
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A palladium inverse crown: synthesis and characterisation
Felix Krämer *a,
Alan R. Kennedya,
Israel Fernández*b and
Robert E. Mulvey*a
*a,
Alan R. Kennedya,
Israel Fernández*b and
Robert E. Mulvey*a
aDepartment of Pure and Applied Chemistry, University of Strathclyde, Glasgow G1 1XL, UK. E-mail: felix.kraemer@strath.ac.uk; r.e.mulvey@strath.ac.uk
bDepartamento de Química Orgánica I, Facultad de Ciencias Químicas and Centro de Innovación en Química Avanzada (ORFEO-CINQA), Universidad Complutense de Madrid, 28040 Madrid, Spain. E-mail: israel@quim.ucm.es
First published on 9th December 2025
Abstract
A unique palladium inverse crown complex in [Cs2(18-crown-6)2]2+ [(PdPtBuPh)6(PPh)]2− is presented where a neutral (PdP)6 ring hosts the dianionic PhP2− guest. Characterised by SC-XRD and high-resolution mass spectrometry, its solution constitution in THF is probed by DOSY and multinuclear NMR with quantitative insight into its bonding by DFT calculations.
The label inverse crown has been applied loosely as initially it was used in the context of inverse crown ‘ethers’.1 In these, oxygen-based anions are encapsulated by cationic metal–ligand cycles due to their topological similarity but with reversed Lewis acidic (alkali metal)/Lewis basic (oxygen) positions compared to those in Nobel Laureate Pederson's conventional crown ether complexes.2 However, the subsequent synthesis of closely related host–guest structures with a variety of different core anions including alkoxides, arenes, hydrides, and metallocenes led to the dropping of the ether label.3 The cationic cycles are generally heterobimetallic with the alkali metal connected to a second metal such as magnesium or zinc via amide bridges.4–6 Construction of such heterobimetallic inverse crowns can involve multiple deprotonative metallation reactions usually with special selectivities that are challenging to do in the absence of the two-metal cooperativities often operating in inverse crown chemistry.7,8
Alkali metal inverse crown structures containing transition metals are scarce (Scheme 1, top). Examples are shown with Fe, Ru, and Os,9 Fe and Cr10 and Mn.11,12 Harder's group has taken this chemistry to an exciting new level with their report of redox-active inverse crowns composed of Mg(0) centres and Na+ ions that have found use in small molecule activation.13 Transition metal-only inverse crowns are often described as metallacrowns and have been summarized in several review articles.14–16 For palladium the metallacrown structures mostly contain Pd(II) centres which are supported with N, O, S or P donor ligands.15
 | ||
| Scheme 1 Reported transition metal alkali metal inverse crown structures (top) and low valent Pd nanoclusters (bottom). | ||
For low valent Pd compounds the transition from metallacrowns to nanoclusters,17 so-called nano sheets18 or nanowires19 is fluid. A key difference is that such compounds consisting of palladium rings or cages built by Pd–Pd bonds supported by auxiliary ligands having no central guest or an additional Pd atom at the centre of the nanocluster (selected examples shown in Scheme 1, bottom).18,20–34
Herein, we present a unique inverse crown complex that has been synthesised fortuitously during our studies of caesium phosphide chemistry.4,35 It has a spectacular structure and unique composition.
During the reaction of Cs(18-crown-6)PtBuPh with commercially available Pd(PPh3)4 (Scheme 2), dark red crystals formed reproducibly in yields of up to 30% in benzene at 60 °C and were identified by SC-XRD methods as the inverse crown 1 with its eye-catching 12-membered host ring Pd6(PtBuPh)6 incorporating a PhP2− fragment as a guest molecule (Fig. 1a–d). The two negative charges are balanced by two fused 18-crown-6 coordinated Cs+ cations.
