
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNative-carboxylate-assisted enantioselective C–H annulations with allenes and 1,3-dienes on ferrocene
Devendra
Parganiha
 ,
Yagya Dutt
Upadhyay
,
Sumit
Khevariya†
,
Pruthviraj Amar
Patil†
,
Svastik
Jaiswal
,
Yogesh
Dhasmana
and
Sangit
Kumar
*
,
Yagya Dutt
Upadhyay
,
Sumit
Khevariya†
,
Pruthviraj Amar
Patil†
,
Svastik
Jaiswal
,
Yogesh
Dhasmana
and
Sangit
Kumar
*
Department of Chemistry, Indian Institute of Science Education and Research Bhopal, Bhopal By-Pass Road, Bhopal, Madhya Pradesh 462066, India. E-mail: sangitkumar@iiserb.ac.in
First published on 9th December 2025
Abstract
Native carboxylate as a directing group in enantioselective C–H activation results in poor stereodiscrimination, due to dynamic ligand exchange during the enantiodetermining concerted metalation–deprotonation (CMD) step. Herein, we present the synthesis of a new class of chiral ferrocene-fused isochroman derivatives from the readily available native carboxylate functionality. The Pd(II)/MPAA-derived enantioselective C–H activation and intermolecular annulation with allenes and 1,3-dienes afforded structurally diverse ferrocene-fused isochromans with up to 80% yield and 96![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 enantioselectivity.
:4 enantioselectivity.
Transition-metal-catalyzed enantioselective C–H activation is a powerful strategy for synthesizing planar chiral ferrocene molecules, with broad applications in both academic research and industrial processes.1 In this regard, the synthetic utility of the native directing group in planar chiral C–H activation has seen very limited development. The lower enthalpic contribution and conformational flexibility due to native functionalities, such as formaldehydes or carboxylates, lead to rapid ligand exchange.2 Therefore, methodology enabling highly enantioselective C–H activation using native functionalities is highly challenging.3 Additionally, a common platform for functional-group-derived enantioselective C–H activation strategy that could enable the synthesis of complex chiral molecules would be highly desirable.4
Achieving high enantioselective discrimination in C-sp3 carboxylate-assisted enantioselective C–H activation is comparatively facile because of the lower rate of dynamic ligand exchange and greater conformational flexibility present around the C-sp3 carboxylate.5 However, in the case of enantioselective C–H activation, the use of C-sp2 attached carboxylate facilitates a rapid ligand exchange equilibrium with chiral ligands, which may lead to racemization (Scheme 1a).2,6 Consequently, to date, enantioselective alkenylation via FcCOOH-directed C–H activation is achieved with an average of 83% enantioselectivity.6a
