalyzed borylative methylation of alkyl iodides using carbon monoxide as the C1 source in 2021. This protocol enables the selective synthesis of either 1,1-diborylalkanes or monoborylalkanes from the same starting unactivated alkyl iodides.56 The reaction selectivity is controlled by the stoichiometry of the reagents and the choice of base. The 1,1-diborylalkane product is formed preferentially when a higher loading of the boron source (B2pin2, 2.5 equiv.) and a lower loading of methyldiethoxysilane (DEMS, 1.2 equiv.) are used in combination with NaOEt as the base. In contrast, a higher loading of methyldiethoxysilane (DEMS, 3.0 equiv) relative to the boron source (B2pin2, 1.2 equiv.) with LiOtBu as the base directs the reaction to the monoborylalkane (Fig. 16(a)). This tunable system provides a versatile strategy for the one-carbon homologation and borylation of alkyl iodides, enabled by a key kinetic factor: the reaction of CuH with alkyl iodides proceeds faster than that of CuBpin. | ||
| Fig. 16 Cu-catalyzed borylative methylation of alkyl iodides & selective mono- or double-carbonylation of alkyl iodides with amines. | ||
Mechanism studies revealed that the reaction is initiated by a CuH-catalyzed formylation of the alkyl iodide. First, a CuH complex is generated from the copper salt, a base, and a silane (DEMS). This CuH species reacts with the alkyl iodide and carbon monoxide, likely via a radical pathway, to form an acyl–copper hydride intermediate A. Subsequent reductive elimination furnishes the key aldehyde intermediate (RCHO), which serves as the central branch point for the divergent pathways.
For diborylmethylation pathway: under conditions with a high concentration of B2pin2 and NaOEt, the aldehyde enters a borylation cycle. A CuBpin species, generated from CuOEt and B2pin2, inserts into the aldehyde's carbonyl to form an α-boryl-oxido-copper complex B. This intermediate reacts with B2pin2 to give an α-oxyboronate species C while regenerating CuBpin. Finally, the α-C–O bond of the α-oxyboronate C is activated by base and B2pin2, acting as a leaving group to deliver the 1,1-diborylalkane product. For monoborylmethylation pathway: when the reaction is run with a higher concentration of the silane hydride source and LiOtBu, the aldehyde intermediate is instead intercepted by another equivalent of CuH. Insertion of CuH into the carbonyl bond affords a copper alkoxide intermediate D. This intermediate then reacts with B2pin2 to generate species E. Finally, the desired monoborylmethylation product is formed via Cu–Bpin exchange with OBpin, concurrently regenerating the Cu–OBpin species for the next catalytic cycle (Fig. 16(a)).
8.3. Cu-catalyzed selective mono- or double-carbonylation of alkyl iodides with amines
A significant goal in carbonylation chemistry is developing catalytic systems for the controllable synthesis of either mono- or double-carbonylation products. While methods for carbonylation exist, achieving tunable selectivity with substrates like alkyl iodides by simply adjusting reaction conditions remains a valuable objective. In 2022, Wu's group explored a copper-catalyzed system that accomplishes the selective mono- or double-carbonylation of alkyl iodides with amines. The reaction outcome can be effectively controlled by modifying the reaction temperature and reagent stoichiometry, allowing for the selective synthesis of amides and α-keto amides (Fig. 16(b)).57For mono-carbonylation (amide formation), optimal results are achieved with a slight excess of alkyl iodide (1.5 equiv.) at 110 °C, yielding the amide exclusively. In contrast, double carbonylation (α-keto amide formation) is favored at 50 °C using an excess of amine (1.7 equiv.), providing the α-keto amide as the major product. This strategy proved general, accommodating diverse alkyl iodides and amines with broad functional group tolerance.
