
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Atom-efficient iron-catalyzed cascade synthesis of pyrroles from nitroarenes under low-pressure conditions
Germán
López Robledo
 ,
Johannes
Fessler
,
Kathrin
Junge
* and
Matthias
Beller
*
,
Johannes
Fessler
,
Kathrin
Junge
* and
Matthias
Beller
*
Leibniz-Institute für Katalyse e.V., Albert-Einstein-Straße 29a, 18059 Rostock, Germany. E-mail: kathrin.junge@catalysis.de, matthias.beller@catalysis.de
First published on 16th December 2025
Abstract
Conventional pyrrole syntheses typically rely on multistep procedures or pre-functionalized substrates. Here, we report a one-pot Paal–Knorr cascade reaction that directly converts nitroarenes into pyrroles. Employing low-pressure hydrogenation in the presence of an iron catalyst, this approach enhances atom economy and sustainability, offering an efficient and environmentally benign alternative to traditional methods.
Pyrroles are key structural motifs present in a diverse array of natural products,1 pharmaceuticals,2,3 and functional materials.4–6 Since its discovery in 1885, the Paal–Knorr reaction, the acid-catalyzed condensation of 1,4-dicarbonyl compounds with primary amines, has remained a cornerstone in the synthesis of substituted pyrroles.7,8 Subsequent refinements have expanded its scope through modified conditions and alternative substrates.9 Nevertheless, traditional pyrrole syntheses frequently rely on multistep sequences or pre-functionalized starting materials,10 which can undermine their overall efficiency, practicality, and sustainability.
In this context, nitroarenes represent attractive starting materials due to their wide availability, low cost, and greater stability compared to the corresponding anilines.11–13 Accordingly, their direct conversion into pyrroles could significantly streamline synthetic routes, provided that mild and chemoselective reduction strategies are available. Among existing reduction methodologies, catalytic hydrogenation using molecular hydrogen (H2) is particularly appealing, offering excellent atom economy and producing only water as a byproduct. However, conventional hydrogenation protocols typically rely on precious metal catalysts and/or require high H2 pressures and elevated temperatures,14 often compromising functional group tolerance and sustainability.
To address these limitations, growing interest has focused on developing selective catalytic systems based on earth-abundant metals, which offer a more sustainable and cost-effective alternative to precious metal catalysts.15 In parallel, the rising demand for greener synthetic methodologies has catalyzed the adoption of cascade strategies, which integrate multiple bond-forming events into a single operational step.16,17 Such approaches improve atom economy, reduce solvent use and waste, and streamline purification processes.
In this regard, a cascade strategy offers an attractive approach for converting nitroarenes to pyrroles via the Paal–Knorr reaction, as it combines selective nitroarene reduction with condensation–cyclization in a single operation, providing a sustainable and atom-efficient alternative to conventional multistep routes (Scheme 1A).
 | ||
| Scheme 1 Recent developments in homogeneous Fe-catalyzed cascade syntheses of pyrroles from nitroarenes: (A) Paal–Knorr cascade via aniline intermediates; (B) first homogeneous Fe-catalyzed example; (C) this work: homogeneous cascade synthesis under low-pressure hydrogenation. | ||
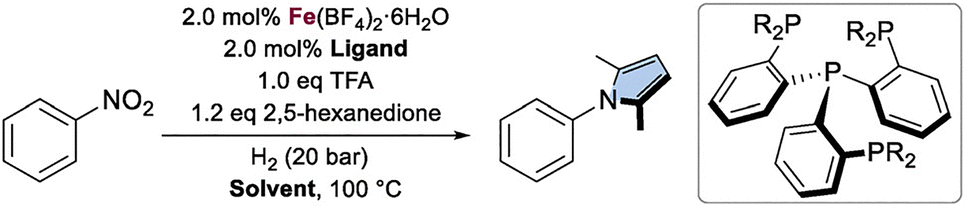
While several cascade syntheses of pyrroles from nitroarenes via the Paal–Knorr reaction have been reported in the past, most rely on heterogeneous catalysts.18–23 Recently, we described the first homogenous non-noble metal catalytic procedure for this transformation (Scheme 1B).24 Shortly after, an alternative approach using a molybdenum catalyst was also described.25 In our previous work, we discovered that the combination of an iron (Fe) precursor with the phosphorous-based ligand tetraphos (tris(2-(diphenylphosphino)ethyl)phosphine) (L1) enabled the efficient reductive condensation of nitroarenes with 1,4-dicarbonyl compounds. While formic acid was selected as the primary hydrogen donor, other hydrogenation conditions were briefly explored, showing limited success across a small set of substrates.