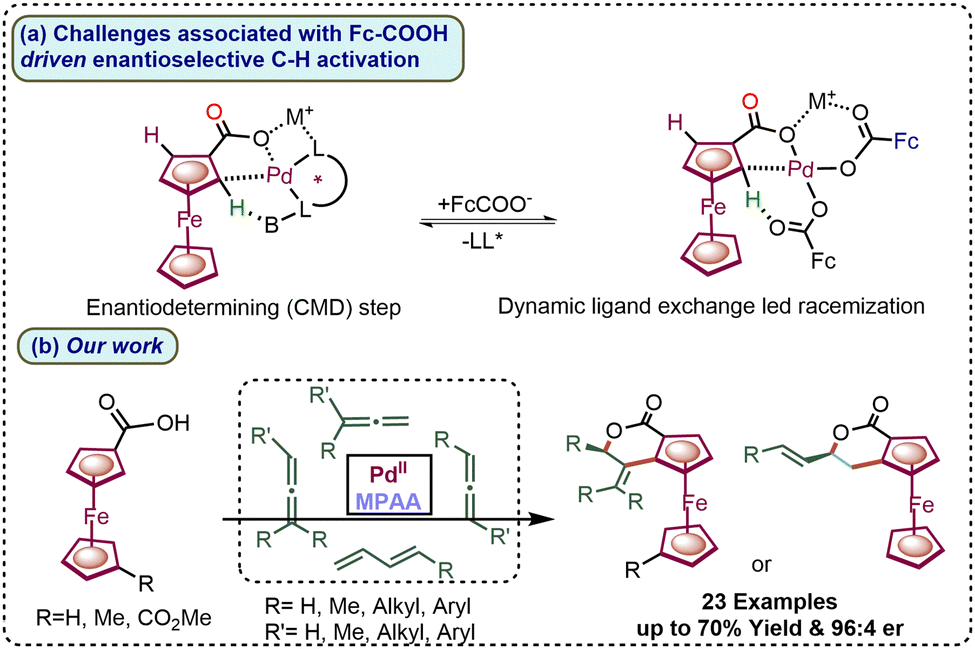 | ||
| Scheme 1 (a) Challenges associated with and the rational development of weak-carboxylate-assisted enantioselective C–H annulations. (b) Our work. | ||
Planar chiral ferrocene-fused heterocycles represent an important class of molecules that have unlocked new dimensions in the utility and versatility of planar chiral ferrocene scaffolds.7 However, the synthetic methodologies for these are very limited, as they depend exclusively on pre-designed tethered directing groups.8 Recently, You and co-workers reported the first Rh-chiral Cp*-enabled amide-directed enantioselective C–H activation followed by intermolecular annulation with alkynes.9
Building on these advances, our group also recently demonstrated an amine-enabled enantioselective C–H activation followed by intermolecular annulation on ferrocene.10 So far, only N-donor directing groups for intermolecular annulations have been reported.9,11 Herein, we report a Pd(II)/MPAA-enabled methodology for enantioselective C–H activation followed by intermolecular annulation with native ferrocene carboxylate. The regio- and enantioselective intermolecular annulation with allenes and 1,3-dienes afforded a new class of structurally diverse ferrocene-fused isochroman derivatives, achieving yields of up to 80% and 96:4 enantioselectivity (Scheme 1b).
We commenced our investigation using ferrocenecarboxylic acid and allene 2i with 10 mol% Pd(OAc)2 as a catalyst, 30 mol% ligand (L1, Fig. 1), Cs2CO3 as a base, and air as the sole oxidant in the solvent THF. The reaction afforded a 40% yield of ferrocene-fused isochroman 3a with up to 82:18 er (Table 1, Entry 1). Therefore, to further increase the yield and enantioselectivity, other solvents were screened under the same reaction conditions (Table 1, entries 2–5). tert-Amyl alcohol was found to be an efficient solvent for enantioselective synthesis, as it provided a better yield (66%) of 3a with up to 85:15 er (Table 1, entry 5). The moderate enantiomeric ratio suggested that the rate of ligand exchange may increase with temperature. Therefore, to suppress the ligand exchange, the temperature was lowered to 35 °C. This resulted in an improved reaction outcome, providing 3a with 70% yield and 90:10 er (Table 1, entry 6). Furthermore, the base Cs2CO3 was replaced with CsF and a slight increment in reaction outcome was observed, affording up to a 75% yield of 3a and 92:8 er (Table 1, entry 7).