9. Conclusions and outlook
This review highlights recent progress in selectivity-controlled carbonylative transformations, demonstrating how strategic manipulation of catalysts and reaction conditions enables divergent syntheses from common substrates. While significant advances have been made, challenges in selectivity-controlled carbonylation persist. The inherent reactivity of substrates still presents a major obstacle. For example, achieving selective C–Cl bond carbonylation over a competing C–I or C–Br bond in the same molecule remains highly challenging. Addressing this issue requires sophisticated catalyst design (especially ligand tuning) or alternative pathways like radical-mediated mechanisms. Furthermore, although many plausible mechanisms have been proposed, detailed mechanistic elucidation still requires deeper investigation. A comprehensive understanding of intermediates, transition states, and how reaction parameters like ligands, bases, or solvents precisely influence reaction pathways is crucial for elevating catalyst design to a more predictive level. Moreover, despite considerable progress in chemo- and regioselectivity, enantioselective variants remain challenging. Future efforts should prioritize asymmetric transformations using chiral ligands or cooperative catalysis to access enantioenriched products. As the field continues to evolve, the ability to “turn left or turn right” through rational catalyst design will remain central to achieving precise control in carbonylative transformations. We anticipate that continued efforts in selectively controlled carbonylation will open new avenues for molecular divergence and advance the future of chemical synthesis.Conflicts of interest
The authors declare no competing financial interest.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Acknowledgements
We are thankful for the financial support provided by the National Natural Science Foundation of China (22302198, 22572190, and 22571291), the National Key R&D Program of China (2023YFA1507500), and the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB1530000).References
- (a) J. Mahatthananchai, A. M. Dumas and J. W. Bode, Angew. Chem., Int. Ed., 2012, 51, 10954–10990 CrossRef CAS PubMed; (b) L. Li, Z. Chen, X. Zhang and Y. Jia, Chem. Rev., 2018, 118, 3752–3832 CrossRef CAS PubMed; (c) C. Nájera, I. P. Beletskaya and M. Yus, Chem. Soc. Rev., 2019, 48, 4515–4618 RSC.
- B. M. Trost, Science, 1983, 219, 245–250 CrossRef CAS PubMed.
- (a) L. Kollár, Modern carbonylation methods, John Wiley & Sons, 2008 CrossRef; (b) J.-B. Peng, H.-Q. Geng and X.-F. Wu, Chem, 2019, 5, 526–552 CrossRef CAS; (c) M. Beller, Catalytic carbonylation reactions, Springer, 2006 CrossRef; (d) C.-S. Kuai, Y. Yuan and X.-F. Wu, Chem, 2025, 11, 102503 CrossRef CAS.
- (a) C. F. J. Barnard, Organometallics, 2008, 27, 5402–5422 CrossRef CAS; (b) X.-F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986–5009 RSC.
- (a) A. Brennführer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114–4133 CrossRef PubMed; (b) T. Xu and H. Alper, J. Am. Chem. Soc., 2014, 136, 16970–16973 CrossRef CAS PubMed; (c) F. Sha and H. Alper, ACS Catal., 2017, 7, 2220–2229 CrossRef CAS.
- (a) E. Monflier and A. Mortreux, J. Mol. Catal., 1994, 88, 295–300 CrossRef CAS; (b) M. Papp, P. Szabó, D. Srankó, G. Sáfrán, L. Kollár and R. Skoda-Földes, RSC Adv., 2017, 7, 44587–44597 RSC; (c) D. Das and B. M. Bhanage, Adv. Synth. Catal., 2020, 362, 3022–3058 CrossRef CAS; (d) L. Lu, F. Qiu, H. Alhumade, H. Zhang and A. Lei, ACS Catal., 2022, 12, 9664–9669 CrossRef CAS.
- (a) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS PubMed; (b) X.-F. Wu, X. Fang, L. Wu, R. Jackstell, H. Neumann and M. Beller, Acc. Chem. Res., 2014, 47, 1041–1053 CrossRef CAS PubMed.
- (a) C. J. O’ Connor, H. S. G. Beckmann and D. R. Spring, Chem. Soc. Rev., 2012, 41, 4444–4456 RSC; (b) I. P. Beletskaya, C. Nájera and M. Yus, Chem. Soc. Rev., 2020, 49, 7101–7166 RSC.
- (a) P. W. N. M. van Leeuwen, P. C. J. Kamer, J. N. H. Reek and P. Dierkes, Chem. Rev., 2000, 100, 2741–2770 CrossRef CAS PubMed; (b) P. Braunstein and N. M. Boag, Angew. Chem., Int. Ed., 2001, 40, 2427–2433 CrossRef CAS; (c) M.-N. Birkholz, Z. Freixa and P. W. N. M. van Leeuwen, Chem. Soc. Rev., 2009, 38, 1099–1118 RSC.
- (a) C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2953–2956 CrossRef CAS; (b) M. S. Nechaev, Organometallics, 2021, 40, 3408–3423 CrossRef CAS; (c) T. Gensch, G. dos Passos Gomes, P. Friederich, E. Peters, T. Gaudin, R. Pollice, K. Jorner, A. Nigam, M. Lindner-D’Addario, M. S. Sigman and A. Aspuru-Guzik, J. Am. Chem. Soc., 2022, 144, 1205–1217 CrossRef CAS PubMed.