Building on these developments and recognizing the advantages of molecular hydrogen as a clean and atom-efficient reductant, we sought to optimize this transformation, offering a more sustainable approach to pyrrole synthesis. Herein, we report a homogeneous, one-pot, cascade reduction-condensation strategy for the synthesis of pyrroles from nitroarenes and 2,5-hexanedione under low-pressure hydrogenation conditions (Scheme 1C). The method exhibits broad functional group tolerance and provides various substituted pyrroles in good to excellent yields (up to 93%).
Guided by our previous results with the Ph-tetraphos (tris(2-(diphenylphosphino)phenyl)phosphine) (L2) system,26 we conducted a preliminary evaluation of a series of structurally related ligands, selecting nitrobenzene as the model substrate, 2,5-hexanedione as the 1,4-dicarbonyl compound, and Fe(BF4)2·6H2O as the Fe source (Table 1, entries 1–4). Compared to the unmodified ligand, the introduction of an electron-donating group (L3) proved slightly detrimental to the reaction, whereas the incorporation of an electron-withdrawing group (L4) resulted in a significant decrease in catalytic activity. Increasing the steric bulk of the ligand (L5) also led to a pronounced negative effect. Ultimately, the initial system outperformed all modified variants, which can be rationalized by considering the steric hindrance associated with the ligand modifications (Ph < 4-MeO-Ph < 4-CF3-Ph < Xylyl).
|
|
|||
|---|---|---|---|
| Entry | Ligand | Solvent | Yield [%] |
| Reaction conditions: Fe(BF4)2·6H2O (0.01 mmol, 2.0 mol%), ligand (0.01 mmol, 2.0 mol%), TFA (0.5 mmol, 1.0 equiv.), nitrobenzene (0.5 mmol, 1.0 equiv.), 2,5-hexanedione (0.6 mmol, 1.2 equiv.), solvent (1.5 mL), H2 (20 bar), 100 °C.a 2 h.b 1 h. Yields were determined by GC-FID analysis using hexadecane as internal standard. t-AmOH: 2-methyl-2-butanol. | |||
| 1a | L2: R = Ph | THF | 84 |
| 2a | L3: R = 4-MeO-Ph | 70 | |
| 3a | L4: R = 4-CF3-Ph | 38 | |
| 4a | L5: R = Xylyl | 27 | |
| 5b | L2 | THF | 71 |
| 6b | 1,4-Dioxane | 19 | |
| 7b | EtOH | 38 | |
| 8b | i-PrOH | 64 | |
| 9b | t-AmOH | 58 | |
| 10b | 2-Butanol | 66 | |
| 11b | 2-Pentanol | 72 | |
| 12b | Dibutyl ether | <5 | |
| 13b | Toluene | <5 | |
| 14b | Cyclohexane | <5 | |
Having identified the optimal ligand for the transformation, we next examined the influence of different Lewis acids. The results of this extensive evaluation–which included sulfonamides, sulfonic acids, mineral acids, halogenated acids, and other common acids—are presented in the SI (Fig. S1 and Table S1). Among the acids tested, the highest yields were obtained with fluorinated analogues such as heptafluorobutyric acid (69%), chlorodifluoroacetic acid (74%), and trifluoroacetic acid (TFA, 98%). Interestingly, no product formation was observed when trichloroacetic acid was used. Attempts employing strong mineral acids, non-halogenated carboxylic acids, or sulfonamides resulted only in poor conversions.
Given the increasing regulatory restrictions on polyfluorinated compounds,27 and aiming for a greener and more sustainable approach, we investigated the effect of acid concentration in a series of sulfonic acids, as they were the second-best performing group (up to 18%) (Fig. S2). This analysis revealed that each acid—as well as TFA—exhibits optimal performance within a narrow and well-defined concentration (pH) range. Although comparably higher yields could be achieved (up to 41% for camphorsulfonic acid), these values remained significantly lower than those obtained with TFA (98%). For this reason, TFA was selected for use in subsequent experiments.
Next, the influence of the solvent was evaluated (Table 1, entries 5–14). Polar solvents such as i-PrOH, 2-butanol, and 2-pentanol, along with THF, provided the best yields. In contrast, product formation was significantly reduced when the reaction was carried out in non-polar solvents such as toluene and cyclohexane. An extended list can be found in Table S2. Since THF proved to be more robust than 2-pentanol, it was selected as the preferred solvent.