 | ||
| Fig. 1 Screening of ligands for ferrocene carboxylate-assisted enantioselective C–H annulation under the optimized conditions (Table 1, entry 7). | ||
|
|
||||
|---|---|---|---|---|
| Entry | Solvent | Temp. | 3a (%) | erc (%) |
| a Reaction conditions: 1a (0.1 mmol), 2a (0.2 mmol), Pd(OAc)2 (0.01 mmol), ligand (0.03 mmol), Cs2CO3 (0.2 mmol), solvent (1 mL), air, T °C, 10 h. b The crude yield of 3a was determined via1H NMR with CH2Br2 as an internal standard. c The enantiomeric ratio (er) of 3a was determined by HPLC analysis. d 1.5 equiv. of CsF was used instead of Cs2CO3. e 0.6 equiv. of L1 was used instead of 0.3 equiv. with 1.8 equiv. CsF. | ||||
| 1 | THF | 60 | 40 | 82:18 |
| 2 | Toluene | 60 | 18 | 60:40 |
| 3 | DMF | 60 | NR | NR |
| 4 | TFE | 60 | NR | NR |
| 5 | t Amyl Alc | 60 | 66 | 85:15 |
| 6 | t Amyl Alc | 35 | 74 | 90:10 |
| 7 | t Amyl Alc | 35 | 75 | 92:8 |
| 8e | t Amyl Alc | 35 | 80 | 93:7 |
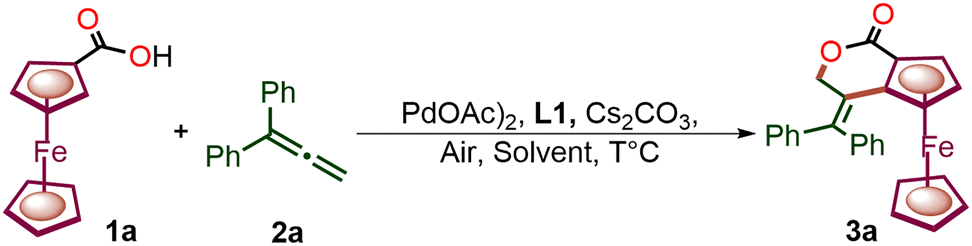
The use of other ligands (L2–L12) was also examined under the optimised reaction conditions (entry 7 and Fig. 1), but no further improvement in the reaction was observed. However, it is worth noting that the presence of the –COOH group in the ligand backbone is crucial for achieving enantioselectivity. Other bidentate ligands (L7–L9, L11) without –COOH were found to be completely unfit to achieve any chirality (Fig. 1). L10 also failed to provide any enantioselectivity due to the possibility of 6-membered palladacycle formation. This suggests that the carboxylate group of the chiral ligand may exhibit a κ2(O,O) mode of binding in the enantio-determining CMD under the reaction conditions.2 Substituting the amino acid for a chiral phosphoric acid (L12) provided an er of only 54:46 (Fig. 1). Furthermore, increasing the loading of Ligand L1 to 0.6 equiv. provided 80% yield and 93:7 er (Table 1, Entry 8).
With the optimized conditions in hand, we next investigated the substrate scope in the developed enantioselective C–H annulation using the native carboxylic acid as a directing group. A wide array of allenes 2b–2k, including mono-, di-, and tri-substituted variants, were subjected to the reaction to evaluate the versatility of the methodology.
Symmetrical α,α-disubstituted allenes 2b–2d bearing aryl and alkyl groups participated efficiently in the annulation, affording the corresponding ferrocenyl-fused isochroman derivatives 3b–3d in good yields (68–70%) with enantioselectivities of 85:15 to 92:8 er (Fig. 2). Encouraged by these results, we turned our attention to unsymmetrical α,α-disubstituted allenes 2e–2h. These substrates also underwent enantioselective C–H annulation to provide fused isochromans 3e–3h in good yields and with improved E/Z selectivity of up to 1:10, along with enantioselectivities ranging from 89:11 to 93:7 er.
 | ||
| Fig. 2 Substrate scope for the ferrocene-fused isochromans 3a–3v. [b] Isolated yields for 3a–3v. Crystal structure of 3d. The absolute configuration (Sp) assigned based on the crystal structure of 3d. | ||
Allene 2i bearing α,γ-di-substitution was also evaluated and provided the desired isochroman 3i in a moderate yield of 62%. Notably, product 3i exhibited an outstanding diastereoselectivity of 20:1 (Fig. 2). Furthermore, trisubstituted allenes 2j and 2k, which are typically considered to be difficult substrates in transition-metal-catalysed allene functionalization, were evaluated.
Gratifyingly, these complex allenes proved to be competent reaction partners, affording the corresponding annulated products (3j, 3k) in moderate yields of 52% and 50%, respectively. Excellent diastereoselectivity of 10:1 and 20:1 dr was observed for 3j and 3k. It is worth noting that the corresponding racemic reactions with these di- and tri-substituted allenes resulted in poor regio- and diastereoselectivity, rendering chromatographic separation of isomers challenging. Therefore, enantiomeric ratios for these racemic products could not be reliably determined.