- (a) C. A. Tolman, J. Am. Chem. Soc., 1970, 92, 2956–2965 CrossRef CAS; (b) C. A. Tolman, Chem. Rev., 1977, 77, 313–348 CrossRef CAS.
- J. B. Peng and X. F. Wu, Angew. Chem., Int. Ed., 2018, 57, 1152–1160 CrossRef CAS PubMed.
- (a) R. I. McDonald, G. Liu and S. S. Stahl, Chem. Rev., 2011, 111, 2981–3019 CrossRef CAS PubMed; (b) Y. Li, D. Wu, H.-G. Cheng and G. Yin, Angew. Chem., Int. Ed., 2020, 59, 7990–8003 CrossRef CAS PubMed; (c) S. K. Dorn and M. K. Brown, ACS Catal., 2022, 12, 2058–2063 CrossRef CAS PubMed.
- (a) R. Franke, D. Selent and A. Börner, Chem. Rev., 2012, 112, 5675–5732 CrossRef CAS PubMed; (b) A. Börner and R. Franke, Hydroformylation: Fundamentals, Processes, and Applications in Organic Synthesis, 2 Volumes, John Wiley & Sons, 2016, vol. 1 CrossRef.
- K. Dong, R. Sang, X. Fang, R. Franke, A. Spannenberg, H. Neumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2017, 56, 5267–5271 CrossRef CAS PubMed.
- (a) S. Cai, H. Zhang and H. Huang, Trends Chem., 2021, 3, 218–230 CrossRef CAS; (b) Y.-H. Yao, H.-Y. Yang, M. Chen, F. Wu, X.-X. Xu and Z.-H. Guan, J. Am. Chem. Soc., 2021, 143, 85–91 CrossRef CAS PubMed; (c) F. Doraghi, M. H. Morshedsolouk, N. Raisi, T. Hosseinifar, M. Noori, B. Larijani and M. Mahdavi, Chem. Rec., 2025, 25, e202500029 CrossRef CAS PubMed.
- P. J. C. van Leeuwen, Hydroformylation, Hydrocarbonylation, Hydrocyanation, and Hydroacylation of Carbon–Carbon Double Bonds, Science of Synthesis, Georg Thieme, Stuttgart, 2011, vol. 1, pp. 477–519 Search PubMed.
- Z.-L. Li, G.-C. Fang, Q.-S. Gu and X.-Y. Liu, Chem. Soc. Rev., 2020, 49, 32–48 RSC.
- F. P. Wu, Y. Yuan, C. Schünemann, P. C. J. Kamer and X. F. Wu, Angew. Chem., Int. Ed., 2020, 59, 10451–10455 CrossRef CAS PubMed.
- (a) D. S. Laitar, P. Müller and J. P. Sadighi, J. Am. Chem. Soc., 2005, 127, 17196–17197 CrossRef CAS PubMed; (b) D. S. Laitar, E. Y. Tsui and J. P. Sadighi, Organometallics, 2006, 25, 2405–2408 CrossRef CAS.
- F. Zhao, J.-X. Xu, F.-P. Wu and X.-F. Wu, J. Catal., 2023, 417, 379–381 CrossRef CAS.
- (a) C.-T. Yang, Z.-Q. Zhang, H. Tajuddin, C.-C. Wu, J. Liang, J.-H. Liu, Y. Fu, M. Czyzewska, P. G. Steel, T. B. Marder and L. Liu, Angew. Chem., Int. Ed., 2012, 51, 528–532 CrossRef CAS PubMed; (b) S. Akiyama, N. Oyama, T. Endo, K. Kubota and H. Ito, J. Am. Chem. Soc., 2021, 143, 5260–5268 CrossRef CAS PubMed.
- Y. Yuan, F. P. Wu, J. X. Xu and X. F. Wu, Angew. Chem., Int. Ed., 2020, 59, 17055–17061 CrossRef CAS PubMed.
- (a) I. Nakamura and Y. Yamamoto, Adv. Synth. Catal., 2002, 344, 111–129 CrossRef CAS; (b) M. Rubin, M. Rubina and V. Gevorgyan, Chem. Rev., 2007, 107, 3117–3179 CrossRef CAS PubMed; (c) J. Zhou, L. Meng, S. Lin, B. Cai and J. Wang, Angew. Chem., Int. Ed., 2023, 62, e202303727 CrossRef CAS PubMed; (d) W. Zheng, B. B. Tan, S. Ge and Y. Lu, J. Am. Chem. Soc., 2024, 146, 5366–5374 CrossRef CAS PubMed.
