
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Expect the unexpected – Li+ migration governing the substitution of 7H-benzo[de]anthracene with main-group fragments
Rajesh
Deka
 *,
Toma
Bhowmick
and
Andreas
Orthaber
*
*,
Toma
Bhowmick
and
Andreas
Orthaber
*
Synthetic Molecular Chemistry, Department of Chemistry Ångström Laboratory, Uppsala University, BOX 523, 75120 Uppsala, Sweden. E-mail: andreas.orthaber@kemi.uu.se; rajesh.deka@kemi.uu.se
First published on 17th December 2025
Abstract
We report electrophile-dependent, site-selective heavier main-group functionalization of the benzanthracene framework, affording a heteroleptic phosphine oxide and phosphaalkene derivative. Combined experimental and DFT studies reveal that kinetic and thermodynamic factors govern this reactivity, providing new insights into main-group incorporation within extended π-conjugated polycyclic aromatic hydrocarbons.
The functionalization of polyaromatic hydrocarbons (PAHs) with heavier main-group elements has emerged as a powerful approach to tune their electronic structures and explore new modes of bonding and reactivity.1–3 Among these, phosphorus stands out for its versatility: both endocyclic and peripheral incorporation, ranging from λ3σ2 low-valent phosphaalkenes4,5 to λ4σ4 phosphonium motifs (Fig. 1A), has produced a diverse array of compounds with remarkable optoelectronic properties.6–9 Over the past decade, researchers have steadily expanded the range of PAH scaffolds and phosphorus building blocks, seeking new combinations that balance structural stability with finely controlled electronic modulation.10,11
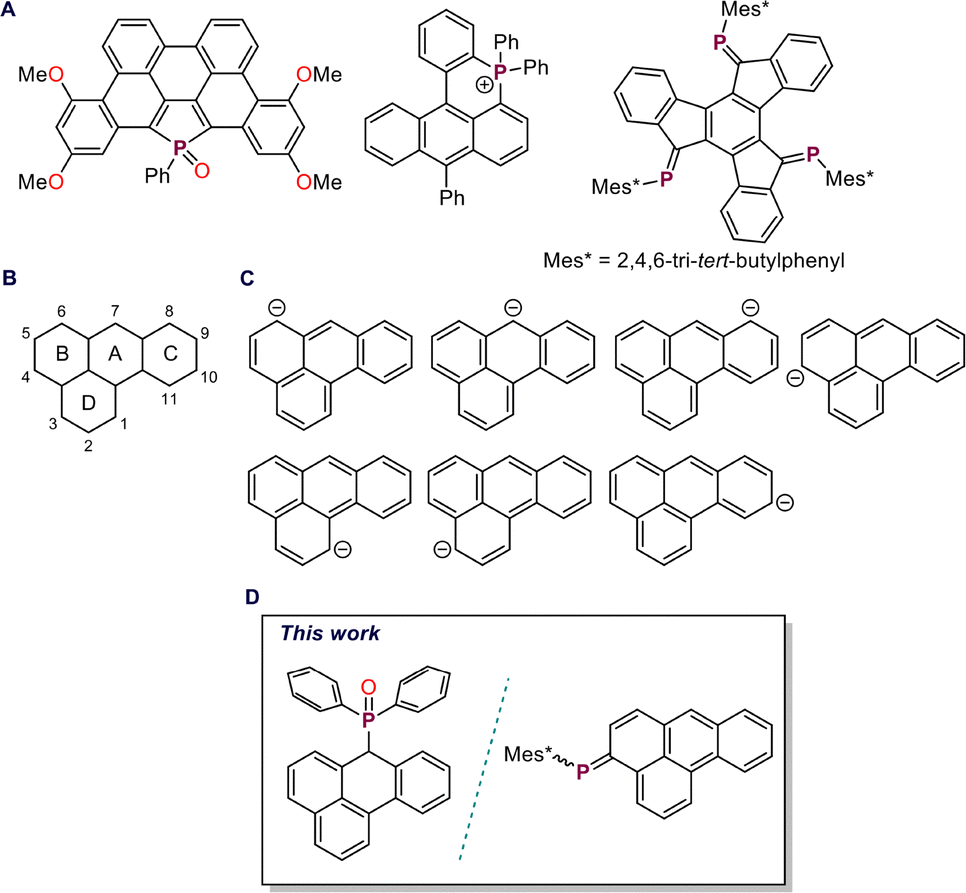 | ||
| Fig. 1 (A) Examples of polyaromatic hydrocarbons bearing endocyclic or exocyclic phosphorus functionalization; (B) numbering scheme for benzanthracene, (C) possible resonance structures of the benzanthracenyl anion; and (D) present work. | ||
Among the various PAH frameworks, benzanthrone remains relatively underexplored despite its attractive photophysical characteristics. This chromophore has found use as a fluorescent probe for amyloid fibrils, as a dye in liquid crystals, and as a green light emitter in OLEDs.12–15 However, progress in its derivatization has been hindered by the inherent complexity of its nucleophilic addition chemistry, which often leads to competing 1,2-, 1,4-, and even 1,8-addition pathways, making selective functionalization challenging (Fig. 1B).16–19 Notably, the parent 7H-benzo[de]anthracene core has not yet been explored for phosphorus functionalization, despite the potential of such modification to generate unique electronic environments and finely tune optoelectronic properties in this extended π-system.