In addition to allenes, phenyl-substituted 1,3-dienes 2l and 2m were also successfully employed in the acid-directed C–H annulation. Both substrates delivered the desired products in moderate yields of up to 62% with a diastereoselectivity of 1.5:1 for 3l and 5:1 for 3m, respectively. Finally, the scope was extended to structurally modified ferrocene carboxylic acids, with methyl-substituted ferrocenyl derivatives delivering ferrocene-fused isochroman products 3n–3q in up to 58% yield with excellent diastereoselectivity of 20:1 and enantioselectivities of up to 93:7 er. Similarly, ester-substituted ferrocenyl substrates underwent annulation to provide 3r–3v in lower yields (25–30%), albeit with consistently high stereoselectivities of up to 95:5 er.
After successfully incorporating diverse allenes and dienes in the enantioselective C–H annulation, we explored post-synthetic transformations of the resulting products. The reduction of compound 3f with LiAlH4 led to the formation of 1,2-keto acid ferrocene derivative 4 in trace amounts. Upon optimization under an oxygen atmosphere over three days, the yield of compound 4 increased to 45% without any loss of enantioselectivity (Scheme 2). Additionally, the ester-functionalized ferrocene-fused isochroman derivative 3r underwent selective hydrolysis with NaOH in EtOH at 60 °C for 6 h to afford product 5.
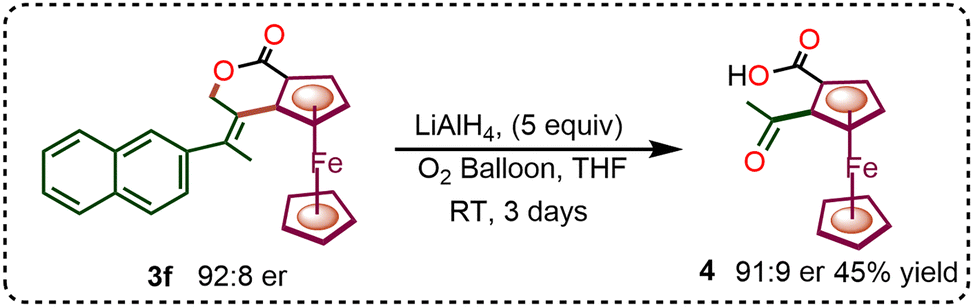 | ||
| Scheme 2 Post-synthetic transformation. | ||
In the proposed plausible catalytic cycle (Scheme 3), initially, ferrocene carboxylic acid 1a, Pd(OAc)2 and ligand L1 with a Cs+ cation form Pd–carboxylate intermediate I, which further undergoes enantioselective C–H activation to generate the chiral palladacycle II. DFT calculations showed an unfavorable transition state within the enantio-determining step having a 12.0 kcal mol−1 higher energy (Scheme 3).
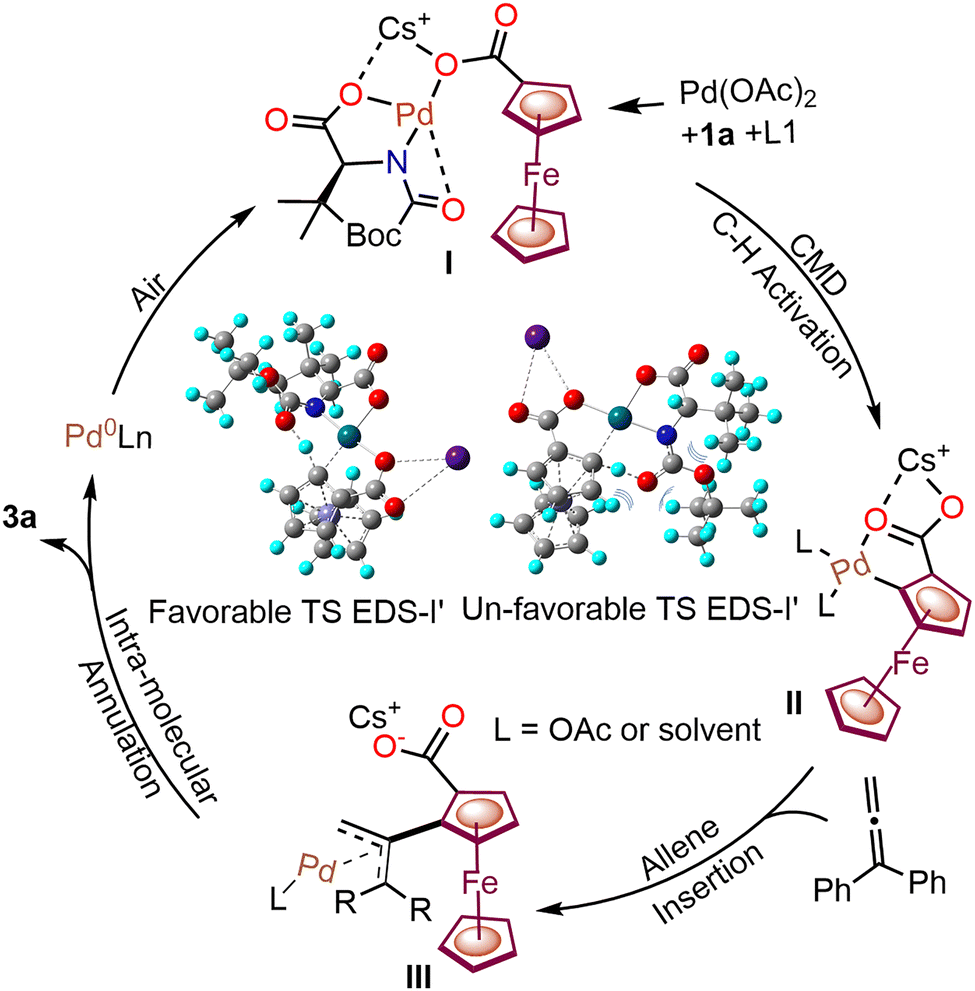 | ||
| Scheme 3 The catalytic cycle for native carboxylate-assisted enantioselective C–H annulations with allenes. Favorable and unfavorable transition states are shown (EDS = enantio-determining step). | ||
The chiral palladacycle II interacts with allenes through π-bond interaction, followed by migratory interaction, leading to the formation of Pd-allyl intermediate III.12 Consequently, intermediate III undergoes intramolecular annulation to afford a chiral ferrocenyl-fused isochroman.