 | ||
| Scheme 2 Reaction of Cs(18-crown-6)PtBuPh with Pd(PPh3)4 leading to the inverse crown complex 1. | ||
 | ||
| Fig. 1 Molecular structure of 1 in the solid state. Hydrogen atoms and solvent molecules are omitted for clarity. (a) Full structure; (b) all organic groups are omitted for clarity; (c) [Pd6(PtBuPh)6(PPh)]2− dianion view from above; (d) [Pd6(PtBuPh)6(PPh)]2− anion sideview. Selected bond distances (Å) and angles (°) are summarised in Table S2 and S3 in the SI. | ||
The crystal structure of 1 is composed of six interconnected Pd(I) atoms separated by six PtBuPh bridges resulting in a [Pd6P6] inverse crown host ring. Within the centre of this ring is a PhP2− dianion incorporated as a so-called guest molecule. The two negative charges are balanced by two 18-crown-6 coordinated Cs+ cations fused via Cs2, with Cs1 bonded to one palladium atom Pd2 (Fig. 1b). The dianionic [(PdPtBuPh)6(PPh)]2− moiety could be described as a system made up of 12 fused rings comprising 6 internal and 6 external (Pd2P) triangles. As can be seen in Table S2 (see SI), the Pd–Pd distances (2.66–2.67 Å) are nearly all the same within experimental error of each other and within the range of those earlier reported.32,36 The Pd–P distances involving the peripheral P centres cover a narrow range [2.258(2)–2.270(2) Å] within the range of reported systems.37,38 Shorter distances [2.235(2) and 2.247(2) Å] are found for Pd2. This is attributed to the additional short Pd2–Cs1 bond [distance of 3.3004(11) Å], which in turn increases the positive partial charge at the Pd atom. Two distinct distances between the palladium atoms and the central phosphorus atom were found. Pd1, Pd3, Pd4 and Pd6 show shorter (approximately, 2.6 Å) bonds whereas Pd2 and Pd5 exhibit longer distances of 2.877(2) Å and 2.726(2) Å respectively, breaking the symmetry of the [Pd6P6] core. This is reflected in the different bond angles between the palladium atoms (Pd4–Pd5–Pd6 and Pd1–Pd2–Pd3; the latter are narrower) and the angles between two Pd atoms with the central P7 atom where P7–Pd3–Pd2 (66.30(5)°) and P7–Pd4–Pd5 (62.27(5)°) are wider compared to the others with approximately 59° (Table S3, SI). The angles between the peripheral phosphorus atoms and the two connected Pd atoms are in a very narrow range around 54°. The dihedral angle between the planes spanned by Pd1–Pd2–Pd3 and Pd4–Pd5–Pd6 is 17° reflecting a slight concave distortion from planarity as drawn (Fig. 1d).
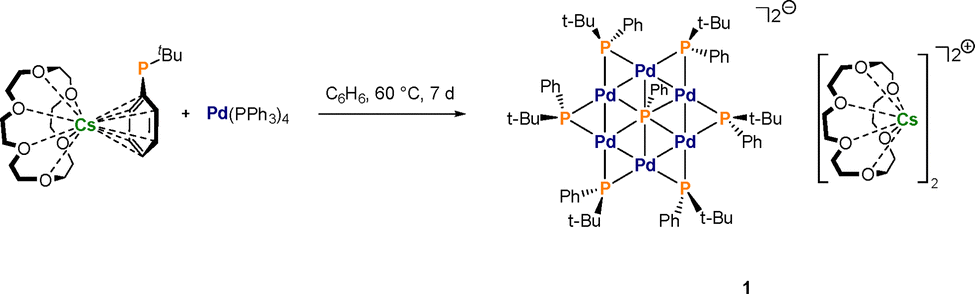
Further proof of the structure was gained by electron spray ionisation high resolution mass spectrometry (ESI-HRMS) giving a signal m/z = 1868.8382 (calc. 1868.84258) associated with the monoanion [CsPd6(PtBuPh)6(PPh)]− (Fig. 2 and Fig. S4 in the SI). In the positive region, a signal at m/z = 397.0625 (calc. 397.06219) was detected for the corresponding Cs(18-crown-6)+ cation (see Fig. S5 in the SI for more details).