One of the most common routes for introduction of peripheral phosphaalkene motifs into PAH commence from its methylene precursor.20 Deprotonation of the methylene proton(s) with a suitable base, followed by reaction with a phosphorus precursor, furnishes the desired phosphorus-functionalized species. In extended PAH frameworks, deprotonation generally generates resonance-stabilized carbanions in which the negative charge is delocalized across the π-conjugated backbone.20–22 Such delocalization can produce multi-site nucleophilic intermediates capable of reacting at distinct positions, each of which can be selectively trapped by electrophiles, leading to regioselective transformations. Such charge migration can thus be harnessed to direct reactivity toward otherwise inaccessible positions. Cornelisse and co-workers demonstrated this behaviour in the 1-hydropyrenyl anion, which can be regarded as a vinyl-substituted phenalenyl anion, where charge delocalization directs substitution predominantly to the 3a or 5 position, depending on the electrophile.22 In the case of the benzanthracenyl anion, a benzo-annulated phenalenyl system, theoretical studies have shown that the negative charge and HOMO coefficients are primarily localized at position 7, with notable contributions at positions 4 and 6 (Fig. 1C).23 Reactions of the benzanthracenyl anion with alkyl halides yielded the 7-substituted product as the major species, whereas vinamidinium salts reveal additional reactivity at positions 3 and 4. Despite these insights, the reactivity of the benzanthracenyl anion toward heavier main-group electrophiles has not been investigated. In this work, we explore its reactivity with different aryl(chloro)phosphines, probing the site-selectivity and electronic consequences of main-group functionalization within the benzanthracene framework (Fig. 1D).
We first targeted the introduction of a phosphine oxide fragment onto the benzanthracene core to examine the site-selectivity of electrophilic phosphorus incorporation. Lithiation of 7H-benzo[de]anthracene (1)23 with n-BuLi at –78 °C, followed by treatment with Ph2PCl and H2O2 yielded the heteroleptic phosphine oxide 2 as the major product (isolated yield 62%), with selective functionalization at the C7-position (Fig. 2A). Compound 2 exhibited a 31P NMR resonance at 32.5 ppm. Minor side products resulting from functionalization at alternative sites were also detected, though their combined yield remained below 10%, as estimated from the 31P NMR spectrum of the crude reaction mixture. The compound crystallizes in the monoclinic space group P21/c, forming red, plate-shaped crystals with a P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond length of 1.491(1) Å (Fig. 2B).