In conclusion, we have presented a Pd(II)/MPAA-enabled methodology for enantioselective C–H activation followed by intermolecular annulation with native ferrocene carboxylate. Regio- and enantioselective intermolecular annulation with allenes and 1,3-dienes afforded a new class of structurally diverse ferrocene-fused isochromans, achieving yields of up to 70% and 96:4 er. The isochroman core is very important for facilitating various applications driven by electron transfer phenomena and is currently being explored in our laboratory.
SK and DP designed the research. DP, YDU, SKK, PP, and SJ synthesized all precursors and chiral ferrocene-fused isochroman derivatives. SK did the post-synthetic transformations. YD performed the DFT computational studies. All authors have approved the final version of the manuscript.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information. Supplementary information: all the data related to the experimental procedures, theoretical calculation data, characterization data, HPLC analysis data, and copies of NMR spectra of the synthesized chiral ferrocene fused isochromans 3a–3v and 4. FAIR data, including the primary NMR FID files, has also been provided as a ZIP file. See DOI: https://doi.org/10.1039/d5cc06160a.CCDC 2498520 (3d) contains the supplementary crystallographic data for this paper.13
Acknowledgements
SK acknowledges DST-ANRF (CRG/2023/002473), New Delhi, and IISER Bhopal for financial support. DP and SJ acknowledge UGC [14/(CSIR-UGC NET DEC 2019)] and UGC [NTA ref No. 201610131472], New Delhi for fellowships. YD thanks DST for INSPIRE fellowship.References
- (a) S. Murai, F. Kakiuchi, S. Sekine, Y. Tanaka, A. Kamatani, M. Sonoda and N. Chatani, Nature, 1993, 366, 529–531 CrossRef CAS; (b) G. Liao, T. Zhang, Z.-K. Lin and B.-F. Shi, Angew. Chem., Int. Ed., 2020, 59, 19773–19786 CrossRef CAS PubMed; (c) R. L. Carvalho, R. G. Almeida, K. Murali, L. A. Machado, L. F. Pedrosa, P. Dolui, D. Maiti and E. N. D. S. Júnior, Org. Biomol. Chem., 2021, 19, 525–547 RSC; (d) R. S. Thombal, P. Yuosef, M. Rubio, D. Lee, D. Maiti and Y. R. Lee, ACS Catal., 2022, 12, 5217–5230 CrossRef CAS; (e) B. B. Zhan, L. Jin and B.-F. Shi, Trends Chem., 2022, 4, 220–235 CrossRef CAS; (f) Q. Zhang, L.-S. Wu and B.-F. Shi, Chem, 2022, 8, 384–413 CrossRef CAS; (g) K. Wu, N. Lam, D. A. Strassfeld, Z. Fan, J. X. Qiao, T. Liu, D. Stamos and J.-Q. Yu, Angew. Chem., Int. Ed., 2024, 63, e202400509 CrossRef CAS PubMed.