 | ||
| Fig. 2 ESI-HRMS of 1 in THF. Experimental (top) and calculated (bottom) isotopic patterns for the fragment of the protonated anion [CsPd6(PtBuPh)6(PPh)]− at m/z = 1868.8382. | ||
1H and 31P{1H} NMR spectra of 1 showed very broad signals of low intensity. However, 1H NMR spectra showed one signal at δ1H = 1.41 ppm for the tBu groups, a multiplet between 6.6–7.1 ppm and a corresponding broad signal at 8.40 ppm for the Ph groups of the terminal phosphides. For the central P bonded phenyl group, a multiplet between 5.50–5.82 ppm was detected. The crown ether in the cation showed a very broad signal at δ1H = 3.11 ppm. The corresponding 31P{1H} NMR shifts were detected at δ31P = 236.2 ppm as a doublet (2JPP = 151.1 Hz) for the terminal phosphorus atoms and a very weak, very broad signal at 229.9 ppm which is attributed to the central P atom. Diffusion ordered spectroscopy (DOSY) methods in THF-d8 revealed that 1 dissociates in solution into a solvent-separated ion pair giving a molecular mass for the anion Mw(exp.) = 2174 g mol−1 differing by only 2% from [Cs(18-crown-6)Pd6(PtBuPh)6(PPh)]− (Mw(calc.) = 2135 g mol−1) and the dication Mw(exp.) = 777 g mol−1 differing by 2% from [Cs2(18-crown-6)2]2+ (Mw(calc.) = 794 g mol−1) determined by the method of Stalke (further details are given in the SI, Section S2).39–41 Having both the anion bearing a Cs(18-crown-6)+ and the fused dication in solution indicates some sort of dynamic exchange of one Cs(18-crown-6)+ fragment.
Density functional theory (DFT) calculations at the dispersion-corrected RI-BP86-D3BJ/def2-SVP level were carried out to gain more insight into the bonding situation in this new type of inverse crown 1. To this end, we focused on the interaction between the central PhP fragment and [Pd(PMe2)]6 core, a model system where the bulky tBu and Ph substituents attached to each phosphorus atom in 1 were replaced by smaller Me groups. Our quantum theory of atom in molecules (QTAIM, Fig. 3) calculations confirm the occurrence of the [Pd6P6] core where the PhP moiety is mainly bonded to four of the six Pd atoms (Fig. 3).
 | ||
| Fig. 3 Contour line diagrams ∇2ρ(r) for [Pd6(PMe2)6(PPh)]2− in the Pt–P–Pd plane. Solid lines connecting the atomic nuclei are bond paths, while the small green spheres indicate the corresponding bond critical points, respectively. | ||
In all cases, the located Pd–P bond critical points feature positive values of ∇2ρ, therefore suggesting donor–acceptor (that is, dative) bonds.42 Moreover, the central Pd−P bonds involving the PhP moiety are significantly weaker than the peripheral Pd–P bonds, as viewed from the computed lower electron density (ρ) and delocalization indices (DI, see Fig. 3) and supported by the computed lower Mayer bond orders (approximately, 0.44 vs 0.82, respectively).