Following this analysis, we explored the effect of other variables on the reaction outcome. The main results are summarized in Table 2. Replacing the original Fe precursor with Fe(acac)2 (Table 2, entry 2) or Fe(OAc)2 (Table 2, entry 3) led to comparable results. In contrast, no product formation was observed when FeBr2 and FeCl2 were used (Table 2, entry 4). Increasing the amount of 2,5-hexanedione (Table 2, entry 5) did not evidence any significant improvement. The addition of molecular sieves to the reaction mixture (Table 2, entry 6) proved detrimental to product formation. Increasing the reaction time to 6 hours led to full conversion and 85% product yield (Table 2, entry 7).
|
|
||
|---|---|---|
| Entry | Deviation from standard conditions | Yield [%] |
| Standard conditions: Fe(BF4)2·6H2O (0.01 mmol, 2.0 mol%), L2 (0.01 mmol, 2.0 mol%), TFA (0.5 mmol, 1.0 equiv.), nitrobenzene (0.5 mmol, 1.0 equiv.), 2,5-hexanedione (0.6 mmol, 1.2 equiv.), THF (1.5 mL), H2 (20 bar), 100 °C, 1 h. Yields were determined by NMR analysis using 1,3,5-trimethoxybenzene as internal standard. | ||
| 1 | None | 59 |
| 2 | Fe(acac)2 as Fe precursor | 52 |
| 3 | Fe(OAc)2 as Fe precursor | 55 |
| 4 | FeCl2 or FeBr2 as Fe precursor | 0 |
| 5 | 1.5 equiv. 2,5-hexanedione | 58 |
| 6 | 3 Å molecular sieves | 35 |
| 7 | Reaction time: 6 h | 85 |
| 8 | 80 °C, 18 h | 83 |
| 9 | 60 °C, 24 h | 66 |
| 10 | 80 °C, 18 h, 1.5 mol% catalyst | 76 |
| 11 | 5 bar H2, 80 °C, 18 h, 0.35 equiv. TFA | 93 |
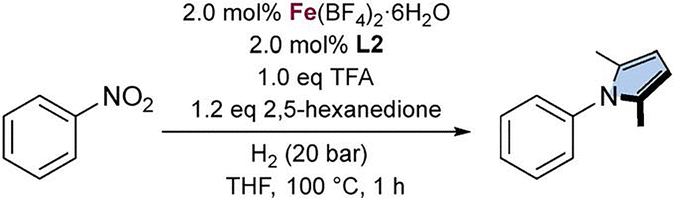
Aiming to achieve the desired transformation under milder conditions, reactions were performed at 80 °C (Table 2, entry 8) and 60 °C (Table 2, entry 9) with extended reaction times. While 80 °C afforded very good yields, the reaction at 60 °C failed to deliver comparable results. Attempts to further reduce the catalyst loading (Table 2, entry 10) also proved unsuccessful. Extended optimization allowed the use of low H2 pressures (5 bar) and reduced TFA loadings (0.35 equiv.) (Table 2, entry 11), which were ultimately selected as the optimized conditions.
With the optimized conditions in hand, we proceeded to evaluate the general applicability of the system (Scheme 2). The reaction of the model substrate (1a) afforded the product in excellent yield (92%). The introduction of a methyl group at the ortho position (2a) led to a pronounced drop in yield (30%), likely due to steric hindrance affecting catalyst coordination. Given the pronounced effect of acid concentration on the reaction outcome, the reaction was repeated using an increased amount of TFA (1.0 equiv.). Under these conditions, the product was isolated in 62% yield, further highlighting the critical role of pH in this transformation. Nitrobenzenes bearing electron-donating groups such as –OH (3a) and –OMe (4a) afforded the desired product in excellent and very good yields, respectively. Notably, sulfur-containing nitroarene 5a, which is known to poison Pd/C during hydrogenation,28 was successfully converted under the present conditions. In general, halogen derivatives (6a–9a) were well tolerated, and no side-products were observed. The reaction also proceeded smoothly in the presence of boronic ester 10a, furnishing the corresponding pyrrole in 70% yield. When an olefin-containing substrate was tested (11a), moderate product formation was observed; importantly, no over-reduction was detected. An alkynyl-nitroarene, 1-ethynyl-4-nitrobenzene (see SI), was detected in 26% yield. However, under the reaction conditions both the starting material and the corresponding pyrrole were not well tolerated and underwent reduction to the respective alkenes and/or hydration products, leading to an overall conversion of 51%. The nitrile-substituted nitroarene (12a) resulted in a poor conversion, likely due to the strong electron-withdrawing nature of the nitrile group, which may hinder reduction, or due to catalyst inhibition.