- (a) T. A. Stephenson, S. M. Morehouse, A. R. Powell, J. P. Heffer and G. Wilkinson, J. Chem. Soc., 1965, 3632–3640 RSC; (b) D. C. Powers and T. Ritter, Nat. Chem., 2009, 1, 302–309 CrossRef CAS PubMed; (c) D. E. Hill, K. L. Bay, Y.-F. Yang, R. E. Plata, R. Takise, K. N. Houk, J.-Q. Yu and D. G. Blackmond, J. Am. Chem. Soc., 2017, 139, 18500–18503 CrossRef CAS PubMed; (d) C. A. Salazar, J. J. Gair, K. Flesch, I. A. Guzei, J. C. Lewis and S. S. Stahl., Angew. Chem., Int. Ed., 2020, 59, 10873–10877 CrossRef CAS PubMed.
- (a) G. Li, L. Wan, G. Zhang, D. Leow, J. Spangler and J.-Q. Yu, J. Am. Chem. Soc., 2015, 137, 4391–4397 CrossRef CAS PubMed; (b) D. A. Strassfeld, C.-Y. Chen and H.-S. Park, et al. , Nature, 2023, 622, 80–86 CrossRef CAS PubMed; (c) S. Saha, J. Das, S. A. Al-Thabaiti, S. M. Albukhari, Q. A. Alsulami and D. Maiti, Catal. Sci. Technol., 2023, 13, 11–12 RSC.
- (a) B.-F. Shi, N. Maugel, Y. H. Zhang and J.-Q. Yu, Angew. Chem., Int. Ed., 2008, 47, 4882–4886 CrossRef CAS PubMed; (b) D. W. Gao, Y. C. Shi, Q. Gu, Z. L. Zhao and S.-L. You, J. Am. Chem. Soc., 2013, 135, 86–89 CrossRef CAS PubMed; (c) L. Chu, K. J. Xiao and J.-Q. Yu, Science, 2014, 346, 451–455 CrossRef CAS PubMed; (d) Q. J. Yao, P. P. Xie, Y. J. Wu, Y. L. Feng, M. Y. Teng, X. Hong and B.-F. Shi, J. Am. Chem. Soc., 2020, 142, 18266–18276 CrossRef CAS PubMed; (e) N. Y. S. Lam, K. Wu and J.-Q. Yu, Angew. Chem., Int. Ed., 2021, 60, 15767–15790 CrossRef CAS PubMed; (f) B. Liu, A. M. Romine, C. Z. Rubel, K. M. Engle and B.-F. Shi, Chem. Rev., 2021, 121, 14957–15074 CrossRef CAS PubMed; (g) J. Grover, G. Prakash, N. Goswami and D. Maiti, Nat. Commun., 2022, 13, 1085 CrossRef CAS PubMed; (h) B. B. Zhan, L. Jin and B.-F. Shi, Trends Chem., 2022, 4, 220–235 CrossRef CAS; (i) J. H. Docherty, T. M. Lister, G. Mcarthur, M. T. Findlay, P. Domingo-Legarda, J. Kenyon, S. Choudhary and I. Larrosa, Chem. Rev., 2023, 123, 7692–7760 CrossRef CAS PubMed.
