
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Organocatalytic peptide coacervates as microreactors for aqueous aldol additions
Zhen
Dai
 ,
Malcolm
Meyn
and
Lucas
Caire da Silva
*
,
Malcolm
Meyn
and
Lucas
Caire da Silva
*
Department of Chemistry, McGill University, Montreal, H3A 0B8, Canada. E-mail: lucas.cairedasilva@mail.mcgill.ca
First published on 22nd December 2025
Abstract
We report a short peptide, PFF-OMe, that self-assembles into coacervate droplets containing a proline-based organocatalyst within their dense phase. The resulting catalyst-bearing coacervates greatly enhance proline-mediated reactions in aqueous environments, offering a minimal and tunable platform for efficient organocatalysis in water. This work demonstrates the potential of self-assembled peptide microreactors as versatile tools for aqueous-phase chemical transformations.
The association of solutes in aqueous media can lead to the formation of two immiscible liquid phases, a phenomenon known as coacervation.1,2 This phase separation arises from a balance between non-covalent intermolecular interactions, which promote condensation, and the retention of water within the dense phase.3,4 The result is a concentrated coacervate phase enriched in the associating molecules, coexisting with a surrounding dilute medium (Fig. 1a).
 | ||
| Fig. 1 Characterization of PFF-OMe coacervates. (a) Structure and LLPS of PFF-OMe. (b) PFF-OMe coacervate droplets and their liquid-like behaviour observed under brightfield microscopy. (c) FRAP recovery profile of PFF-OMe coacervates. Curve represents average normalized fluorescence intensity with shaded area of 1 standard deviation above and below. (d) Size distribution of PFF-OMe coacervates. (e) Phase diagram of PFF-OMe. (f) Reversible LLPS of PFF-OMe. 2.2 µL of 1 M HCl or 1 M NaOH was added in each cycle. All cases: PFF-OMe (9.2 mg mL−1) and 0.1 M HEPES pH 8.5 buffer. | ||
Dispersions of coacervate droplets in water offer a versatile platform for compartmentalized chemistry. The unique chemical environment within the coacervate phase can be tailored to selectively enrich specific reactants and catalysts.5 Unlike simple oil-in-water emulsions or micelles, coacervates form larger, membraneless compartments with tunable composition, ionic strength, and polarity. This enables the selective partitioning of hydrophobic species, and charged and polar molecules including enzymes, nucleic acids, and metal complexes.6–8 Coacervates can stabilize a wide range of chemo- and biocatalysts along with their substrates, and facilitate access to transition states, thereby accelerating chemical reactions.9–12 Moreover, coacervates have emerged as a greener alternative to organic solvents, enabling chemical reactions in aqueous media under mild conditions.13,14
Catalysis within polyelectrolyte-based coacervates has been extensively explored using charged catalysts. Notable examples include enzymes and carbon dots, which are sequestered into the coacervate phase via electrostatic interactions.15,16 These systems enable reactions by co-localizing catalysts and benefit from the intrinsic stabilizing and crowding effects of coacervates, allowing catalysts to remain active in aqueous media. Catalysis in peptide-based coacervates has also been demonstrated, with a key advantage being the high tunability of the microenvironment through amino acid composition.8,17,18 Our group has shown that hydrophobic catalysts can be efficiently sequestered within peptide-derived coacervates and employed as microreactors in aqueous conditions.13
Despite their versatility, coacervate microreactors face a fundamental limitation: catalyst loading and retention are typically governed by passive partitioning.19 This constraint limits the amount of catalyst that can be incorporated. An alternative is to tether the catalyst directly to the phase-separating scaffold, ensuring maximum enrichment. However, this “catalyst-in” architecture remains largely unexplored.