O bond length of 1.491(1) Å (Fig. 2B).
 | ||
| Fig. 2 Synthesis of heteroleptic phosphine oxide 2 and its solid state structure. Condition: (i) n-BuLi, −78 °C, THF; (ii) Ph2PCl, −78 °C; (iii) H2O2, r.t. CH2Cl2, CH3OH; (B) solid-state structure of 2. | ||
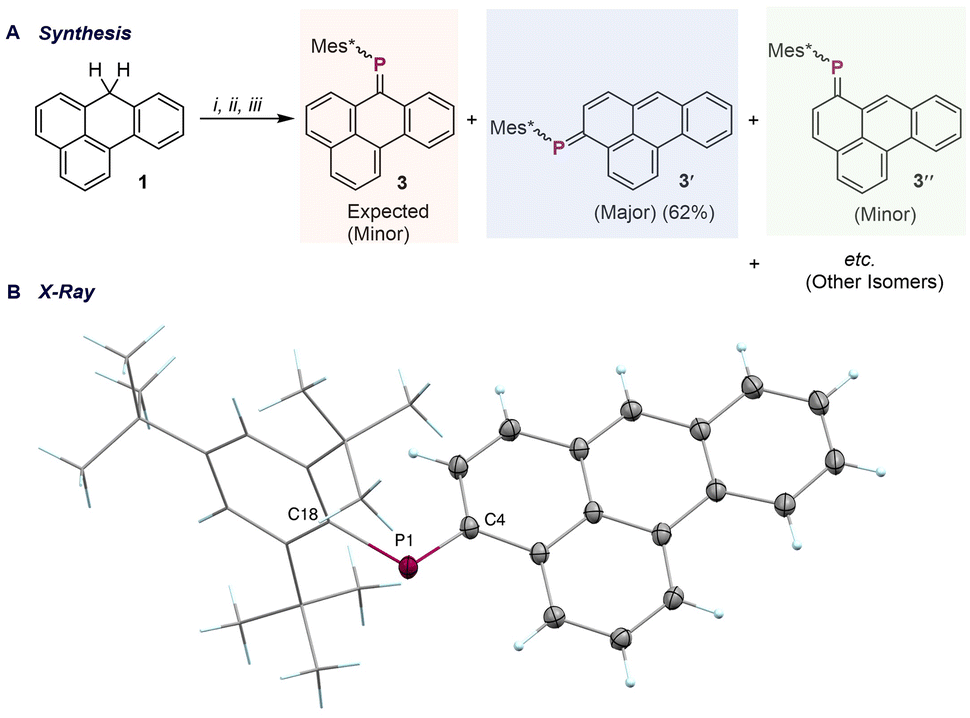
Encouraged by the selective formation of 2, we next sought to introduce a phosphaalkene moiety, as the incorporation of PC unit(s) into PAHs is an effective means of tuning their HOMO–LUMO gap. 1-Li, prepared in an identical way, followed by treatment with Mes*PCl2 (Mes* = 2,4,6-tri-tert-butylbenzene), afforded a mixture of four chlorophosphine intermediates, as indicated by 31P NMR signals at δ = 84.1, 77.3, 76.6, and 75.1 ppm, with the 84.1 ppm species being predominant (Fig. S8, SI). Subsequent addition of 1,5-diazabicyclo[4.3.0]non-5-ene (DBN) resulted in the formation of phosphaalkene species (Fig. 3A). The crude 31P NMR spectrum displayed a set of resonances between δ = 240–255 ppm, characteristic of PC–containing species. Chromatographic purification (silica gel, pentane![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene = 96:4) yielded a fraction exhibiting four distinct 31P NMR resonances (δ = 254.1, 252.7, 242.6, and 241.5 ppm, with 242.6 ppm as the prominent species; Fig. S9, SI), which we attribute to positional isomers and E/Z-stereoisomers formed during DBN-mediated HCl elimination. Further separation using preparative thin-layer chromatography (PTLC) was attempted; however, due to the similar chromatographic properties of all the isomers, they migrated as a broad, indistinct band, highlighting the inherent challenges in their purification. Crystallization by gas-phase diffusion of hexane into a THF solution of the mixture produced red crystalline material, from which phosphaalkene 3′ (δ = 242.9 ppm; Fig. S6, SI) was isolated by macroscopic separation. Solid-state structural analysis revealed that the phosphaalkene unit is incorporated at the C4-position (vide infra), rather than the anticipated C7-position of 1. Notably, compound 3′ underwent isomerization under ambient light, yielding an almost 1:1 mixture of E- and Z-isomers over six hours, as determined by 31P NMR spectroscopy, with the Z-isomer exhibiting a resonance at 253.2 ppm (Fig. S7, SI). A similar ambient light-induced isomerization process has been previously reported for phosphaalkenes derived from the meta-terphenyl scaffold.24 In the present benzanthracene system, both facile E/Z-isomerization and the possibility of substitution at up to seven distinct positions (Fig. 1C) significantly complicate the purification process. Following the isolation of crystals of compound 3′, further crystallization from the mother liquor yielded crystals of a second regioisomer, 3″. Solid-state structural analysis of 3″ indicated incorporation of the phosphaalkene unit at the C6-position (Fig. S10, SI). Although the crystal quality did not permit a detailed analysis of bond metrics, bond connectivity was unambiguously established. The solid-state structure of 3′ revealed a planar benzanthracene core (Fig. 3B), with a PC bond length of 1.714(1) Å and a C–PC bond angle of 105.41(6)°, which are in excellent agreement with values reported for related phosphaalkene derivatives.25–27