 | ||
Scheme 2 Substrate scope for the cascade synthesis of pyrroles from nitroarenes. Reaction conditions: a![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Fe(BF4)2·6H2O (0.01 mmol, 2.0 mol%), L2 (0.01 mmol, 2.0 mol%), TFA (0.175 mmol, 0.35 equiv.), nitrobenzene (0.5 mmol, 1.0 equiv.), dicarbonyl (0.6 mmol, 1.2 equiv.), THF (1.5 mL), H2 (5 bar), 80 °C, 18 h; bTFA (0.5 mmol, 1.0 equiv.); cFe(BF4)2·6H2O (0.025 mmol, 5.0 mol%), L2 (0.025 mmol, 5.0 mol%). Isolated yields are shown. Fe(BF4)2·6H2O (0.01 mmol, 2.0 mol%), L2 (0.01 mmol, 2.0 mol%), TFA (0.175 mmol, 0.35 equiv.), nitrobenzene (0.5 mmol, 1.0 equiv.), dicarbonyl (0.6 mmol, 1.2 equiv.), THF (1.5 mL), H2 (5 bar), 80 °C, 18 h; bTFA (0.5 mmol, 1.0 equiv.); cFe(BF4)2·6H2O (0.025 mmol, 5.0 mol%), L2 (0.025 mmol, 5.0 mol%). Isolated yields are shown. | ||
We next examined acyl-substituted nitrobenzenes (13a–15a), which generally afforded moderate yields (40–60%). Here, higher yields were obtained when substrate 13a was tested at an increased catalyst loading (from 40% to 66%). Similar improvements are likely achievable for other low-yielding substrates, although higher catalyst loadings would be required. Notably, an aldehyde-containing substrate, 4-nitrobenzaldehyde (see SI) failed to react, and no product formation was detected.
To further assess the scope of the methodology, three nitroarenes bearing heterocycles were evaluated. Nitrogen-containing heteroarenes, such as 6-nitroquinoline (16a), were not tolerated under the reaction conditions (18% yield). A similar behavior was observed for 3-nitropyrrole, which showed approximately 20% conversion by GC-FID analysis (see SI). In contrast, 5-nitrobenzofuran (17a) afforded the desired pyrrole in 78% yield. Notably, the sulfur-containing heterocycle 5-nitrobenzo[b]thiophene (18a) was not reduced.
Finally, we examined the reaction when 2,5-hexanedione was replaced with other dicarbonyl sources. Using 2,5-dimethoxytetrahydrofuran as a succinaldehyde surrogate (19a) afforded moderate yields of the corresponding pyrrole (50%). The reaction with 1-phenylpentane-1,4-dione (20a) provided very good yields (80%), whereas a more challenging dicarbonyl, 1,4-diphenylbutane-1,4-dione (21a), gave moderate yields (56%). Overall, the scope and limitations of the reaction across diverse nitroarene substrates and dicarbonyl sources were evaluated.
In summary, we have developed an atom-economical cascade synthesis of pyrroles directly from nitroarenes. Using an iron-based catalyst in combination with a phosphine ligand, the transformation proceeds under mild conditions and low hydrogen pressure, delivering pyrroles in good to excellent yields. This sustainable protocol demonstrates comparable efficiency to our previous methodology while offering improved potential for industrial implementation.
GLR: conceptualization, methodology, validation, investigation, writing – original draft, writing – review & editing, visualization. JF: conceptualization, methodology, validation, investigation, writing – review & editing. KJ: writing – review & editing, supervision, project administration, funding acquisition. MB: writing – review & editing, supervision, project administration, funding acquisition.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information is available. See DOI: https://doi.org/10.1039/d5cc05916j.Acknowledgements
The authors thank PD Dr Wolfgang Baumann and Susann Buchholz for NMR measurements. GLR is also grateful to Dr Rafal Kusy, Sebastian Ahrens, Faral Akhtar, and Fairoosa Poovan for proofreading and valuable discussions.References
- N. Singh, S. Singh, S. Kohli, A. Singh, H. Asiki, G. Rathee, R. Chandra and E. A. Anderson, Org. Chem. Front., 2021, 8, 5550–5573 RSC.
- B. H. Ganesh, A. G. Raj, B. Aruchamy, P. Nanjan, C. Drago and P. Ramani, ChemMedChem, 2024, 19, e202300447 CrossRef PubMed.