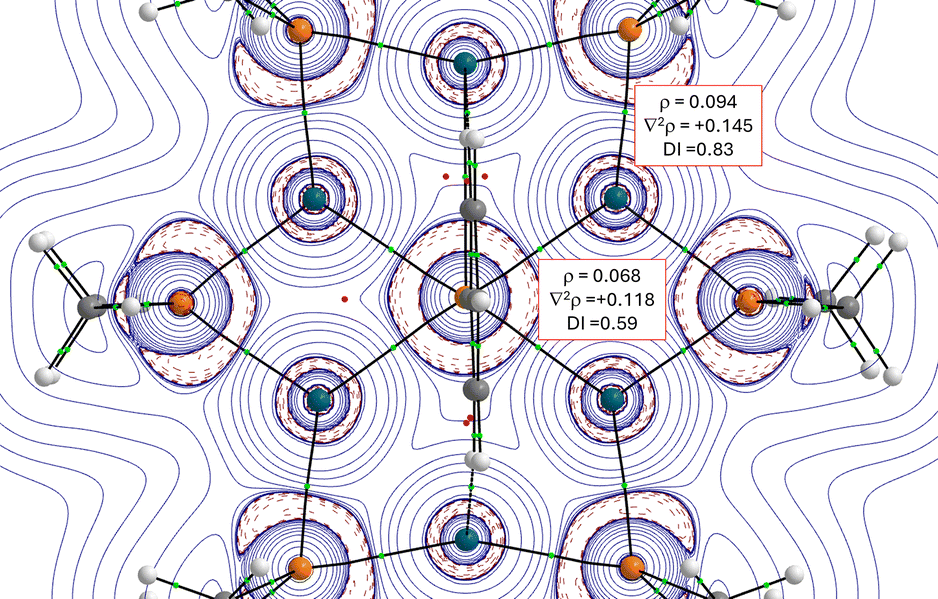
More quantitative insight into the central Pd–P bonds can be gained by means of the energy decomposition analysis (EDA) method (see details in the SI). To this end, we analysed the interaction between the central PhP fragment and [Pd(PMe2)]6 core using two possible fragmentation schemes, namely (i) [Pd6P6] and PhP2−, as closed-shell singlets (which agrees with the computed almost neutral natural charge of the palladium atoms, ranging from −0.01 to −0.16e) and (ii) [Pd6P6]2− and neutral phosphinidene PhP, either in their triplet or closed/open-singlet states (Table S5 in the SI). Our calculations indicate that the [Pd6P6]/PhP2− fragmentation constitutes a reasonable description of the bonding situation in [Pd6(PMe2)6(PPh)]2−, which is consistent with the positive ∇2ρ and natural charges commented above. Despite that, the fragmentation involving the charged, closed-shell [Pd6P6]2− core and neutral, open-shell singlet PhP cannot be ruled out as it exhibits a comparable ΔEorb strength.43 Indeed, this particular bonding situation has been found in related systems.33 In any case, it is found that the interaction between the [Pd6(PMe2)6] core and PhP fragments is relatively strong (ΔEint ≈ −200 kcal mol−1), which is not surprising since it involves four Pd⋯P interactions simultaneously (that is, approximately −50 kcal mol−1 per Pd−P bond). Partitioning of the ΔEint into its energy contributors suggests that the electrostatic interactions dominate over the orbital interactions (ΔEelstat contributing 69% to the total ΔEint for the [Pd6P6]/PhP2− fragmentation, which is expected given the charged nature of the PhP2− fragment). Further partitioning of the ΔEorb term with the natural orbital for chemical valence (NOCV) extension of the EDA method indicates that there exist three main orbital interactions between the [Pd6P6] core and PhP2− fragments, all of them involving donor–acceptor interactions from lone-pairs located at the PhP2− moiety to the [Pd6P6] core (Fig. 4). The stronger interactions occur from the donation of the p-type lone pairs at the phosphorus atom to the LUMO and LUMO+2 of the core (ρ1 and ρ2), while the third, weaker interaction (ρ3) involves the s-type lone pair at P to the LUMO+1 of the core, a vacant orbital involving the central Pd4 moiety (for the corresponding NOCV deformation densities involving the alternative [Pd6P6]2−/PhP fragmentations, see also Fig. S8 in the SI).