:toluene = 96:4) yielded a fraction exhibiting four distinct 31P NMR resonances (δ = 254.1, 252.7, 242.6, and 241.5 ppm, with 242.6 ppm as the prominent species; Fig. S9, SI), which we attribute to positional isomers and E/Z-stereoisomers formed during DBN-mediated HCl elimination. Further separation using preparative thin-layer chromatography (PTLC) was attempted; however, due to the similar chromatographic properties of all the isomers, they migrated as a broad, indistinct band, highlighting the inherent challenges in their purification. Crystallization by gas-phase diffusion of hexane into a THF solution of the mixture produced red crystalline material, from which phosphaalkene 3′ (δ = 242.9 ppm; Fig. S6, SI) was isolated by macroscopic separation. Solid-state structural analysis revealed that the phosphaalkene unit is incorporated at the C4-position (vide infra), rather than the anticipated C7-position of 1. Notably, compound 3′ underwent isomerization under ambient light, yielding an almost 1:1 mixture of E- and Z-isomers over six hours, as determined by 31P NMR spectroscopy, with the Z-isomer exhibiting a resonance at 253.2 ppm (Fig. S7, SI). A similar ambient light-induced isomerization process has been previously reported for phosphaalkenes derived from the meta-terphenyl scaffold.24 In the present benzanthracene system, both facile E/Z-isomerization and the possibility of substitution at up to seven distinct positions (Fig. 1C) significantly complicate the purification process. Following the isolation of crystals of compound 3′, further crystallization from the mother liquor yielded crystals of a second regioisomer, 3″. Solid-state structural analysis of 3″ indicated incorporation of the phosphaalkene unit at the C6-position (Fig. S10, SI). Although the crystal quality did not permit a detailed analysis of bond metrics, bond connectivity was unambiguously established. The solid-state structure of 3′ revealed a planar benzanthracene core (Fig. 3B), with a PC bond length of 1.714(1) Å and a C–PC bond angle of 105.41(6)°, which are in excellent agreement with values reported for related phosphaalkene derivatives.25–27
 | ||
| Fig. 3 (A) Synthesis of benzanthracene-based phoshphaalkene. Conditions: (i) n-BuLi, −78 °C; (ii) Mes*PCl2, −78 °C, THF; (iii) DBN, r.t.; (B) solid-state structure of E-3′. Thermal displacement ellipsoids are set at 50% probability levels, and Mes* unit is shown as wireframe for clarity. | ||
To rationalize the observed regioselectivity, we systematically examined the energetic and orbital variations associated with counterion positions above 1− using computational methods.28 The relative energies of the Li+-bound anions were calculated in the gas phase, with continuum solvation (THF-PCM), and using explicit solvation models, and compared with K+-bound anions. The computed energy differences varied notably between models. The free-energy differences were consistently smaller for K+, consistent with its larger ionic radius and weaker K+–arene interaction. In all cases, rings C and D were least favoured (Table S3, SI). For K+, all methods indicated a slight preference for ring A, albeit by only 0.1 (g.p.), 1.1 (THF-PCM), and 0.5 (2× THF) kJ mol−1. In contrast, Li+ exhibited more pronounced preferences: the continuum model favoured binding over ring B (ΔG ≈ 2 kJ mol−1), while gas-phase and explicitly solvated models favoured ring A by 9.5 and 6.0 kJ mol−1, respectively. Despite the relatively short Li+–arene distances, the highest molecular orbitals exhibited only minor bias toward the ring over which the cation was positioned (Fig. S16 SI), precluding strong orbital-controlled stereoselectivity. On the other hand, Mulliken charges vary substantially depending on the cation location (Fig. S17 SI).