- (a) J. Das, D. K. Mal, S. Maji and D. Maiti, ACS Catal., 2021, 11, 4205–4229 CrossRef CAS; (b) S. Dutta, T. Bhattacharya, F. J. Geffers, M. Bürger, D. Maiti and D. B. Werz, Chem. Sci., 2022, 13, 2551–2573 RSC; (c) E. L. Lucas, N. Y. S. Lam, Z. Zhuang, H. S. S. Chan, D. A. Strassfeld and J.-Q. Yu, Acc. Chem. Res., 2022, 55, 537–550 CrossRef CAS PubMed; (d) D. Q. Phan and J.-Q. Yu, J. Am. Chem. Soc., 2025, 147, 36999–37004 CrossRef CAS PubMed.
- (a) Y. Huang, C. Pi, X. Cui and Y. Wu, Adv. Synth. Catal., 2020, 362, 1385–1390 CrossRef CAS; (b) A.-L. Jiang, G. Zhou, B.-Y. Jiang, T. Zhou, X.-T. Xu and B.-F. Shi, Org. Lett., 2024, 26, 5670–5675 CrossRef CAS PubMed.
- (a) O. Bernardo, S. González-Pelayo and L. A. López, Eur. J. Inorg. Chem., 2021, e202100911 Search PubMed; (b) J. Mahrholdt, E. Kovalski, M. Korb, A. Hildebrandt, V. Vrček and H. Lang, Eur. J. Inorg. Chem., 2021, 578–589 CrossRef CAS; (c) S. R. Mohanty, N. Prusty, T. Nanda, P. S. Mahulkar and P. C. Ravikumar, Org. Chem. Front., 2024, 11, 540–575 RSC.
- (a) M. Ogasawara, S. Watanabe, K. Nakajima and T. Takahashi, J. Am. Chem. Soc., 2010, 132, 2136–2137 CrossRef CAS PubMed; (b) D. W. Gao, Q. Yin, Q. Gu and S.-L. You, J. Am. Chem. Soc., 2014, 136, 4841–4844 CrossRef CAS PubMed.
- Q. Wang, Y.-H. Nie, C.-X. Liu, W.-W. Zhang, Z.-J. Wu, Q. Gu, C. Zheng and S.-L. You, ACS Catal., 2022, 12, 3083–3093 CrossRef CAS.
- (a) R. A. Thorat, D. Parganiha, S. Jain, V. Choudhary, B. Shakir, K. Rohilla, R. K. Jha and S. Kumar, Org. Lett., 2025, 27, 552–558 CrossRef PubMed; (b) D. Parganiha, R. A. Thorat, A. D. Dhumale, Y. D. Upadhyay, R. K. Jha, S. Raju and S. Kumar, Chem. Sci., 2025, 16, 700–708 RSC.
- (a) X. Vidal, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2019, 141, 1862–1866 CrossRef CAS PubMed; (b) J. M. González, B. Cendón, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2021, 143, 3747–3752 CrossRef PubMed; (c) J. M. González, X. Vidal, M. A. Ortuño, J. L. Mascareñas and M. Gulías, J. Am. Chem. Soc., 2022, 144, 21437–21442 CrossRef PubMed; (d) P.-F. Qian, Y.-X. Wu, J.-H. Hu, J.-H. Chen, T. Zhou, Q.-J. Yao, Z.-H. Zhang, B.-J. Wang and B.-F. Shi, J. Am. Chem. Soc., 2025, 147, 10791–10802 CrossRef CAS PubMed.
- (a) D. Zhao, B. Xu and C. Zhu, Nat. Commun., 2023, 14, 3308 CrossRef CAS PubMed; (b) J. Zhang and C. Zhu, Angew. Chem., Int. Ed., 2025, 64, e202512977 CrossRef CAS PubMed.
- CCDC 2498520: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2pvxfx.
Footnote |
| † SKK and PP contributed equally. |
| This journal is © The Royal Society of Chemistry 2026 |