Here, we introduce an ultrashort peptide-based coacervate that incorporates its own organocatalyst. Specifically, we introduce PFF-OMe (L-Pro-L-Phe-L-Phe with a C-terminal methyl ester), in which the N-terminal proline functions as a covalently embedded catalyst, while the phenylalanine dyad drives liquid–liquid phase separation.20 PFF-OMe spontaneously forms coacervate droplets in water, effectively concentrating the proline catalyst within the dense phase and enhancing proline-mediated organocatalysis in aqueous media. We designed the PFF-OMe peptide to be structurally simple and readily synthesized. Its phase behavior was characterized using turbidity measurements and microscopy. At a concentration of 9.2 mg mL−1, increasing the pH above 8 resulted in a marked rise in turbidity and the formation of micron-sized spherical droplets (Fig. 1b). These droplets exhibited a unimodal size distribution, with a median diameter of 0.9 µm (Fig. 1d), placing them at the lower end of the size range typically reported for short peptide coacervates (1–10 µm).13,17 Over the course of several seconds, the droplets slowly merge and wet glass surfaces, confirming their liquid nature (Fig. 1b). Addition of a non-ionic surfactant (Pluronic F-108) suppressed droplet wetting and coalescence. Phase separation of PFF-OMe occurs at a pH slightly above the pKa of its secondary amine (8.7), consistent with previously reported FF-OMe coacervates (Fig. 1e and Fig. S1). This suggests that phase separation is driven by reversible deprotonation of the secondary amine, which increases the peptide's hydrophobicity. Cycling the pH between 7 and 9, below and above the critical pH, leads to the formation and dissolution of PFF-OMe coacervates (Fig. 1f). Tests with urea and 1,6-hexanediol showed that the hydrophobic effect and H-bonding were the main interactions driving coacervation (Fig. S2 and S3).21 On the other hand, π–π interactions could not be observed using Raman spectroscopy (Fig. S4).22 PFF-OMe coacervates were also highly temperature stable and could be heated to up to 80 °C (Fig. S5).
Addition of salt to the system enhanced phase separation, lowering the critical pH and increasing turbidity at a constant pH (Fig. 1e). This effect is attributed to peptide dehydration and electrostatic screening, which reduces repulsive interactions between peptides.23,24 Further increases in pH led to progressively greater phase separation, with maximum turbidity observed around pH 9.5 (Fig. 1e). Coacervates at pH > 10 showed a markedly reduced lifetime attributed to hydrolysis of the methyl ester group, yielding the more soluble PFF-OH peptide and leading to coacervate dissolution (Fig. S6). PFF-OMe coacervates showed a hydrophobic environment. Nonpolar Nile red readily and uniformly partitioned into the coacervate droplets, whereas more water-soluble, negatively charged dyes such as fluorescein and calcein exhibited preferential accumulation at the droplet interface (Fig. 2a), indicative of a nonpolar core. This hydrophobic character is consistent with phase separation driven by peptide deprotonation, yielding the uncharged, nonpolar PFF-OMe species. To further probe the coacervate polarity, we employed Prodan, a solvatochromic dye, as a molecular polarity sensor. When encapsulated within PFF-OMe coacervates, Prodan exhibited a fluorescence emission peak at 457 nm (Fig. S7), representing a blue shift from its emission in water (524 nm) and closely matching its emission in acetonitrile (462 nm).25 These results reinforce the notion that PFF-OMe coacervates possess organic solvent-like properties. We next investigated the viscosity of the coacervate phase using fluorescence recovery after photobleaching (FRAP). Rhodamine B fluorescence recovered smoothly following photobleaching, and the viscosity was calculated to be 36.8 Pa s using the Stokes–Einstein equation (Fig. 