These results indicate that the benzanthracenyl anion is inherently amenable to electrophilic functionalization. However, the small differences in electronic structure and the shallow potential energy surface associated with counterion positioning might still provide control over stereoselectivity. As such, product formation could be both kinetically and thermodynamically controlled, resulting in a complex reactivity landscape for this deceptively simple PAH fragment.
To experimentally validate the computational findings and assess whether temperature and cation identity could be utilized to steer product selectivity, we conducted reactions under varying conditions using both lithium and potassium bases. Neither LiHMDS nor KHMDS effectively deprotonated 1 at low temperatures, preventing direct comparison with the low-temperature reactivity of n-BuLi; hence, we opted to study the silazide bases at room temperature. In contrast to the low-temperature n-BuLi reaction, deprotonation of 1 with LiHMDS or KHMDS followed by treatment with Mes*PCl2 produced chlorophosphine intermediates with 31P NMR resonances at 76.8, 75.6, and 74.7 ppm as the major peaks (Table S1 and Fig. S8, SI for further details). Upon addition of DBN, both LiHMDS and KHMDS reactions yielded a similar distribution of phosphaalkene species with compounds exhibiting 31P NMR resonance of 256.5 and 255.1 ppm appeared as the predominant products. These results indicate that temperature significantly influence Li migration, leading to distinct product distributions when using Li bases; however, at room temperature, both Li and K bases resulted in similar product distributions. Based on the experimental observations of different chlorophosphine intermediates, we infer that the regioselectivity is determined at the initial step rather than a result of subsequent sigmatropic P-migration.
In order to explore the impact of the phosphalkene incorporation on the electronic structure, we recorded the UV-vis spectrum of the E/Z mixture of 3′. The spectrum features two prominent low-energy absorptions at 455 and 475 nm, together with a band at 340 nm (Fig. S13, SI). The low energy transitions are in the expected range for phosphaalkenes conjugated with large(r), coplanar aromatic substituents, involving various π–π* transitions, with the exocyclic PC units act as a strong acceptor motif. In comparison, the phosphine oxide 2 shows the typical benzanthracene bands around 350 nm alongside a featureless low energy transition (ca. 410 nm), illustrating the superior acceptor character and excellent conjugation achieved by phosphaalkene functionalization.
The redox behaviour of the phosphaalkene was examined by cyclic voltammetry in THF/[nBu4N][PF6]. A mixture of E/Z-3′ shows two reversible reductions at E1/2 = –1.20 V and –1.35 V vs. Ag/Ag+ (Fig. 4B), which are attributed to the independent phosphaalkene/phosphaalkenyl radical couples of the E- and Z-isomers. The configurationally induced differences in their electronic environments are consistent with the LUMO energy difference of 0.1 eV for the two isomers (vide infra). Both reduction potentials are less negative than those reported for fluorenyl-based phosphaalkene, reflecting the extended conjugation of the benzanthracene core and the increased acceptor nature of the systems.26
 | ||
| Fig. 4 (A) Highest occupied molecular orbitals of E-3′. (B) Cyclic voltammogram of compound E-/Z-3′ (2 mM in THF, scan rate 50 mV S−1). For further details see SI. | ||
Finally, we explored how phosphaalkene substitution at different positions influences the electronic structure of the benzanthracene framework using DFT calculations. Additionally, we examined whether the E- and Z-isomers exhibit significant differences in their frontier molecular orbital energies (Fig. 4A and Table S4; SI). The optimized structure of E-3′ (functionalized at C4) aligns closely with the experimental solid-state geometry, featuring an almost planar carbon backbone and a slight out-of-plane distortion of the exocyclic PC moiety (PC–C–C torsion = 175.5°). The corresponding Z-isomer displays a marginally greater distortion (168.7°). Both isomers exhibit delocalized frontier orbitals spanning the benzanthracene core and the PC bond, with HOMO–LUMO gaps of 6.44 eV (E) and 6.54 eV (Z). Interestingly, this difference arises primarily from the shift in the LUMO energy. In contrast, the regioisomers E-/Z-3 exhibit significantly greater distortion of the carbon framework, resulting in pronounced out-of-plane twisting of the PC bond, reflected by torsion angles of 136.1° and 141.1° for the E- and Z-3 isomers, respectively. Yet, the PC bond lengths remain remarkably consistent, while the HOMO–LUMO gaps increase to 7.17 eV and 7.10 eV for the E- and Z-3 isomers, respectively. The C6-substituted isomer 3″ exhibits only minor structural distortion, with HOMO–LUMO gaps of 6.56 eV (E) and 6.51 eV (Z), values closely matching those of 3′.