- (a) J. Zhou, Q. Yang, C. S. Lee and J. Wang, Angew. Chem., Int. Ed., 2022, 61, e202202160 CrossRef CAS PubMed; (b) Y.-S. Zhu, Y.-L. Guo, Y.-Y. Zhu and B. Su, J. Am. Chem. Soc., 2024, 146, 32283–32291 CrossRef CAS PubMed.
- F. P. Wu and X. F. Wu, Chem. Sci., 2022, 13, 4321–4326 RSC.
- Y. Yuan and X.-F. Wu, Green Carbon, 2024, 2, 70–80 CrossRef CAS.
- Y. Yuan, Y. Zhang, J.-X. Xu and X.-F. Wu, CCS Chem., 2022, 5, 1866–1875 CrossRef.
- B. Wang, C. Shen and K. Dong, Org. Lett., 2024, 26, 3628–3633 CrossRef CAS PubMed.
- (a) B. El Ali, J. Tijani, A. El-Ghanam and M. Fettouhi, Tetrahedron Lett., 2001, 42, 1567–1570 CrossRef CAS; (b) N. Iranpoor, H. Firouzabadi, A. Riazi and K. Pedrood, J. Organomet. Chem., 2016, 822, 67–73 CrossRef CAS.
- H. J. Ai, W. Lu and X. F. Wu, Angew. Chem., Int. Ed., 2021, 60, 17178–17184 CrossRef CAS PubMed.
- Y. Luo, X. Wang, Q. Liu, Y. He, J. Li, S. Luo and Q. Zhu, Green Chem., 2023, 25, 1120–1127 RSC.
- (a) Z. Xiao, N. C. Waters, C. L. Woodard, Z. Li and P.-K. Li, Bioorg. Med. Chem. Lett., 2001, 11, 2875–2878 CrossRef CAS PubMed; (b) S. Dakshanamurthy, M. Kim, M. L. Brown and S. W. Byers, Bioorg. Med. Chem. Lett., 2007, 17, 4551–4556 CrossRef CAS PubMed.
- D. Ding, G. Zhu and X. Jiang, Angew. Chem., Int. Ed., 2018, 57, 9028–9032 CrossRef CAS PubMed.
- J. Liu, J. Yang, W. Baumann, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2019, 58, 10683–10687 CrossRef CAS PubMed.
- J. Liu, J. Yang, C. Schneider, R. Franke, R. Jackstell and M. Beller, Angew. Chem., Int. Ed., 2020, 59, 9032–9040 CrossRef CAS PubMed.
- (a) J.-K. Cheng and T.-P. Loh, J. Am. Chem. Soc., 2015, 137, 42–45 CrossRef CAS PubMed; (b) K.-F. Zhang, K.-J. Bian, C. Li, J. Sheng, Y. Li and X.-S. Wang, Angew. Chem., Int. Ed., 2019, 58, 5069–5074 CrossRef CAS PubMed; (c) Q. Dherbassy, S. Manna, F. J. T. Talbot, W. Prasitwatcharakorn, G. J. P. Perry and D. J. Procter, Chem. Sci., 2020, 11, 11380–11393 RSC; (d) Q.-C. Shan, Y. Zhao, S.-T. Wang, H.-F. Liu, X.-H. Duan and L.-N. Guo, ACS Catal., 2024, 14, 2144–2150 CrossRef CAS.
- C.-S. Kuai, Y. Wang, T. Yang and X.-F. Wu, J. Am. Chem. Soc., 2025, 147, 7950–7964 CrossRef CAS PubMed.
- (a) T. Henkel, R. M. Brunne, H. Müller and F. Reichel, Angew. Chem., Int. Ed., 1999, 38, 643–647 CrossRef CAS; (b) J. M. Herrmann and B. König, Eur. J. Org. Chem., 2013, 7017–7027 CrossRef CAS.
- (a) H. A. van Kalkeren, S. H. A. M. Leenders, C. R. A. Hommersom, F. P. J. T. Rutjes and F. L. van Delft, Chem. – Eur. J., 2011, 17, 11290–11295 CrossRef CAS PubMed; (b) B. T. Sargent and E. J. Alexanian, Angew. Chem., Int. Ed., 2019, 58, 9533–9536 CrossRef CAS PubMed.