1c and Fig. S8). This value is substantially higher than that of previously reported diphenylalanine-based coacervates; for example, Hamachi et al.'s cationic dipeptide coacervates exhibited a viscosity of 4.08 Pa s.26 This nine-fold difference in viscosity is likely attributable to the permanent positive charge of the cationic peptide, which diminishes attractive intermolecular interactions compared to the neutral PFF-OMe. To evaluate the catalytic potential of PFF-OMe coacervates, we selected the aldol addition between cyclohexanone and 4-nitrobenzaldehyde as a model reaction (Fig. 3a). We first assessed whether the presence of organic reagents would affect coacervate stability. Due to its low solubility, 4-nitrobenzaldehyde formed a dispersion upon stirring in water, without visibly altering the coacervate phase. In contrast, cyclohexanone, being more water-soluble, induced a notable increase in sample turbidity upon addition to coacervate dispersions. This elevated turbidity persisted for over two hours, suggesting changes in droplet properties, likely size and/or refractive index, due to cyclohexanone uptake (Fig. 2b and Fig. S9). Microscopic analysis revealed a unimodal size distribution with a median droplet diameter of 1.1 µm, approximately 20% larger than in the absence of cyclohexanone (Fig. 2b). The distribution also broadened significantly, with some droplets reaching diameters up to 5 µm, indicating substantial swelling and fusion of coacervates in the presence of cyclohexanone. A significant drop in fluorescence recovery half-life, an indirect measure of viscosity, is observed upon cyclohexanone uptake, suggesting accelerated droplet fusion in the less viscous coacervates (Table S1).27 To confirm the incorporation of cyclohexanone into the coacervate phase, we performed confocal Raman spectroscopy. The spectrum of cyclohexanone-containing PFF-OMe coacervates revealed characteristic peaks at 653.0, 753.1, 833.5, and 1705 cm−1 matching those of pure cyclohexanone as well as peaks at 1587 and 1599 cm−1, consistent with PFF-OMe coacervates alone (Fig. 2c). Corresponding sets of peaks were also observed when comparing with aqueous cyclohexanone solutions (Fig. S10). These results confirm that both the PFF-OMe peptide and cyclohexanone coexist within the coacervate droplets. Partitioning experiments also confirmed that both reagents favour the coacervate phase over the dilute phase (Table S2). We next examined the colloidal stability of PFF-OMe coacervates under stirring. In the presence of both Pluronic F-108 and cyclohexanone, the coacervate suspensions remained turbid, and droplets were observed under microscopy for up to one week (Fig. 2d), confirming their long-term stability under reaction conditions. To evaluate catalytic activity, we performed the aldol addition between cyclohexanone and 4-nitrobenzaldehyde within PFF-OMe droplets. Cyclohexanone concentrations were kept within its aqueous solubility to avoid emulsion formation. Using 10 mol% PFF-OMe at pH 8, the reaction reached 45% conversion in 2 hours, with a moderate diastereoselectivity of 87![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :13 (anti:syn) (Fig. 3a). Reactions with FF-OMe coacervates lacking proline showed lower conversion and reduced diastereoselectivity, indicating that the proline is essential for both catalytic activity and stereocontrol. This aligns with previous findings that peptide coacervates alone can enhance aldol reaction rates.17 To assess the contribution of the coacervate environment, we used proline methyl ester (P-OMe), a soluble catalyst that does not form coacervates. Under identical conditions, P-OMe yielded minimal conversion, highlighting the importance of the hydrophobic coacervate microenvironment. Combining FF-OMe coacervates with free P-OMe led to a slight improvement in reaction rate, but conversion remained