In summary, we have established that the benzanthracene scaffold supports electrophile-dependent, site-selective incorporation of heavier main-group fragments. While the phosphine oxide was predominantly installed at the desired C7-position, employing standard phosphaalkene synthetic protocols led to predominant incorporation at the C4-position. The contrasting behaviour highlights the sensitivity of this π-extended framework to the nature of the electrophile and to the underlying charge distribution of the benzanthracenyl anion. Systematic variation of the base and reaction temperature, supported by DFT calculations, reveals that both kinetic and thermodynamic factors contribute to the observed product distribution. The shallow energy landscape arising from counterion migration and cation–π interactions renders several accessible reaction sites, with the steric and electronic characteristics of the P-electrophile ultimately steering the observed selectivity.
Despite the growing interest in exploring unique PAH scaffolds for main-group functionalization to access tailored electronic properties and reactivity, the benzanthracene framework has remained largely unexplored. The present study provides new insights into the reactivity of the benzanthracenyl anion toward heavier main-group halides, revealing an electrophile-dependent reactivity pattern within the core. These findings highlight the delicate interplay between cation position and charge distribution that governs site selectivity. Given the inherent electronic and structural complexity of the benzanthracenyl anion, our results advance the understanding of main-group functionalization in extended π-systems, where cation migration plays a crucial role, and pave the way for the design of next-generation main-group–modified polycyclic aromatic hydrocarbons.
Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the supplementary information (SI). Supplementary information: experimental details, spectral data and cartesian coordinates for the DFT-optimised geometries. See DOI: https://doi.org/10.1039/d5cc05878c.CCDC 2465161 and 2465162 contain the supplementary crystallographic data for this paper.29a,b
Acknowledgements
The authors thank the Swedish Research council (A. O. project 2017-03727, 2021-03658), and Olle Engkvists Stiftelse for their financial support. The computations were enabled by resources in project NAISS 2023/5-355 provided by the National Academic Infrastructure for Supercomputing in Sweden (NAISS) at UPPMAX, funded by the Swedish Research Council through grant agreement no. 2022-06725.Notes and references
- R. Szucs, P. A. Bouit, L. Nyulaszi and M. Hissler, ChemPhysChem, 2017, 18, 2618–2630 CrossRef CAS PubMed.
- F. Vidal and F. Jäkle, Angew. Chem., Int. Ed., 2019, 58, 5846–5870 CrossRef CAS PubMed.
- T. Delouche, M. Hissler and P.-A. Bouit, Coord. Chem. Rev., 2022, 464, 214553 CrossRef CAS.
- R. Deka, S. Chattopadhyay and A. Orthaber, Dalton Trans., 2025, 54, 3113–3117 RSC.
- R. Deka, M. A. Ansari, S. Chattopadhyay, R. Lomoth, A. Thapper and A. Orthaber, Angew. Chem., Int. Ed., 2024, 63, e202406076 CrossRef CAS PubMed.
- F. Riobé, R. Szűcs, P.-A. Bouit, D. Tondelier, B. Geffroy, F. Aparicio, J. Buendía, L. Sánchez, R. Réau, L. Nyulászi and M. Hissler, Chem. – Eur. J., 2015, 21, 6547–6556 CrossRef PubMed.
- E. Regulska and C. Romero-Nieto, Dalton Trans., 2018, 47, 10344–10359 RSC.
- A. Belyaev, B.-K. Su, Y.-H. Cheng, Z.-Y. Liu, N. M. Khan, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2022, 61, e202115690 CrossRef CAS PubMed.