 | ||
| Fig. 4 Plot of the individual components of the deformation densities Δr and the associated orbitals of the fragments for [Pd6(PMe2)6(PPh)]2−. The color code of the charge flow in the deformation densities is red → blue and the eigenvalues Δx give the size of the charge flow. All data have been computed at the ZORA-BP86-D3BJ/TZ2P//RI-BP86-D3BJ/def2-SVP level. | ||
In summary, we have fortuitously discovered an example of a palladium inverse crown 1 consisting of a [Pd6P6] ring incorporating a PhP2− guest, which to the best of our knowledge is unique. The two negative charges are balanced by two fused 18-crown-6 coordinated Cs+ cations. Multinuclear NMR spectroscopic studies revealed the existence of the inverse crown structure in solution forming a solvent-separated ion pair. The composition of 1 was further proven by high resolution mass spectrometry. Quantum chemical methods including QTAIM and EDA-NOCV calculations indicate that the interaction between the [Pd6P6] core and the PhP2− host is strong, indicative of its stability in solution, and is mainly electrostatic with a significant contribution of donor–acceptor orbital interactions from lone-pairs at the P atom of the PhP2− to vacant molecular orbitals of the core. From the DFT calculations, although the [Pd6P6]/PhP2− fragmentation seems a reasonable description of the bonding situation in 1, the alternative description as a phosphinidine Pd6 nanocluster stabilized by six PPhtBu ligands cannot be fully dismissed. The report of this structure opens up an exciting new chapter in inverse crown chemistry from both an alkali metal-transition metal and phosphide bridge-phosphide core perspective.
Author contributions
F. K. conducted the experiments and wrote the manuscript with the input of all authors. A. R. K. solved and modelled the solid-state structure. I. F. conducted quantum chemical calculations and R. E. M. wrote the introduction and supervised the project.Conflicts of interest
There are no conflicts to declare.Data availability
Data that support the findings of this study are openly available in Pureportal.strath.ac.uk at https://doi.org/10.15129/3d704252-8748-41d5-8170-a85f3910b1cd, reference number 316661626.The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc06236e.
CCDC 2492602 contains the supplementary crystallographic data for this paper.44
Acknowledgements
We acknowledge Dr Graeme J Anderson and Dr Jessica Bame running the Mass Spectrometry Facility of the University of Strathclyde. R. E. M. thanks the Leverhulme Trust for generous funding via RPG-2023-248. F. K. is grateful for a Walter-Benjamin-Fellowship from Deutsche Forschungsgesellschaft (Project No. 535206405). Results were obtained using the ARCHIE-WeSt High Performance Computer (https://www.archie-west.ac.uk) at the University of Strathclyde (Grant code EP/K000586/1). I. F. is grateful for financial support from the Spanish MICIU/AEI/10.13039/501100011033 (Grant PID2022-139318NB-I00).References
- A. R. Kennedy, R. E. Mulvey, B. A. Roberts, R. B. Rowlings and C. L. Raston, Chem. Commun., 1999, 353–354, 10.1039/A809681C.
- C. J. Pedersen, J. Am. Chem. Soc., 1967, 89, 7017–7036 CrossRef.
- S. D. Robertson, M. Uzelac and R. E. Mulvey, Chem. Rev., 2019, 119, 8332–8405 CrossRef.
- R. E. Mulvey, Organometallics, 2006, 25, 1060–1075 CrossRef.
- R. E. Mulvey, Chem. Commun., 2001, 1049–1056, 10.1039/B101576L.
- A. R. Kennedy, R. E. Mulvey and R. B. Rowlings, J. Am. Chem. Soc., 1998, 120, 7816–7824 CrossRef.
- A. J. Martínez-Martínez, D. R. Armstrong, B. Conway, B. J. Fleming, J. Klett, A. R. Kennedy, R. E. Mulvey, S. D. Robertson and C. T. O’Hara, Chem. Sci., 2014, 5, 771–781 RSC.