lower than with PFF-OMe coacervates. This suggests that covalent tethering of the catalyst to the coacervate scaffold is key to achieving high local catalyst concentration. When the reaction was performed below the critical pH for coacervate formation, conversion was negligible, indicating that PFF-OMe is a poor homogeneous catalyst. These results confirm that both the coacervate environment and the anchored proline moiety are essential for optimal catalysis. Lowering the catalyst loading led to poor performance (Table S3). Rapid stirring was essential for optimal catalytic performance, with a drop in conversion at lower stirring speeds (Table S4). Extending the reaction time to 8 hours resulted in 90% conversion and an 88:12 diastereomeric ratio, demonstrating efficient and stereoselective catalysis within a practical timeframe. Further lowering the catalyst loading led to incomplete conversion (Table S5). In comparison, Yolacan et al. reported 82% yield using PFF-OMe as a homogeneous catalyst in acidic aqueous conditions with benzoic acid, but required 72 hours at the same catalyst loading, underscoring that this coacervate-based approach is a viable alternative method to traditional homogeneous catalysis.28 To confirm that the aldol reaction was taking place inside PFF-OMe coacervates, the reaction was also attempted using a fluorogenic substrate. An increase in fluorescence signal was observed inside the droplets over time, showing that the reaction was indeed localized within coacervates (Fig. S11).29
:13 (anti:syn) (Fig. 3a). Reactions with FF-OMe coacervates lacking proline showed lower conversion and reduced diastereoselectivity, indicating that the proline is essential for both catalytic activity and stereocontrol. This aligns with previous findings that peptide coacervates alone can enhance aldol reaction rates.17 To assess the contribution of the coacervate environment, we used proline methyl ester (P-OMe), a soluble catalyst that does not form coacervates. Under identical conditions, P-OMe yielded minimal conversion, highlighting the importance of the hydrophobic coacervate microenvironment. Combining FF-OMe coacervates with free P-OMe led to a slight improvement in reaction rate, but conversion remained lower than with PFF-OMe coacervates. This suggests that covalent tethering of the catalyst to the coacervate scaffold is key to achieving high local catalyst concentration. When the reaction was performed below the critical pH for coacervate formation, conversion was negligible, indicating that PFF-OMe is a poor homogeneous catalyst. These results confirm that both the coacervate environment and the anchored proline moiety are essential for optimal catalysis. Lowering the catalyst loading led to poor performance (Table S3). Rapid stirring was essential for optimal catalytic performance, with a drop in conversion at lower stirring speeds (Table S4). Extending the reaction time to 8 hours resulted in 90% conversion and an 88:12 diastereomeric ratio, demonstrating efficient and stereoselective catalysis within a practical timeframe. Further lowering the catalyst loading led to incomplete conversion (Table S5). In comparison, Yolacan et al. reported 82% yield using PFF-OMe as a homogeneous catalyst in acidic aqueous conditions with benzoic acid, but required 72 hours at the same catalyst loading, underscoring that this coacervate-based approach is a viable alternative method to traditional homogeneous catalysis.28 To confirm that the aldol reaction was taking place inside PFF-OMe coacervates, the reaction was also attempted using a fluorogenic substrate. An increase in fluorescence signal was observed inside the droplets over time, showing that the reaction was indeed localized within coacervates (Fig. S11).29
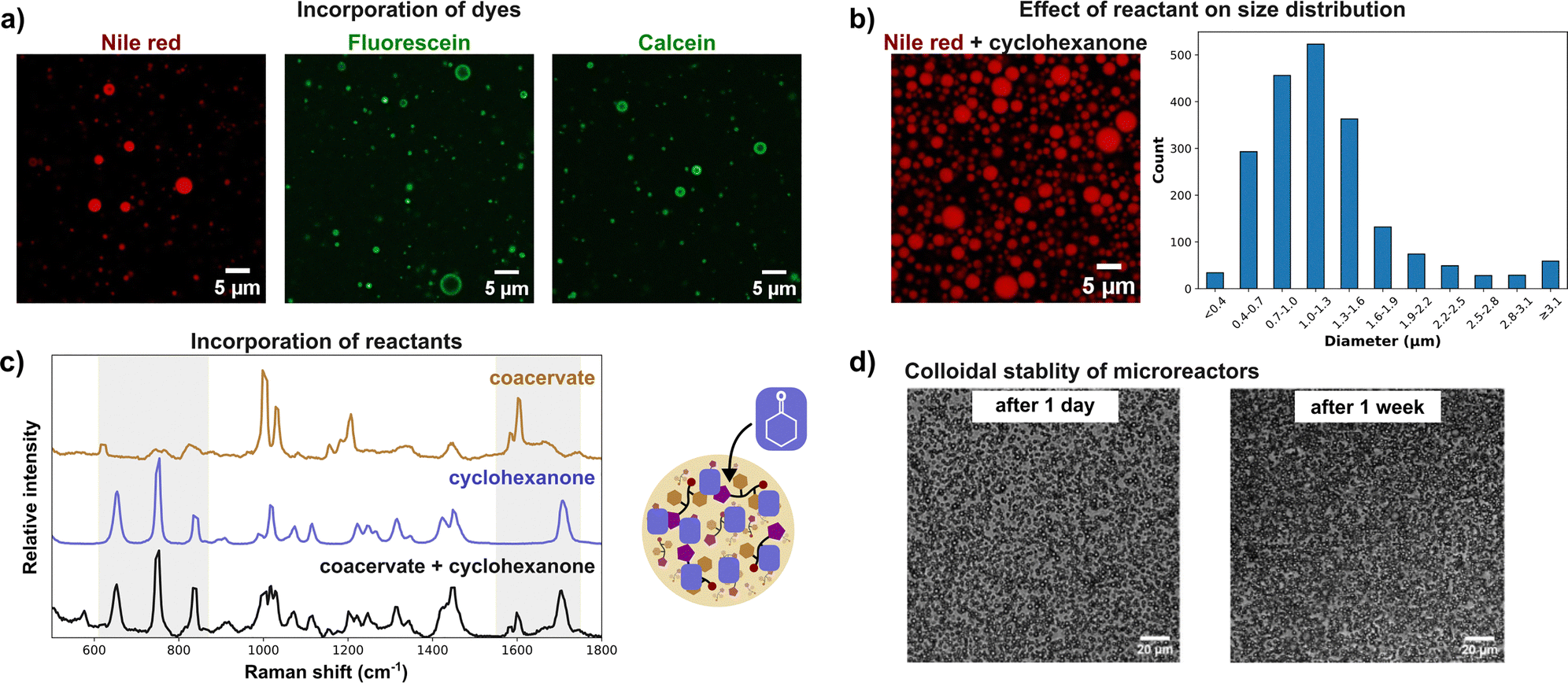 | ||