- X.-W. Gu, Y.-H. Zhao and X.-F. Wu, Nat. Commun., 2024, 15, 9412 CrossRef CAS PubMed.
- (a) Y.-K. Liu, X.-W. Gu and X.-F. Wu, Org. Lett., 2025, 27, 1316–1321 CrossRef CAS PubMed; (b) Y.-K. Liu, X.-W. Gu, Y.-H. Zhao and X.-F. Wu, J. Catal., 2025, 443, 115956 CrossRef CAS.
- X.-W. Gu, Y.-H. Zhao and X.-F. Wu, Chem. Sci., 2024, 15, 19970–19976 RSC.
- Y. Wang, Y. Xu, X. Qi, L.-C. Wang, C. Xu, G. Huang and X.-F. Wu, Nat. Commun., 2025, 16, 6305 CrossRef CAS PubMed.
- M. B. Li, A. K. Inge, D. Posevins, K. P. J. Gustafson, Y. Qiu and J. E. Backvall, J. Am. Chem. Soc., 2018, 140, 14604–14608 CrossRef CAS PubMed.
- (a) J. Tjutrins and B. A. Arndtsen, Chem. Sci., 2017, 8, 1002–1007 RSC; (b) T. Itoh, Y. Kanzaki, Y. Shimizu and M. Kanai, Angew. Chem., Int. Ed., 2018, 57, 8265–8269 CrossRef CAS PubMed; (c) Z. Li, L. Zhang, M. Nishiura, G. Luo, Y. Luo and Z. Hou, J. Am. Chem. Soc., 2020, 142, 1966–1974 CrossRef CAS PubMed; (d) K. Morisaki, H. Morimoto and T. Ohshima, ACS Catal., 2020, 10, 6924–6951 CrossRef CAS.
- (a) Y. Wu, L. Hu, Z. Li and L. Deng, Nature, 2015, 523, 445–450 CrossRef CAS PubMed; (b) A. Patra, S. Mukherjee, T. K. Das, S. Jain, R. G. Gonnade and A. T. Biju, Angew. Chem., Int. Ed., 2017, 56, 2730–2734 CrossRef CAS PubMed.
- F. P. Wu and X. F. Wu, Angew. Chem., Int. Ed., 2020, 60, 695–700 CrossRef PubMed.
- Z. Li, L. Zhang, M. Nishiura and Z. Hou, ACS Catal., 2019, 9, 4388–4393 CrossRef CAS.
- B. Lu, M. Xu, X. Qi, M. Jiang, W. J. Xiao and J. R. Chen, J. Am. Chem. Soc., 2022, 144, 14923–14935 CrossRef CAS PubMed.
- (a) A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318–3326 CrossRef CAS; (b) A. Schoenberg and R. F. Heck, J. Org. Chem., 1974, 39, 3327–3331 CrossRef CAS.
- R. R. Xu, C. S. Kuai and X. F. Wu, ChemistryEurope, 2025, 3, e202400105 CrossRef CAS.
- (a) L.-J. Cheng and N. P. Mankad, Chem. Soc. Rev., 2020, 49, 8036–8064 RSC; (b) L.-J. Cheng and N. P. Mankad, Acc. Chem. Res., 2021, 54, 2261–2274 CrossRef CAS PubMed.
- (a) A. Rudolph and M. Lautens, Angew. Chem., Int. Ed., 2009, 48, 2656–2670 CrossRef CAS PubMed; (b) N. Kambe, T. Iwasaki and J. Terao, Chem. Soc. Rev., 2011, 40, 4937–4947 RSC.
- (a) Y. Yuan, F.-P. Wu, A. Spannenberg and X.-F. Wu, Sci. China: Chem., 2021, 64, 2142–2153 CrossRef CAS; (b) Y. Yuan, Y. Zhang, W. Li, Y. Zhao and X. F. Wu, Angew. Chem., Int. Ed., 2023, 62, e202309993 CrossRef CAS PubMed; (c) Y. Yuan, Y. Zhang and X. F. Wu, Nat. Commun., 2024, 15, 6705 CrossRef CAS PubMed.
- F. P. Wu and X. F. Wu, Angew. Chem., Int. Ed., 2021, 60, 11730–11734 CrossRef CAS PubMed.
- F. Zhao, H. J. Ai and X. F. Wu, Angew. Chem., Int. Ed., 2022, 61, e202200062 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2026 |