- P. C. Andrews, A. R. Kennedy, R. E. Mulvey, C. L. Raston, B. A. Roberts and R. B. Rowlings, Angew. Chem., Int. Ed., 2000, 39, 1960–1962 CrossRef.
- P. C. Andrikopoulos, D. R. Armstrong, W. Clegg, C. J. Gilfillan, E. Hevia, A. R. Kennedy, R. E. Mulvey, C. T. O’Hara, J. A. Parkinson and D. M. Tooke, J. Am. Chem. Soc., 2004, 126, 11612–11620 CrossRef PubMed.
- P. Alborés, L. M. Carrella, W. Clegg, P. García-Álvarez, A. R. Kennedy, J. Klett, R. E. Mulvey, E. Rentschler and L. Russo, Angew. Chem., Int. Ed., 2009, 48, 3317–3321 CrossRef.
- A. R. Kennedy, J. Klett, R. E. Mulvey, S. Newton and D. S. Wright, Chem. Commun., 2008, 308–310, 10.1039/B714880A.
- V. L. Blair, L. M. Carrella, W. Clegg, J. Klett, R. E. Mulvey, E. Rentschler and L. Russo, Chem. – Eur. J., 2009, 15, 856–863 CrossRef PubMed.
- J. Maurer, L. Klerner, J. Mai, H. Stecher, S. Thum, M. Morasch, J. Langer and S. Harder, Nat. Chem., 2025, 17, 703–709 CrossRef.
- M. Ostrowska, I. O. Fritsky, E. Gumienna-Kontecka and A. V. Pavlishchuk, Coord. Chem. Rev., 2016, 327–328, 304–332 CrossRef.
- G. Mezei, C. M. Zaleski and V. L. Pecoraro, Chem. Rev., 2007, 107, 4933–5003 CrossRef PubMed.
- J. J. Bodwin, A. D. Cutland, R. G. Malkani and V. L. Pecoraro, Coord. Chem. Rev., 2001, 216–217, 489–512 CrossRef.
- Q. Liu and L. Zhao, Chin. J. Chem., 2020, 38, 1897–1908 CrossRef CAS.
- Q. You, X.-L. Jiang, W. Fan, Y.-S. Cui, Y. Zhao, S. Zhuang, W. Gu, L. Liao, C.-Q. Xu, J. Li and Z. Wu, Angew. Chem., Int. Ed., 2024, 63, e202313491 CrossRef CAS.
- K. Nakamae, Y. Takemura, B. Kure, T. Nakajima, Y. Kitagawa and T. Tanase, Angew. Chem., Int. Ed., 2015, 54, 1016–1021 CrossRef CAS PubMed.
- J. Dubrawski, J. C. Kriege-Simondsen and R. D. Feltham, J. Am. Chem. Soc., 1980, 102, 2089–2091 CrossRef CAS.
- E. G. Mednikov, N. K. Eremenko, V. A. Mikhailov, S. P. Gubin, Y. L. Slovokhotov and Y. T. Struchkov, J. Chem. Soc., Chem. Commun., 1981, 989–990, 10.1039/C39810000989.
- D. Fenske, H. Fleischer and C. Persau, Angew. Chem., Int. Ed. Engl., 1989, 28, 1665–1667 CrossRef.
- A. D. Burrows, J. C. Machell and D. M. P. Mingos, J. Am. Chem. Soc., Dalton Trans., 1992, 1991–1995, 10.1039/DT9920001991.
- N. T. Tran and L. F. Dahl, Angew. Chem., Int. Ed., 2003, 42, 3533–3537 CrossRef PubMed.
- J. Chen, L. Liu, L. Weng, Y. Lin, L. Liao, C. Wang, J. Yang and Z. Wu, Sci. Rep., 2015, 5, 16628 CrossRef PubMed.
- S. L. Benjamin, T. Krämer, W. Levason, M. E. Light, S. A. Macgregor and G. Reid, J. Am. Chem. Soc., 2016, 138, 6964–6967 CrossRef PubMed.