- G. Li Petri, V. Spano, R. Spatola, R. Holl, M. V. Raimondi, P. Barraja and A. Montalbano, Eur. J. Med. Chem., 2020, 208, 112783 CrossRef PubMed.
- M. Krzeszewski, D. Gryko and D. T. Gryko, Acc. Chem. Res., 2017, 50, 2334–2345 CrossRef PubMed.
- Y. Liu and F. Wu, Nanoscale Adv., 2023, 5, 3606–3618 RSC.
- A. Deronzier and J. C. Moutet, Acc. Chem. Res., 1989, 22, 249–255 CrossRef.
- C. Paal, Ber. Dtsch. Chem. Ges., 1885, 18, 367–371 CrossRef.
- L. Knorr, Ber. Dtsch. Chem. Ges., 1885, 18, 299–311 CrossRef.
- J. Li, J. Sun, Y. Wang, J. Liu and H. Cheng, ACS Sustainable Chem. Eng., 2024, 13, 571–582 CrossRef.
- V. Bhardwaj, D. Gumber, V. Abbot, S. Dhiman and P. Sharma, RSC Adv., 2015, 5, 15233–15266 RSC.
- M. Orlandi, D. Brenna, R. Harms, S. Jost and M. Benaglia, Org. Process Res. Dev., 2016, 22, 430–445 CrossRef.
- C. W. Cheung, M. Leendert Ploeger and X. Hu, Chem. Sci., 2018, 9, 655–659 Search PubMed.
- A. Capperucci, M. Clemente, A. Cenni and D. Tanini, ChemSusChem, 2023, 16, e202300086 CrossRef PubMed.
- D. Formenti, F. Ferretti, F. K. Scharnagl and M. Beller, Chem. Rev., 2019, 119, 2611–2680 CrossRef PubMed.
- V. Zubar, A. Dewanji and M. Rueping, Org. Lett., 2021, 23, 2742–2747 CrossRef PubMed.
- E. S. Beach, Z. Cui and P. T. Anastas, Energy Environ. Sci., 2009, 2, 1038–1049 RSC.
- K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., Int. Ed., 2006, 45, 7134–7186 CrossRef PubMed.
- Y. Lin, F. Wang, E. Ren, F. Zhu, Q. Zhang and G.-P. Lu, J. Catal., 2022, 416, 39–46 CrossRef.
- P. Ryabchuk, T. Leischner, C. Kreyenschulte, A. Spannenberg, K. Junge and M. Beller, Angew. Chem., Int. Ed., 2020, 59, 18679–18685 CrossRef PubMed.
- Z. Gong, Y. Lei, P. Zhou and Z. H. Zhang, New J. Chem., 2017, 41, 10613–10618 RSC.
- A. Serrano-Maldonado, E. Martin and I. Guerrero-Ríos, Eur. J. Inorg. Chem., 2019, 2863–2870 CrossRef.
- X. Yu, M. Miao, S. Huo, X. Tang, L. Ni, S. Liu and L. Wang, ACS Appl. Mater. Interfaces, 2024, 16, 16363–16372 CrossRef PubMed.
- J. Luis del Río-Rodríguez, A. Tiscareño-Ferrer, S. Gutiérrez-Tarriño and P. Oña-Burgos, ChemCatChem, 2025, 17(22), e00811 CrossRef.
- J. Fessler, K. Junge and M. Beller, Chem. Sci., 2023, 14, 11374–11380 RSC.
- S. Gómez-Gil, S. Suárez-Pantiga, M. R. Pedrosa and R. Sanz, Adv. Synth. Catal., 2024, 367, e202401170 CrossRef.
- G. Wienhofer, M. Baseda-Kruger, C. Ziebart, F. A. Westerhaus, W. Baumann, R. Jackstell, K. Junge and M. Beller, Chem. Commun., 2013, 49, 9089–9091 RSC.
- J. C. DeWitt, J. Gluge, I. T. Cousins, G. Goldenman, D. Herzke, R. Lohmann, M. Miller, C. A. Ng, S. Patton, X. Trier, L. Vierke, Z. Wang, S. Adu-Kumi, S. Balan, A. M. Buser, T. Fletcher, L. S. Haug, A. Heggelund, J. Huang, S. Kaserzon, J. Leonel, I. Sheriff, Y. L. Shi, S. Valsecchi and M. Scheringer, Environ. Sci. Technol. Lett., 2024, 11, 786–797 CrossRef PubMed.
- R. Xiong, W. Ren, Z. Wang and M. Zhang, ChemCatChem, 2020, 13, 548–552 CrossRef.
| This journal is © The Royal Society of Chemistry 2026 |