| Fig. 2 Uptake of molecules by PFF-OMe coacervates and colloidal stability. (a) Uptake of dyes inside PFF-OMe coacervates. Images taken under confocal microscopy for samples containing PFF-OMe. Nile red, fluorescein or calcein (0.01 mM). (b) Swelling of PFF-OMe coacervates upon addition of cyclohexanone. Samples contained 6% cyclohexanone by volume. 0.01 mM Nile red. (c) Uptake of cyclohexanone into PFF-OMe coacervates. Confocal Raman microscopy of PFF-OMe coacervates. (d) Stability of PFF-OMe dispersions under stirring. Brightfield microscopy. All cases: PFF-OMe (9.2 mg mL−1) and 0.1 M HEPES pH 8.5 buffer. | ||
 | ||
| Fig. 3 PFF-OMe microreactors for aldol addition. Catalysis and controls. Reaction conditions: 0.6 mmol cyclohexanone, 0.2 mmol nitrobenzaldehyde, 0.1 M HEPES pH 8.5 buffer. aDetermined via1H NMR (Fig. S6). bNo buffer was added. cNot determined. Diastereomeric ratio (anti/syn) were determined via1H NMR. Enantiomeric excess (e.e.) was determined via chiral HPLC. | ||
We then explored the substrate scope of the aldol reaction catalyzed by PFF-OMe coacervates (Fig. S12). For the model reaction, an 89% isolated yield was obtained with a diastereomeric ratio of 86:14 and 71% enantiomeric excess (e.e.). For other substrates, the reaction time was extended to 24 h to allow for consistent comparisons between substrates with varying reactivity. The reaction tolerated 2-nitro and 3-nitrobenzaldehyde, yielding similarly high conversions and moderate diastereo- and enantioselectivity, indicating that ortho and meta substitution on the aromatic ring is compatible. Electron-deficient aldehydes such as 4-cyano- and 4-chlorobenzaldehyde also served as effective substrates, although the yield for the latter was slightly reduced, likely due to the weaker electron-withdrawing nature of the chloro group. Regarding aldol donors, cyclopentanone was a competent substrate, though it favoured syn selectivity, and moderate enantioselectivity was observed only for the minor anti product. In contrast, acetone led to low yield and poor stereoselectivity, likely due to its high-water solubility, which favours partitioning into the dilute aqueous phase rather than the coacervate interior where the catalyst resides. Overall, hydrophobic ketones and electron-deficient aldehydes were the most suitable substrates, affording high yields but moderate enantioselectivity. To test enantioselectivity, the (D,L,L)-PFF-OMe peptide with inverted proline chirality was synthesized. However, it aggregates in water, precluding its use as a catalytic coacervate.
In summary, we have developed an organocatalytic coacervate system based on the structurally simple and readily accessible PFF-OMe peptide. This component forms micron-sized coacervates with a viscous liquid and nonpolar liquid interior capable of encapsulating hydrophobic dyes and organic reagents. PFF-OMe coacervates function as effective microreactors for aldol addition reactions in aqueous media, with both the nonpolar coacervate environment and the tethered proline catalyst being essential for optimal activity.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc06002h.Acknowledgements
We acknowledge support from the Fonds de recherche du Québec (FRQ; New Academics; Master's #345594), the Natural Sciences and Engineering Research Council of Canada (NSERC; Discovery; CGS-M), the Centre en Chimie Verte et Catalyse (CCVC), and the Canada Foundation for Innovation (CFI).References
- N. A. Yewdall, A. A. M. André, T. Lu and E. Spruijt, Curr. Opin. Colloid Interface Sci., 2021, 52, 101416 CrossRef CAS.
- Z. Lin, T. Beneyton, J.-C. Baret and N. Martin, Small Methods, 2023, 7, 2300496 CrossRef CAS.
- C. E. Sing and S. L. Perry, Soft Matter, 2020, 16, 2885–2914 RSC.
- S. F. Banani, H. O. Lee, A. A. Hyman and M. K. Rosen, Nat. Rev. Mol. Cell Biol., 2017, 18, 285–298 CrossRef CAS PubMed.
- R. Harris, N. Berman and A. Lampel, Chem. Soc. Rev., 2025, 54, 4183–4199 RSC.