- C. Jandl, K. Öfele and A. Pöthig, Organometallics, 2017, 36, 4348–4350 CrossRef.
- M. Teramoto, K. Iwata, H. Yamaura, K. Kurashima, K. Miyazawa, Y. Kurashige, K. Yamamoto and T. Murahashi, J. Am. Chem. Soc., 2018, 140, 12682–12686 CrossRef PubMed.
- T. N. Hooper, S. Lau, W. Chen, R. K. Brown, M. Garçon, K. Luong, N. S. Barrow, A. S. Tatton, G. A. Sackman, C. Richardson, A. J. P. White, R. I. Cooper, A. J. Edwards, I. J. Casely and M. R. Crimmin, Chem. Sci., 2019, 10, 8083–8093 RSC.
- T. Ishikawa, A. Kawamura, T. Sugawa, R. Moridaira, K. Yamamoto and T. Murahashi, Angew. Chem., Int. Ed., 2019, 58, 15318–15323 CrossRef PubMed.
- K. Shimamoto and Y. Sunada, Chem. – Eur. J., 2019, 25, 3761–3765 CrossRef PubMed.
- A. W. Cook, P. Hrobárik, P. L. Damon, G. Wu and T. W. Hayton, Inorg. Chem., 2020, 59, 1471–1480 CrossRef PubMed.
- K. Breitwieser, M. Bevilacqua, S. Mullassery, F. Dankert, B. Morgenstern, S. Grandthyll, F. Müller, A. Biffis, C. Hering-Junghans and D. Munz, Adv. Sci., 2024, 11, 2400699 CrossRef PubMed.
- X. Liu, J. N. McPherson, C. E. Andersen, M. S. B. Jørgensen, R. W. Larsen, N. J. Yutronkie, F. Wilhelm, A. Rogalev, M. Giménez-Marqués, G. Mínguez Espallargas, C. R. Göb and K. S. Pedersen, Nat. Commun., 2024, 15, 1177 CrossRef PubMed.
- F. Krämer, M. H. Crabbe, A. R. Kennedy, C. E. Weetman, I. Fernández and R. E. Mulvey, Chem. – Eur. J., 2025, e02127 CrossRef PubMed.
- T. Murahashi and H. Kurosawa, Coord. Chem. Rev., 2002, 231, 207–228 CrossRef.
- M. Montgomery, H. M. O’Brien, C. Méndez-Gálvez, C. R. Bromfield, J. P. M. Roberts, A. M. Winnicka, A. Horner, D. Elorriaga, H. A. Sparkes and R. B. Bedford, Dalton Trans., 2019, 48, 3539–3542 RSC.
- N. Jeddi, N. W. J. Scott, T. Tanner, S. K. Beaumont and I. J. S. Fairlamb, Chem. Sci., 2024, 15, 2763–2777 RSC.
- R. Neufeld and D. Stalke, Chem. Sci., 2015, 6, 3354–3364 RSC.
- S. Bachmann, B. Gernert and D. Stalke, Chem. Commun., 2016, 52, 12861–12864 RSC.
- S. Bachmann, R. Neufeld, M. Dzemski and D. Stalke, Chem. – Eur. J., 2016, 22, 8462–8465 CrossRef PubMed.
- For example, see: S. Shaik, D. Danovich, J. M. Galbraith, B. Braïda, W. Wu and P. C. Hiberty, Angew. Chem., Int. Ed., 2020, 59, 984–1001 CrossRef PubMed and references therein.
- For ΔEorb as a criterion for bonding situation, see for instance: G. Frenking, I. Fernández, N. Holzmann, S. Pan, I. Krossing and M. Zhou, JACS Au, 2021, 1, 623–645 CrossRef PubMed , and references therein.
- CCDC 2492602: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pnrjn.
| This journal is © The Royal Society of Chemistry 2026 |