- A. Belyaev, Y.-H. Cheng, Z.-Y. Liu, A. J. Karttunen, P.-T. Chou and I. O. Koshevoy, Angew. Chem., Int. Ed., 2019, 58, 13456–13465 CrossRef CAS PubMed.
- M. A. Shameem, J. A. L. Wells, A. K. Gupta and A. Orthaber, Chem. – Eur. J., 2023, 29, e202300563 Search PubMed.
- M. A. Ansari, R. Deka, A. Thapper and A. Orthaber, Angew. Chem., Int. Ed., 2024, 64, e202415684 CrossRef PubMed.
- B. Lu, J. Xu, C. Fan, H. Miao and L. Shen, J. Phys. Chem. B, 2009, 113, 37–48 Search PubMed.
- M.-X. Yu, L.-C. Chang, C.-H. Lin, J.-P. Duan, F.-I. Wu, I.-C. Chen and C.-H. Cheng, Adv. Funct. Mater., 2007, 17, 369–378 CrossRef CAS.
- I. Grabchev, I. Moneva, E. Wolarz and D. Bauman, Dyes Pigm., 2003, 58, 1–6 CrossRef CAS.
- G. Gorbenko, V. Trusova, E. Kirilova, G. Kirilov, I. Kalnina, A. Vasilev, S. Kaloyanova and T. Deligeorgiev, Chem. Phys. Lett., 2010, 495, 275–279 CrossRef CAS.
- R. Umeda, T. Namba, T. Yoshimura, M. Nakatsukasa and Y. Nishiyama, Tetrahedron, 2013, 69, 1526–1531 CrossRef CAS.
- M. Debeaux, K. Brandhorst, P. G. Jones, H. Hopf, J. Grunenberg, W. Kowalsky and H.-H. Johannes, Beilstein J. Org. Chem., 2009, 5, 31 Search PubMed.
- C. F. H. Allen, Can. J. Chem., 1973, 51, 382–387 Search PubMed.
- C. F. H. Allen and S. C. Overbaugh, J. Am. Chem. Soc., 1935, 57, 740–744 Search PubMed.
- A. Decken, C. J. Carmalt, J. A. C. Clyburne and A. H. Cowley, Inorg. Chem., 1997, 36, 3741–3744 CrossRef CAS PubMed.
- K. Müllen, Angew. Chem., Int. Ed. Engl., 1987, 26, 204–217 CrossRef.
- M. A. Hempenius, C. Erkelens, P. P. J. Mulder, H. Zuilhof, W. Heinen, J. Lugtenburg and J. Cornelisse, J. Org. Chem., 1993, 58, 3076–3084 CrossRef CAS.
- J. T. M. van Dijk, B. J. van de Panne, A. C. Bleeker, J. Lugtenburg and J. Cornelisse, Tetrahedron, 1996, 52, 2647–2662 Search PubMed.
- V. B. Gudimetla, A. L. Rheingold, J. L. Payton, H.-L. Peng, M. C. Simpson and J. D. Protasiewicz, Inorg. Chem., 2006, 45, 4895–4901 CrossRef CAS PubMed.
- A. El Nahhas, M. A. Shameem, P. Chabera, J. Uhlig and A. Orthaber, Chem. – Eur. J., 2017, 23, 5673–5677 CrossRef CAS PubMed.
- Y. V. Svyaschenko, A. Orthaber and S. Ott, Chem. – Eur. J., 2016, 22, 4247–4255 CrossRef CAS PubMed.
- R. Deka, J. Steen, M. F. Hilbers, W. G. Roeterdink, A. Iagatti, R. Xiong, W. J. Buma, M. Di Donato, A. Orthaber and S. Crespi, Angew. Chem., Int. Ed., 2025, 64, e202419943 Search PubMed.
- J. S. Lee, S. A. Krasnokutski and D.-S. Yang, J. Chem. Phys., 2011, 134 Search PubMed.
- (a) CCDC 2465161: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nr6bz; (b) CCDC 2465162: Experimental Crystal Structure Determination, 2025 DOI:10.5517/ccdc.csd.cc2nr6c0.
| This journal is © The Royal Society of Chemistry 2026 |