- U. Capasso Palmiero, C. Paganini, M. R. G. Kopp, M. Linsenmeier, A. M. Küffner and P. Arosio, Adv. Mater., 2022, 34, 2104837 CrossRef CAS.
- I. B. A. Smokers, E. Lavagna, R. V. M. Freire, M. Paloni, I. K. Voets, A. Barducci, P. B. White, M. Khajehpour and E. Spruijt, J. Am. Chem. Soc., 2025, 147, 25692–25704 CrossRef CAS.
- S. Cao, P. Zhou, G. Shen, T. Ivanov, X. Yan, K. Landfester and L. Caire da Silva, Nat. Commun., 2025, 16, 2407 CrossRef CAS.
- I. B. A. Smokers, B. S. Visser, A. D. Slootbeek, W. T. S. Huck and E. Spruijt, Acc. Chem. Res., 2024, 57, 1885–1895 CrossRef CAS PubMed.
- K. Lv, A. W. Perriman and S. Mann, Chem. Commun., 2015, 51, 8600–8602 RSC.
- A. F. Mason, B. C. Buddingh’, D. S. Williams and J. C. M. van Hest, J. Am. Chem. Soc., 2017, 139, 17309–17312 CrossRef CAS PubMed.
- J. Wang, M. Abbas, J. Wang and E. Spruijt, Nat. Commun., 2023, 14, 8492 CrossRef CAS PubMed.
- S. Cao, T. Ivanov, J. Heuer, C. T. J. Ferguson, K. Landfester and L. Caire da Silva, Nat. Commun., 2024, 15, 39 CrossRef CAS.
- S. Singh, C. Rao, C. K. Nandi and T. K. Mukherjee, ACS Appl. Nano Mater., 2022, 5, 7427–7439 CrossRef CAS.
- B. Saini, S. Singh and T. K. Mukherjee, ACS Appl. Mater. Interfaces, 2021, 13, 51117–51131 CrossRef CAS.
- Y. Chen, M. Yuan, Y. Zhang, S. Liu, X. Yang, K. Wang and J. Liu, Chem. Sci., 2020, 11, 8617–8625 RSC.
- M. Abbas, W. P. Lipiński, K. K. Nakashima, W. T. S. Huck and E. Spruijt, Nat. Chem., 2021, 13, 1046–1054 CrossRef CAS PubMed.
- D. Q. P. Reis, S. Pereira, A. P. Ramos, P. M. Pereira, L. Morgado, J. Calvário, A. O. Henriques, M. Serrano and A. S. Pina, Nat. Commun., 2024, 15, 9368 CrossRef CAS PubMed.
- M. I. Jacobs, E. R. Jira and C. M. Schroeder, Langmuir, 2021, 37, 14323–14335 CrossRef CAS.
- B. List, R. A. Lerner and C. F. Barbas, J. Am. Chem. Soc., 2000, 122, 2395–2396 CrossRef CAS.
- M. Poudyal, K. Patel, L. Gadhe, A. S. Sawner, P. Kadu, D. Datta, S. Mukherjee, S. Ray, A. Navalkar, S. Maiti, D. Chatterjee, J. Devi, R. Bera, N. Gahlot, J. Joseph, R. Padinhateeri and S. K. Maji, Nat. Commun., 2023, 14, 6199 CrossRef CAS.
- A. A. Profit, V. Felsen, J. Chinwong, E.-R. E. Mojica and R. Z. B. Desamero, Proteins: Struct., Funct., Bioinf., 2013, 81, 690–703 CrossRef CAS.
- M. Abbas, W. P. Lipiński, J. Wang and E. Spruijt, Chem. Soc. Rev., 2021, 50, 3690–3705 RSC.
- M. Bogunia and M. Makowski, J. Phys. Chem. B, 2020, 124, 10326–10336 CrossRef CAS.
- G. Weber and F. J. Farris, Biochemistry, 1979, 18, 3075–3078 CrossRef CAS PubMed.
- R. Kubota, S. Torigoe and I. Hamachi, J. Am. Chem. Soc., 2022, 144, 15155–15164 CrossRef CAS.
- T. Lu and E. Spruijt, J. Am. Chem. Soc., 2020, 142, 2905–2914 CrossRef CAS.
- D. G. Yilmaz, F. Aydogan and C. Yolacan, J. Heterocycl. Chem., 2022, 59, 1169–1179 CrossRef CAS.
- L. E. Greene, R. Lincoln, K. Krumova and G. Cosa, ACS Omega, 2017, 2, 8618–8624 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2026 |